

Abstract
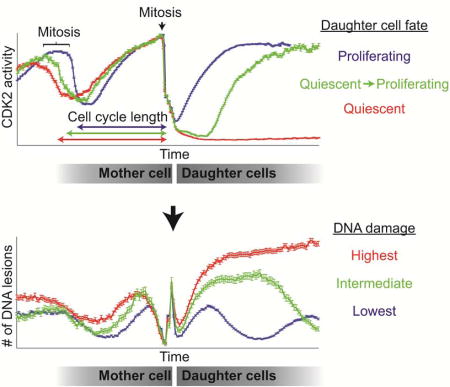
Mammalian cells have two fundamentally different states – proliferative and quiescent – but our understanding of how and why cells switch between these states is limited. We previously showed that actively proliferating populations contain a subpopulation that enters quiescence (G0) in an apparently stochastic manner. Using single-cell time-lapse imaging of CDK2 activity and DNA damage, we now show that endogenous replication stress in the previous (mother) cell cycle prompts p21-dependent entry of daughter cells into quiescence immediately after mitosis. Furthermore, the amount of time daughter cells spend in quiescence is correlated with the extent of inherited damage. Our study thus links replication errors in one cell cycle to the fate of daughter cells in the subsequent cell cycle. More broadly, this work reveals that entry into quiescence is not purely stochastic but has a strong deterministic component arising from a memory of events that occurred in the previous generation(s).
Arora et al. find that unresolved DNA replication errors in mother cells are passed on to daughter cells, prompting entry of daughter cells into a temporary quiescence whose duration is correlated with the extent of inherited damage. The authors thereby uncover a key source of heterogeneity in cell-cycle duration.
INTRODUCTION
In mammalian systems, the reversible switch between proliferation and quiescence is a vital component of tissue homeostasis. Quiescence is a non-proliferative state from which cells can emerge and resume proliferation once they receive appropriate cues. Pathways controlling the proliferation-quiescence decision are defective in most, if not all, human tumors (Hanahan and Weinberg, 2000), demonstrating the importance of this key decision point. Despite this, our understanding of how and why cells switch between proliferation and quiescence is limited.
Quiescence, also referred to as G0, is stimulated when cells are faced with unfavorable environmental conditions such as growth factor deprivation or loss of adhesion (Coller et al., 2006). During quiescence, Cyclin-Dependent Kinase (CDK) activity is turned off and the retinoblastoma protein (Rb) is un- or hypo-phosphorylated. In this state, Rb inhibits cell-cycle progression by binding to E2F and inhibiting its transcriptional activity. Cells exiting quiescence commit to cell-cycle re-entry at the so-called Restriction Point, after which the cell cycle progresses independently of mitogen stimulation (Pardee, 1974; Zetterberg and Larsson, 1985). Stimulation of mitogen-starved quiescent cells causes activation of Cyclin D/CDK4/6, which initiates phosphorylation of Rb, leading to activation of E2F–mediated transcription. Cyclin E, whose transcription is stimulated by E2F, forms a complex with CDK2 to further phosphorylate Rb, establishing a positive-feedback loop and passage through the Restriction Point (Massague, 2004; Trimarchi and Lees, 2002). In contrast, knowledge of the control mechanisms governing entry into quiescence is limited, in large part due to the lack of tools for identifying quiescent cells in a mixed population, and the difficulty of distinguishing them from cells experiencing a G1 or G1/S checkpoint arrest.
We previously established a non-transformed human mammary epithelial cell line (MCF10A) stably expressing a CDK2 activity sensor (Figure S1A) and a Histone 2B nuclear marker (Spencer et al., 2013). Using time-lapse imaging and custom MATLAB scripts to track CDK2 activity in thousands of cells through several cell cycles, we previously identified divergent cycling behavior in multiple types of mammalian cells. While CDK2 activity steadily increases after mitosis in a majority of newly born cells (“CDK2 cells”), a subset of cells lack CDK2 activity and enter a transient quiescence (“CDK2low cells”), representing 20–30% of MCF10A cells in full growth media, ((Spencer et al., 2013) and Figure 1A, left). We define CDK2low cells as those having CDK2 activity < 0.55 for at least for 4 hr after mitosis, and refer to them as G0 or quiescent cells in this work. We ruled out the possibility that CDK2low cells are senescent as < 1% of asynchronously growing MCF10A cells stained positive for senescence associate β-galactosidase activity (whereas 20–30% of MCF10A cells are CDK2low; Figure S1B). Additionally, ~50% of the CDK2low population, or 10–15% of the total population, remained quiescent for a finite period and later emerged from quiescence by building up CDK2 activity to re-enter the cell cycle (hereafter we refer to these CDK2low→inc cells as CDK2emerge cells). Entry into the quiescent CDK2low state was dependent on increased levels of the CDK inhibitor, p21 (Figure 1A, center), since p21−/− MCF10A cells rarely entered the CDK2low (or the CDK2emerge) state (Spencer et al., 2013). As all cells in the population are exposed to the same growth and nutrient conditions, the root cause of this apparently stochastic, spontaneous entry into quiescence remains unknown.
Figure 1. Immunofluorescence reveals a correlation between low phospho-Rb, high p21, and the presence of 53BP1 foci.

A. Single-cell traces of CDK2 activity aligned computationally to the time of anaphase (left panel). Traces were colored red if the CDK2 activity (Cytoplasmic/nuclear ratio of the sensor) was below 0.55 at 4 hr after anaphase and through the end of the movie (CDK2low) and were colored green if the CDK2 activity was ≤ 0.55 at 4 hr after anaphase but rose above 0.55 thereafter (CDK2low→inc, hereafter referred to as CDK2emerge); otherwise traces were colored blue (CDK2inc). At the end of the imaging period, p21 and phospho-Rb-Serine 807/811 levels were visualized by immunofluorescence and plotted as histograms for CDK2inc (blue) or CDK2low (red) cells. For simplicity, only 150 randomly selected CDK2 activity traces are shown in the left panel whereas the immunofluorescence data is aggregated across 10 replicate wells to obtain 743 (p21) or 751 (phospho-Rb) cells.
B. Immunofluorescence images of RPE cells labeled with EdU and then stained with Hoechst and the following antibody combinations: 1) anti-53BP1 with anti-phospho-Rb-S807/811 (top panel), 2) anti-53BP1 with anti-p21 (middle panel), and 3) anti-p21 with anti-phospho-Rb-S807/811 (bottom panel). For the top two panels, dashed ovals mark EdU-negative cells with 53BP1 foci whereas in the bottom panel, the dashed ovals mark cells that are EdU negative and stain exclusively for either p21 or phospho-Rb. Images are representative of three biological replicates. Scale bars are 50µm.
C. Example of automated detection of 53BP1 foci using a custom MATLAB script. Foci identified using the script are circled in yellow. A cell with foci that are below the threshold set for detection (see Supplementary Methods for details) is marked by a yellow arrow.
Proliferating cells routinely experience DNA damage arising from natural processes such as DNA replication or from reactive oxygen species generated during cell metabolism. DNA lesions, DNA-protein complexes (such as transcription machinery or DNA repair proteins), and DNA secondary structures can impair the progression of DNA replication forks (Zeman and Cimprich, 2014). If stalled replication forks fail to restart, or collapse, stretches of un-replicated DNA will accumulate (Durkin and Glover, 2007). Severe DNA replication stress can lead to activation of checkpoints and cause cells to arrest prior to mitosis (Errico and Costanzo, 2012; Jones and Petermann, 2012). However, it is becoming increasingly evident that local low-level replication stress does not always lead to checkpoint activation and cell-cycle arrest (Mankouri et al., 2013). In fact, cells can tolerate moderate levels of replication stress and proceed through mitosis even in the presence of partial checkpoint activation (Koundrioukoff et al., 2013). Transmission of the under-replicated regions of the genome through anaphase generates DNA lesions (El Achkar et al., 2005) which can be repaired by POLD3-dependent DNA synthesis in mitosis (Minocherhomji et al., 2015). A second backup system involves protection of the lesions by formation of 53BP1 nuclear bodies, visible by immunofluorescence as large bright foci in newly born daughter cells, until their repair (Harrigan et al., 2011; Lukas et al., 2011). Markedly, 53BP1foci are observed in 21% of non-transformed G1 cells (Harrigan et al., 2011), implying a higher rate of DNA replication errors than previously thought. Indeed, one or two stalled forks are expected to occur in every S phase in HeLa cells (Al Mamun et al., 2016; Moreno et al., 2016). What is the consequence, if any, on the subsequent cell cycle given this high transmission of replication errors from mothers to daughters?
Since the fraction of newly born cells with 53BP1 foci (21%; (Harrigan et al., 2011)), bears a striking similarity to the fraction of newly born MCF10A cells passing through the CDK2low state (20–30%; (Spencer et al., 2013)) we set out to test the hypothesis that unresolved replication stress inherited from the previous cycle is a major cause of entry into the quiescent CDK2low state after mitosis. Using live-cell sensors for cell-cycle progression and DNA damage combined with time-lapse imaging and single-cell tracking, we find that cells ‘spontaneously’ entering quiescence indeed have increased DNA damage, and that the length of time spent in quiescence is related to the extent of inherited damage. We further show that this replication stress-associated entry into quiescence is dependent on p21 and that addition of low-level replication stress increases the fraction of quiescent daughter cells. Thus, we have uncovered a deterministic basis for what previously appeared to be stochastic entry of a subset of cells into a temporary quiescence following mitosis. In doing so, we identify a key source of heterogeneity in G1 and cell-cycle durations.
RESULTS
Cells that ‘spontaneously’ enter quiescence have markers of DNA lesions
Due to the well-documented role of p21 in arresting the cell cycle in response to DNA damage, we hypothesized that the ‘spontaneously’ quiescent CDK2low cells might have inherited DNA damage from the previous cell cycle. To test this hypothesis, we first examined the correlation between endogenous (naturally occurring) DNA lesions and spontaneous quiescence in three non-transformed human cell lines: MCF10A cells, Hs68 foreskin fibroblasts, and RPE-hTERT retinal epithelial cells. Using unperturbed, asynchronously cycling cells, we assessed the presence of DNA lesions by analyzing 53BP1 localization. 53BP1, which is a widely accepted marker of DNA lesions, forms distinct foci at sites of unrepaired DNA lesions (Anderson et al., 2001; Rappold et al., 2001; Schultz et al., 2000). Preliminary visual analysis revealed the presence of large, high intensity 53BP1 foci (often referred to as “nuclear bodies”) in 15–20% of G1 cells, in accordance with prior observations (Harrigan et al., 2011). Given that CDK2low cells exit mitosis with high levels of p21 and low levels of phospho-Rb ((Spencer et al., 2013) and Figure 1A center and right), the CDK2low quiescent subpopulation can be identified via fixed-cell immunofluorescence as the phospho-Rblow/p21high subpopulation. Presence of the large 53BP1 foci correlated strongly with increased levels of p21 and reduced Rb phosphorylation (top and middle panel, Figure 1B). Cells with high p21 levels also had reduced phosphorylation of the Rb protein (bottom panel, Figure 1B). These cells had a 2N DNA content and were EdU-negative (not in S phase), revealing that they were in G0 or G1, and thus that the 53BP1 foci were likely a result of replication stress inherited from the previous cell cycle, rather than due to damage associated with ongoing DNA replication.
To further quantify this observation, we developed custom MATLAB scripts to detect the number of 53BP1 foci per cell in thousands of single cells, along with the levels of Rb phosphorylation and total p21 protein. To detect only the replication stress-associated 53BP1 foci in G0/G1, we set the foci detection parameters such that only large 53BP1 “nuclear bodies” were detected (Figure 1C, yellow circles), as opposed to other tiny foci (Figure 1C, yellow arrow). Both phospho-Rb and p21 signals in G0/G1 cells (2N DNA content, EdU-negative; Figure S2) are strongly or moderately bimodally distributed, respectively, indicating two subpopulations among 2N DNA content cells (Figures 2A and 2B, top row, black histograms). About 20–30% of MCF10A cells with 2N DNA content had high p21 and low phospho-Rb, comparable to the 20–30% CDK2low we typically observe by time-lapse microscopy using the CDK2 sensor (Spencer et al., 2013). Strikingly, when we examined the subset of 2N DNA content cells containing one-or-more 53BP1 foci, we found that this subset of cells was dramatically enriched for high levels of p21 (46–76% p21high depending on the cell line; Figures 2A, bottom row, red histograms, and 2C, red bars) and low phospho-Rb (55–66% phospho-Rblow depending on the cell line; Figures 2B, bottom row, red histograms, and 2D, red bars). In contrast, the subset of cells containing no 53BP1 foci had reduced levels of p21 and increased levels of phospho-Rb (Figures 2A and 2B, middle rows, blue histograms, and 2C and 2D, blue bars).
Figure 2. Cells with 53BP1 foci tend to be in a phospho-Rblow/p21high/CDK2low state, and phospho-Rblow/p21high cells tend to have 53BP1 foci.

A & B. Histograms showing bimodal distribution of p21 or phospho-Rb-S807/811 in G0/G1 MCF10A (left), Hs68 (center) or RPE-hTERT (right) cells with either no (“53BP1 foci”, blue) or at least one 53BP1 focus (“53BP1 foci+”, red). The distribution of the phospho-Rb and p21 signal in all G0/G1 cells is shown for comparison (black). Data shown are aggregated from four (MCF10A) or eight (Hs68 & RPE) replicate wells to obtain well-populated histograms. The number of cells analyzed for p21 histograms are 25,817 (MCF10A), 25,366 (Hs68) and 20,829 (RPE) and for the phospho-Rb histograms are 28,249 (MCF10A), 22,708 (Hs68) and 19,579 (RPE). Dashed green lines indicate the cutoffs for p21high and phospho-Rblow. For p21, the cutoff was based on the distribution seen in S-phase cells (p21 is degraded in S-phase and hence all S-phase cells are p21low, data not shown). For phospho-Rb, the cutoff was based on the approximate “saddle” point between the two peaks observed in the “All G0/G1” histogram. Percentage of cells to the right or left of the dashed green lines are indicated.
C & D. Probability of a cell being in the CDK2low state given that it has DNA lesions. G0/G1 cells were first classified as 53BP1+ (red) or 53BP1 (blue) based on the presence or absence of 53BP1 foci, followed by calculation of the fraction of cells with high p21 or low phospho-Rb based on the cutoffs marked by dashed lines in (A) and (B).
E & F. Probability of a cell having DNA lesions given that it is in the CDK2low state. G0/G1 cells were first classified as p21high (red) or p21low (blue), or phospho-Rblow (red) or phospho-Rbhigh (blue) based on the cutoffs marked by dashed lines in (A) and (B), followed by calculation of the fraction of cells with at least one 53BP1 focus.
Since hypo-phosphorylation of Rb can be used as marker for the CDK2low state, we also determined the converse – the fraction of cells with hypo-phosphorylated Rb that also harbor 53BP1 foci. We find that the probability that a cell has DNA lesions given that it is in the quiescent CDK2low state (P(DNA lesions | CDK2low)) is 0.57, 0.34, and 0.54, using phospho-Rb data from MCF10A, Hs68, and RPE cells, respectively (Figure 2E and 2F)). Thus, averaging across the three cell types, 53BP1 foci are present in 48% of the cells passing through the quiescent CDK2low state.
Single-cell tracking of CDK2 activity and DNA lesions reveals that damage in mother cells directs the proliferation-quiescence decision of daughter cells
To determine a direct temporal correlation between endogenously arising DNA lesions and the fate (G0/CDK2low or G1/CDK2inc) a cell takes after mitosis, we established a three-color MCF10A cell line expressing an mCherry-tagged fragment of the 53BP1 protein (mCherry-BP1) (Dimitrova et al., 2008) along with the mVenus-tagged CDK2 activity sensor (DHB-Ven) and the nuclear marker, H2B–mTurquoise (Figure 3A). The mCherry-BP1 fragment consists of amino acids 1220–1711 of the 53BP1 protein, lacking most of the functional domains of 53BP1, and was shown to not affect DNA damage repair in cells expressing the fusion protein (Dimitrova et al., 2008). This setup allows us to simultaneously monitor cell-cycle progression (or lack thereof) and the presence of DNA lesions. Consistent with our fixed cell data, we observed large mCherry-BP1 foci in ~30% of untreated cells.
Figure 3. Live-cell imaging of mCherry-BP1 foci and CDK2 activity in MCF10A cells.

A. Time-lapse images of CDK2 activity (DHB-Ven) and a marker of DNA lesions (mCherry-BP1) in a CDK2low (left) or CDK2inc (right) MCF10A cell. The first image (0 hr) represents the time of metaphase. Scale bars are 10µm.
B. Examples of single-cell traces of CDK2 activity (green, left y-axis) and number of mCherry-BP1 foci (red, right y-axis) detected during the 48 hr time-lapse imaging. Black circles denote the time of anaphase. Top panel: CDK2low cells; bottom panel: CDK2inc cells. The leftmost panels represent traces of cells shown in Figure 3A.
Using time-lapse imaging, we tracked CDK2 activity and the number of 53BP1 foci in 5700 individual MCF10A cells for 48 hr. Preliminary observation showed that MCF10A cells taking a CDK2inc path showed few 53BP1 foci after mitosis, whereas 53BP1 foci in the CDK2low cells were detectable for as long as the cells remained quiescent (Figures 3A and 3B). Note that while all four of the CDK2low cells shown in Figure 3B have mCherry-BP1 foci after mitosis, significant foci are present before mitosis in two of the four cells, suggesting that DNA lesions can already exist during the previous G2 but may also first arise during mitosis.
To quantify this observation, we first computationally sorted the cells based on the average number of mCherry-BP1 foci in a 4 hr window after mitosis (G0 or G1 phase of the cell cycle). We then determined the average CDK2 activity of the top 5% and bottom 5% of cells from this list (orange and cyan traces respectively, right panel, Figure 4A). The cells at the bottom 5% of the list had zero mCherry-BP1 foci during this window, thus representing cells without DNA lesions, whereas the top 5% are cells with the largest number of DNA lesions during this window. Cells in the top 5% of mCherry-BP1 foci (Figure 4A, top right panel) had significantly lower average CDK2 activity after mitosis as compared to cells without any foci (Figure 4A, bottom right panel). Indeed, among cells in the top 5% of mCherry-BP1 foci, 62% were CDK2low, whereas among cells in the bottom 5% of mCherry-BP1 foci, only 24% were in the CDK2low state (Figure 4B). Similarly, the top 5% of cells with mCherry-BP1 foci in the previous S phase or the previous G2 phase (Figure 4A top center and middle panels) also had significantly lower average CDK2 activity during these windows as well as after mitosis (Figure 4A bottom center and middle panels) and were more likely to be in the quiescent CDK2low state after mitosis (Figure 4B). These results show that replication errors or DNA lesions in the mother cell cycle increase the likelihood that daughter cells enter the quiescent CDK2low state after mitosis.
Figure 4. Daughter cells that enter the CDK2low or CDK2emerge state after mitosis have increased mCherry-BP1 foci and arise from mothers with a prolonged cell cycle and increased foci.

A. Cells were first sorted computationally based solely on the average number of mCherry-BP1 foci during a specified time window (gray vertical bar), and then the average CDK2 activity was calculated for the top 5% and bottom 5% of the cells in that list. Top panels show the average number of mCherry-BP1 foci, and bottom panels show average CDK2 activity during the specified cell cycle phase: i) −6 to −11 hr before anaphase (~S phase, left panel) ii) −1 to −6 hr before anaphase (~G2 phase, middle panel) and iii) 1 to 5 hr after anaphase (~G0/G1 phase, right panel). Error bars represent standard error of mean, where n=225 (S-phase), 253 (G2), or 275 (G1) cells in the bottom 5th and top 5th percentile for the indicated time-window; data are aggregated from 16 replicate wells. The H-value plot (purple, bottom panel) indicates time points where the CDK2 activity is significantly different (H=1) between cells in the top 5% and bottom 5% of mCherry-BP1 foci, based on a two sample t-test with a p-value of 0.05.
B. Percent of cells that enter the CDK2low state among cells in the top 5% or bottom 5% of mCherry-BP1 foci as classified as in (A). Error bars represent standard error of the mean based on the number of cells in (A).
C. Cells were first classified as CDK2inc (blue), CDK2emerge 4–7hr (cyan), CDK2emerge 7–10hr (green),or CDK2low (red) based on their CDK2 activity after anaphase, regardless of the number of mCherry-BP1 foci. Traces were classified as CDK2low if the CDK2 activity (Cytoplasmic/nuclear ratio of the sensor) was ≤ 0.55 at 4 hr after anaphase and for the remainder of the imaging period. Traces were classified as CDK2emerge 4–7hr or CDK2emerge 7–10hr if the CDK2 activity was ≤ 0.55 at 4 hr after anaphase and rose above 0.55 4–7 hr or 7–10 hr after anaphase, respectively; otherwise traces were classified as CDK2inc. Note that although only cells emerging from the CDK2low state 4–7 hr or 7–10 hr after anaphase are included here, cells can emerge from the CDK2low state at all different times, ranging from several hours to several days (Spencer et al., 2013). For simplicity, only 150 randomly selected traces from each subpopulation are shown.
D. Median CDK2 activity of the four subpopulations described in (C). Error bars represent standard error of the mean where n=2387 CDK2inc cells, 778 CDK2emerge 4–7hr cells, 455 CDK2emerge 7–10hr cells, 1082 CDK2low cells, derived from aggregated data from 16 replicate wells.
E. For cells in each group described in (C), the average number of mCherry-BP1 foci per cell was calculated at each frame of the movie and plotted relative to time of anaphase. The estimated cell cycle phases are depicted at the bottom, based on previously reported lengths of each cell-cycle phase in MCF10A cells (Spencer et al., 2013). Note that the mCherry-BP1 signal is unreliable during mitosis due to cell rounding and chromosomal condensation. Error bars represent standard error of the mean as in (D).
F. Overlay of the median CDK2 activity (black trace) and mean number of mCherry-BP1 foci (cyan or green trace) in the CDK2emerge 4–7hr and CDK2emerge 7–10hr cells depicted in (C). The time that cells spend in the quiescent CDK2low state prior to emerging is marked in black as “G0”. The vertical dashed lines represent the time at which the average number of foci per cell begins to fall. Error bars represent standard error of the mean as in (D) and (E).
As an additional test of the relationship between 53BP1 foci and CDK2 activity, we first classified the cells based on the CDK2 activity after mitosis, regardless of the number of the 53BP1 foci (Figure 4C and Figure S3B). Strikingly, the average mother cell-cycle length for CDK2low daughters was longer than the average mother cell-cycle length of CDK2emerge daughters, which was longer than the average mother cell-cycle length of CDK2inc daughters (Figure 4D). We then calculated the average number of 53BP1 foci in the CDK2inc and CDK2low subpopulations, as well as in two selected populations of CDK2emerge cells that remained quiescent for a short period but later emerged from quiescence either 4–7 hr or 7–10 hr after mitosis (cyan and green traces, respectively, Figure 4C–E and Figure S3D). In all four subpopulations of cells, the average number of foci dropped during S-phase of both the mother and daughter cell cycles, presumably due to repair of the existing foci from the preceding G1. A false spike in the mCherry-BP1 signal is observed around the time of anaphase due to inaccurate detection of BP1 foci resulting from cell rounding and chromosomal condensation during mitosis. CDK2low cells had much higher and more persistent mCherry-BP1 foci relative to the CDK2inc cells after mitosis (Figure 4E). Similar results were also observed in RPE-hTERT cells expressing the CDK2 sensor, mCherry-BP1, and H2B–mTurquoise (Figure S3). CDK2emerge cells had intermediate numbers of mCherry-BP1 foci, which gradually resolved after the CDK2 activity started rising and the CDK2emerge cells re-entered the cell cycle (Figure 4F). Thus, foci are not resolved while cells are in the quiescent CDK2low state, but only after cells are in late G1 or S phase. Together, these results support the hypothesis that cells inheriting DNA replication stress from the previous cell cycle enter a quiescent CDK2low state after mitosis. Furthermore, comparison of the number of inherited foci in cells that emerge from the CDK2low state early, later, or not at all (CDK2emerge 4–7hr, CDKemerge 7–10hr, CDK2low, respectively) reveals that the timing of escape from quiescence is correlated with the extent of inherited replication stress.
Notably, mothers of CDK2low and CDK2emerge daughters also showed higher number of mCherry-BP1 foci, as compared to mothers of CDK2inc daughters (Figure 4E, negative x-axis values). Similar results were also seen in RPE-hTERT cells (Figure S3). This difference was especially prominent during the mother cell S and G2 phase (~ −1 to −10 hr) and to a lesser extent in the mother cell G1 (~ −10hr and earlier). Given that mothers of CDK2low and CDK2emerge daughters had a longer cell cycle as compared to mothers of CDK2inc daughters (Figure 4D), it is likely that mothers of CDK2low and CDK2emerge daughters also passed through the CDK2low state themselves when they were born. Furthermore, given the demonstrated correlation between inheritance of DNA lesions and passage through the CDK2low state, we can further speculate that these slow-cycling mother cells inherited replication stress from the grandmother generation. Taken together, these results suggest that cells that enter quiescence after mitosis not only begin the new cell cycle with higher amounts of replication stress-related DNA damage, but also had increased levels of DNA lesions/replication stress in the previous cell cycle.
Since CDK2low cells showed higher numbers of mCherry-BP1 foci in the previous G2, we next asked if signaling events leading to entry into quiescence after mitosis could already be detected in the previous G2. To do this, we quantified the distribution of p21 and phospho-Rb in G2/M cells (4N DNA, EdU-negative, Figure S2) and found, surprisingly, that a subpopulation of cells with high p21 and low phospho-Rb was already discernible at the end of the previous cell cycle (Figures 5A and 5B, black histograms).
Figure 5. G2/M cells with DNA lesions have increased p21 and reduced Rb phosphorylation.

A & B. Histograms showing distribution of p21 or phospho-Rb-S807/811 in G2/M MCF10A (left) or Hs68 (right) cells with either no (“53BP1 foci”, blue) or at least one 53BP1 focus (“53BP1 foci+”, red). The distribution of the phospho-Rb and p21 signal in all G2/M cells is shown for comparison (black). Dashed vertical green lines denote the cutoff for calculating the percent p21high or phospho-Rblow as in Figure 2; the percentage of cells to the right or left of the green lines is indicated. The number of cells analyzed is the same as in Figure 2A and 2B.
C & D. Probability of a cell having increased p21 or reduced Rb phosphorylation given that it has DNA lesions in G2/M phase. G2/M cells were first classified as 53BP1+ (red) or 53BP1 (blue) based on the presence or absence of 53BP1 foci, followed by calculation of the fraction of cells with high p21 or low phospho-Rb.
E & F. Probability of a cell having DNA lesions given that it has high p21 or low Rb phosphorylation in G2/M phase. G2/M cells were first classified as p21high (red) or p21low (blue), or phospho-Rbhigh (blue) or phospho-Rblow (red), based on the cutoffs marked by dashed lines in (A) and (B), followed by calculation of the fraction of cells with at least one 53BP1 focus.
We next used immunofluorescence analysis of p21, phospho-Rb, and 53BP1 to determine whether p21 upregulation is enriched specifically in G2 cells with 53BP1 foci as compared to G2 cells without any detectable foci. While it is not possible to distinguish between G2 and M cells using only DNA content and EdU, we speculate that the majority of the cells identified as 53BP1 foci+ are in G2, as full length 53BP1 protein does not form foci on mitotic chromosomes (Giunta et al., 2010). As seen with G0/G1 cells, the subset of G2 cells containing one-or-more 53BP1 foci was enriched for high levels of p21 (58–64% p21high depending on the cell line; Figure 5A, red histograms, and 5C, red bars). In contrast, only 16–31% of the cells without any detectable 53BP1 foci had increased p21 levels (Figure 5A, blue histograms, and 5C, blue bars). Similar enrichment of cells with hypo-phosphorylated Rb was seen in cells with one-or-more 53BP1 foci (Figure 5B, red histograms, and 5D, red bars). Of the three cell lines that we tested, both MCF10A and Hs68 had comparable responses to the presence of 53BP1 foci during G2, while RPE cells showed only a modest upregulation of p21 and no change in Rb phosphorylation (Figure S4). We also determined the converse – the fraction of G2/M cells with increased p21 (p21high) or hypo-phosphorylated Rb (phospho-Rblow) that have one-or-more 53BP1 foci. About 21–45% of cells with high p21 levels and 50% of the cells with hypo-phosphorylated Rb were also positive for 53BP1 foci (Figure 5E and 5F, red bars). Together, the live-cell and immunofluorescence data from G2 cells argue that the signaling events leading to quiescence of daughter cells can be detected in at least a subset of mother cells.
Addition of low-level replication stress in the previous cell cycle causes entry into quiescence after mitosis
Previous work has demonstrated that the presence of large 53BP1 foci in G1 cells is due to replication errors from the previous S phase (Harrigan et al., 2011; Lukas et al., 2011). Furthermore, these studies showed that treatment with low-dose Aphidicolin, an inhibitor of replicative polymerases that mimics physiological fork stalling and replication stress (Glover et al., 1984), increases the fraction of cells with large 53BP1 foci, as well as the number of foci per cell. We therefore hypothesized that if endogenous unrepaired replication errors are indeed a trigger for entry into CDK2low state, introducing additional low-level replication stress by treatment with low-dose Aphidicolin (0.2–0.4µM) would promote entry of additional cells into the CDK2low state.
We first imaged asynchronous MCF10A cells for 12 hr in the absence of any drug, which allowed us to ascertain each cell’s cell-cycle stage at the time of treatment with Aphidicolin. We then added 0.4µM Aphidicolin and imaged for an additional 48 hr (Figure 6A). For analysis, we selected only those cells that were in G1 when Aphidicolin was added (i.e. CDK2inc cells that had undergone anaphase 2–4 hr prior to Aphidicolin treatment; Figure 6B). Addition of Aphidicolin in G1 did not affect G1 progression, but significantly slowed S phase progression (presumably due to slowing of replication fork progression). This can be seen as a plateauing of CDK2 activity for about 8–10 hr at CDK2 activity = 1, which we previously showed corresponds to the start of S-phase (Spencer et al., 2013). Consequently, the length of the ongoing cell cycle increased significantly (Figures S5A). As anticipated, this low-level replication stress was not a strong enough signal to block mitosis, as 98% of cells eventually underwent cell division (Supplementary movies S1 and S2). Strikingly, however, 84% of drug-treated cells (vs. 17% of untreated control cells) completing mitosis after receiving Aphidicolin in G1 of the previous cell cycle entered the quiescent CDK2low state immediately after completion of mitosis (red traces, Figure 6B).
Figure 6. Introduction of low-level replication stress causes entry into the CDK2low state after mitosis.

A. Experimental timeline. MCF10A cells were imaged for 12 hr prior to addition of DMSO or APH, then imaged for another 48 hr after drug addition, and then prepared for immunofluorescence.
B. & E. Single cell traces of CDK2 activity in wild-type or p21−/− MCF10A cells treated with DMSO or 0.4µM Aphidicolin (APH). Only cells that underwent anaphase in a 2–4 hr window prior to drug addition are included (n=390 (WT+DMSO), 115 (WT+APH), 356(p21−/− + DMSO), 125 (p21−/− + APH); data are aggregated from 6 replicate wells for each condition. A black vertical bar represents the time of drug addition. Traces were colored blue (CDK2inc) if the CDK2 activity was above the threshold (y=0.55) at 4 hr after the first mitosis (M) subsequent to drug addition; otherwise traces were colored red (CDK2low). The number of traces that could not be classified as either CDK2inc or CDK2low is shown in Figure S5D.
C. Percentage of MCF10A cells with at least one 53BP1 focus after 48 hr treatment with either DMSO or 0.4µM APH. Error bars represent standard error of mean. * denotes p-value of 0.009 calculated using two-tailed Student’s t-test with equal variance.
D. Histograms of p21 levels measured by immunofluorescence in cells fixed at the end of 48hr of imaging in the presence of DMSO (gray) or 0.4µM APH (red).
Cells that had already completed DNA replication and were in G2 phase at the time of Aphidicolin addition proceeded normally through mitosis and G1 (without entry into the CDK2low state) to the next S phase, where they were then temporarily delayed (Figure S5B). The vast majority of these cells nevertheless completed mitosis, and then immediately entered the quiescent CDK2low state (Figure S5B). This confirms that Aphidicolin has to be present during S phase in order to trigger entry into the quiescent CDK2low state after mitosis. Consistent with previous findings (Harrigan et al., 2011; Lukas et al., 2011), the fraction of daughter cells with replication stress and presenting with 53BP1 foci was significantly increased with Aphidicolin treatment (Figure 6C). We obtained similar results when we used low-dose Hydroxyurea (300µM) to induce replication stress (Figure S5C). These data suggest that low levels of replication stress that allow bypass of the surveillance machinery in mother cells are detected by newborn daughter cells, triggering entry into G0/quiescence.
p21 controls replication stress-associated entry into quiescence
Spontaneous entry into the CDK2low state in asynchronously cycling MCF10A cells is regulated by p21, and cells lacking p21 rarely enter the CDK2low state (Spencer et al., 2013). To test the importance of p21 in replication stress-associated cellular quiescence, we first measured p21 levels in Aphidicolin-treated cells at the end of a 48 hr incubation. Indeed, p21 is strongly upregulated in cells treated with Aphidicolin as compared to control (Figure 6D). To determine if replication stress-associated entry into the CDK2low state is dependent on p21, we imaged p21−/− MCF10A cells expressing the CDK2 activity sensor treated with the same low dose of Aphidicolin (0.4µM). As observed in wild-type cells, treatment of p21−/− G1-phase cells with 0.4µM Aphidicolin resulted in a plateauing of CDK2 activity and a lengthening of the ongoing cell cycle (Figure 6E, bottom panel). This result indicates that p21 is not required for the plateau in CDK2 activity or the prolonged S phase seen in response to low-dose Aphidicolin. Contrary to their wild-type counterparts, however, 60% of p21−/− cells could not be classified as CDK2inc/low because the cells either remained arrested in G2 (and did not undergo another mitosis) or died during mitosis (Figure S5D). This indicates that p21−/− cells are particularly sensitive to the introduction of additional replication stress. Notably, of the p21−/− cells that successfully completed mitosis, only 17% entered the CDK2low state (vs. 84% of wild-type cells; Figure 6E vs. 6B). Thus, although additional p21−/− cells entered the CDK2low state in presence of Aphidicolin, the fraction of cells that responded was five-fold lower compared to wild type cells. Furthermore, the majority of the p21−/− cells that entered the CDK2low state remained quiescent only briefly and quickly re-entered another round of cell cycle within 10 hr after mitosis. Given the molecular mechanism of Aphidicolin, it is likely that the majority of the p21−/− cells inherited considerable DNA replication errors but still entered the CDK2inc proliferative state and committed to another cell cycle (Figure 6E). Consistent with this result, asynchronously cycling p21−/− cells did not display hypo-phosphorylated Rb even though they had 53BP1 foci (Figure S5E). These data indicate that without p21, cells experiencing replication stress cannot pause their cell cycle by entering quiescence after mitosis.
DISCUSSION
Heterogeneity in cell-cycle duration has long been attributed to variability in G1 duration (Prescott, 1968) but the source of this variability has not been identified. In the past, this heterogeneity has been attributed to stochastic noise, although recent studies point to a more deterministic model of cell cycle length variation (Sandler et al., 2015). We previously showed that proliferating cells in full-growth media contain a subpopulation of cells that passes through a transient quiescence, thereby contributing to heterogeneity in cell-cycle duration. In this work, we leveraged this population heterogeneity to examine the origin of this ‘spontaneously’ quiescent subpopulation. We found that entry into quiescence is not stochastic, but actually has a strong deterministic component – on average, about half of the transits through ‘spontaneous’ quiescence can be explained by the presence of 53BP1 foci. Specifically, we find that DNA lesions arising spontaneously due to unresolved replication stress in mother cells trigger a p21-dependent entry of daughter cells into quiescence after mitosis. Furthermore, we show that variability in the timing of escape from quiescence is related to the extent of inherited DNA damage. Thus, while the proximal cause of entry into the quiescent CDK2low state has a deterministic component, a key upstream trigger for entry into the CDK2low state arises at least in part from stochastic errors during DNA replication. A similar conclusion was also reached by (Barr et al., 2017).
The study of endogenous, naturally occurring DNA damage and its effect on cellular proliferation has been technically challenging. Most studies in the field rely on biochemical measurements on populations of cells, making it difficult to detect a small subset of cells with low-level damage. Single-cell approaches such as immunofluorescence or comet assays do aid in uncovering the underlying heterogeneity but provide only one snapshot in time, and are unable to link endogenous damage to future proliferation outcomes. Longitudinal time-lapse studies of single cells are therefore critical for linking information about DNA damage to later cell-cycle responses.
Indeed, an emerging idea in the cell-cycle field is that events in the previous cell cycle control the proliferation-quiescence decision in the subsequent cell cycle. For example, a prolonged mitosis can adversely affect the proliferative capacity of daughter cells even after the mitotic checkpoint is satisfied and cell division is complete (Uetake and Sluder, 2010). As a second example, we and others have shown that actively proliferating cells assess the level of mitogenic signals in G2/M of the preceding cell cycle and integrate this information to choose between proliferation and quiescence after mitosis (Hitomi and Stacey, 1999; Naetar et al., 2014; Spencer et al., 2013). In contrast, the classic Restriction Point model postulates that cell-cycle commitment occurs exclusively in mid- to late-G1 depending on the presence or absence of mitogens, and is marked by the transition from hypo- to hyper-phosphorylated Rb (Pardee, 1974; Weinberg, 1995; Zetterberg and Larsson, 1985). In the present work, we demonstrate that daughter cells are born with either hypo- or hyper-phosphorylated Rb depending on the presence or absence of unrepaired DNA lesions, respectively. This bifurcation is also evident during the G2 phase, where cells with detectable 53BP1 foci have higher p21 and lower phosphorylated Rb, compared to cells that do not have foci. Additionally, using live-cell imaging we show that cells that enter quiescence after mitosis had increased endogenous DNA damage in the previous cell cycle. Taken together, our data indicate that cells not only assess the availability of mitogens, but also overall cell health (including unresolved DNA lesions), in the previous cell cycle. In light of these data, we favor the idea that the phosphorylation state of Rb serves not just as a metric of mitogen sufficiency, but also as a metric of overall cell health, and that cells with sufficient mitogens and without detectable problems remain in a post-Restriction Point state from the previous G2 all the way through the entire subsequent cell cycle.
In addition to increased DNA damage, the mother cell cycle of CDK2low daughters was longer than the mother cell cycle of CDK2emerge daughters, which was longer than the mother cell cycle of CDK2inc daughters. While the CDK2low and CDK2inc subpopulations can inter-convert, they do retain some “memory” of their previous decision – a cell that passed through the CDK2low state in the previous generation shows a bias towards taking the same fate in the subsequent generation (Spencer 2013). In light of the current data, one explanation for why cells that pass through a CDK2low state are likely to do so again after re-entering the cell cycle and undergoing mitosis could be that the DNA damage incurred in the grandmother cell is not fully resolved in the mother cell and is passed on to the daughter cells.
It is well known that the presence of DNA damage during G1 activates checkpoints to effect a G1 or a G1/S arrest (Bartek and Lukas, 2001a, b; Shaltiel et al., 2015), typically detected by flow cytometry as an accumulation of cells with 2N DNA content. However, the exact location in the cell cycle of these arrested cells is unclear, as it is not possible using DNA content to distinguish cells in G0/quiescence, from cells in early- or mid-G1, from cells at the G1/S boundary. Furthermore, since several key proteins (e.g. p21 and p53) are shared between the signaling pathways for G0/quiescence, G1-arrest, and G1/S-arrest, it is difficult to use these proteins in isolation for distinguishing between these arrest states. In contrast, single-cell time-lapse microscopy of CDK2 activity is well-suited to making these distinctions. Using Cdt1 degradation and EdU incorporation as markers for entry into S-phase, we previously showed that the CDK2 sensor has a cytoplasmic/nuclear ratio of ~1 at the start of S-phase, and a ratio of <0.55 when MCF10A cells are in a G0/quiescent state (Spencer et al., 2013). Here we use this information to show that endogenous or mild Aphidicolin-induced replication stress in mother cells causes daughter cells to arrest specifically in a G0/quiescent state immediately after mitosis.
During our analysis, we observed that a fraction of cells in the phospho-Rblow state did not have any detectable 53BP1 foci (43%/66%/46% for MCF10A/Hs68/RPE1 cells, respectively; Figure 2F, 100 % minus the indicated percentages). Indeed, the presence of 53BP1 foci only explains about half of the transits (averaged over the three cell lines) through the phospho-Rblow state. This indicates that while unresolved replication stress contributes to cellular quiescence in the subsequent cell cycle, it is not the exclusive cause of entry into the CDK2low state. We speculate that quiescent cells lacking 53BP1 foci represent cells experiencing other stresses that cannot be detected by 53BP1 staining. These likely include other cell-cycle errors not studied here such as chromosomal segregation errors or a delay in progression through mitosis, recently shown to cause upregulation of p53 and p21 (Fong et al., 2016; Lambrus et al., 2016; Meitinger et al., 2016).
Conversely, we also noticed cells with 53BP1 foci that were not in the quiescent phospho-Rblow state (44%/34%/45% for MCF10A/Hs68/RPE1 cells, respectively; Figure 2B, cells to the right of the vertical dashed line, and Figure 2D, 100% minus the indicated percentages). We speculate that these cells may have left the CDK2low state and re-entered the cell cycle to repair their damage, a notion supported by the temporal relationship between a buildup of CDK2 activity and subsequent disappearance mCherry-BP1 foci in Figure 4F.
Multiple checkpoints exist to ensure that, once replication starts, the entire genome is faithfully replicated before chromosomes are segregated in mitosis (Bartek and Lukas, 2001a; Longhese et al., 2003). However, the observation that 20–30% of cells with 2N DNA content have 53BP1 foci suggests the existence of a fidelity “floor” for DNA replication. Consistent with this, recent work showed that organisms with gigabase size genomes, such as mammals, experience a high probability of low-level replication stress (~1–2 stalled forks per S phase), and that the frequency of errors is directly correlated with the density and positioning of replication origins (Al Mamun et al., 2016; Moreno et al., 2016). Such low-level replication stress is too weak to cause global activation of checkpoint pathways and G2/M arrest, allowing the damage to be transmitted through mitosis to daughter cells (Koundrioukoff et al., 2013). The fact that genome stability is nevertheless maintained in the vast majority of cells suggests that cells must possess a post-replicative mechanism that allows for resolution of these errors. We propose that cells experiencing replication stress that is not strong enough to elicit a checkpoint response in G2/M enter a temporary quiescence immediately after mitosis (potentially triggered by DNA lesions created when unreplicated genomic regions are pulled apart in anaphase (Gelot et al., 2015)). The quiescent state may then allow cells to prepare for DNA damage repair in S phase, thereby maintaining genomic stability.
The three non-transformed cell types used in this study all have an intact DNA damage response. An interesting question is whether cancer cells would differ in their ability to enter the CDK2low state in response to replication stress, and we speculate that this would depend on their ability to induce p21 appropriately. Consistent with this idea, p21−/− cells are limited in their ability to enter the quiescent CDK2low state spontaneously (Spencer et al., 2013), as well as upon addition of low-level replication stress (Figure 6E). The inability to enter the CDK2low state might cause cells to be particularly sensitive to additional stresses, potentially leading to aneuploidy. Indeed, p21−/− cells are known to have increased aneuploidy compared to their wild type counterparts (Barboza et al., 2006; Marques-Torrejon et al., 2013; Shen et al., 2005). Consistent with this observation, p21-deficient mice also show increased frequency of spontaneous tumors as compared to wild-type mice (Martin-Caballero et al., 2001). The data presented in this work support the role of p21 as a tumor suppressor and as a safeguard of genome stability by limiting the expansion of daughter cells suffering from inherited replication stress. Paradoxically, a recent report showed that prolonged p53-independent overexpression of p21 can also be a cause of replication stress via upregulation of replication licensing machinery (Galanos et al., 2016). Thus, p21 could act as a double-edged sword depending on the mutational status of the p53 protein. With new technologies to track quiescence dynamically in single cells, it may now be possible to determine to what extent a bypass of quiescence in the presence of stress is a precursor and/or a hallmark of cancer. Such findings could contribute to the development of biomarkers to identify cancer cells that may be particularly sensitive to chemotherapeutic agents due to their inability to enter quiescence in response to stress.
EXPERIMENTAL PROCEDURES
Cell culture and reagents
MCF10A human mammary epithelial cells were maintained in DMEM/F12 (ThermoFisher) supplemented with 5% horse serum (Invitrogen), 20ng/ml epidermal growth factor (EGF, Sigma-Aldrich), 0.5 mg/ml hydrocortisone (Sigma-Aldrich, St. Louis, MO), 100ng/ml cholera toxin (Sigma-Aldrich) and 10 µg/ml insulin (Invitrogen). Hs68 primary human foreskin fibroblasts were cultured in DMEM with 10% FBS and penicillin/streptomycin, and RPE-hTERT retinal epithelial cells were cultured in DMEM/F12 with 10% FBS and penicillin/streptomycin. For live-cell time-lapse imaging, phenol-red free DMEM/F12 was used. MCF10A, Hs68 and parental RPE-hTERT cell lines were purchased from ATCC. Aphidicolin (#A0781) and Hydroxyurea (#H8627) were purchased from Sigma-Aldrich.
Constructs
MCF10A cells expressing the CDK2 sensor (DHB-mVenus) and tagged histone H2B (H2B–mTurquoise) are as described in (Spencer et al., 2013). The MCF10A DHB-mVenus-H2B–mTurquoise cells were further transduced with mCherry-BP1 lentivirus (construct courtesy of Dr. Galit Lahav). RPE-hTERT mCherry-BP1 cells (cell line courtesy of Dr. James Orth) were transduced with DHB-mVenus and H2B–mTurquoise lentivirus. Cells expressing all three fluorescent proteins were sorted using FACS.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde, washed twice with PBS and incubated with a blocking/permeabilization buffer (10% FBS, 1% BSA, 0.1% TX-100, 0.01% NaN3) for an hour at room temperature. Primary antibody staining was carried out overnight at 4°C and visualized using secondary antibodies conjugated to Alexa Flour 488 or Alexa Flour 546. Where indicated, cells were incubated in media containing 10µM EdU for 15 mins, and then fixed and processed according to manufacturer’s instructions (ThermoFisher #C10340). Images were acquired on an IXMicro microscope (Molecular Devices) with a 10X 0.45NA objective. Antibodies used in this study are: 53BP1 (BD Biosciences #612522), p21 Waf1/Cip1 (CST #2947), phospho-Rb (Ser807/811) (CST #8516), Alexa Flour 488 Goat anti-mouse (ThermoFisher A-11001) and Alexa Flour 546 Goat anti-rabbit (ThermoFisher A-11035).
Time-lapse microscopy
Cells were plated at least 24 hr prior to imaging in phenol red-free full-growth media in a 96-well plate (Greiner bio-one #655090) such that the density would remain sub-confluent until the end of the imaging period. For experiments that required drug addition, cells were first “pre-imaged” for ~12 hr in full growth media without any drugs. At the time of drug addition, 50% of the media in the wells was replaced with media containing a 2X drug concentration and cells were imaged for additional 48 hr in presence of the drug. Images were acquired every 12 min on an IXMicro microscope (Molecular Devices) with a 10X 0.45NA objective. Total light exposure time was kept under 300 ms for each time point. Cells were imaged in a humidified, 37°C chamber at 5% CO2. Cell tracking was done using custom MATLAB scripts, as in (Cappell et al., 2016). See Supplementary Methods for details.
Supplementary Material
MCF10A cells expressing the CDK2 sensor (DHB-Venus) were imaged every 12 min for 60 hr with a 10X 0.45NA objective. Cells were imaged for 12 hr in full-growth media and then for an additional 48 hr in full-growth media containing DMSO. Scale bar is 50µm (first frame).
MCF10A cells expressing the CDK2 sensor (DHB-Venus) were imaged every 12 min for 60 hr with a 10X 0.45NA objective. Cells were imaged for 12 hr in full-growth media and then for an additional 48 hr in full-growth media containing 0.4µM Aphidicolin. Scale bar is 50µm (first frame).
Figure 7. Schematic of key inputs that determine the proliferative or quiescent fate of a cell after mitosis.

Cells assess their environment (presence or absence of mitogens (Spencer et al., 2013)), as well as stresses such as endogenous replication stress (this manuscript), during a “decision window” at the end of the previous cell cycle and choose between two alternate fates after mitosis – proliferative/CDK2inc or quiescent/CDK2low
Highlights.
Growing cell populations contain a quiescent subpopulation of unknown origin
This quiescence is triggered when DNA damage in mother cells is passed on to daughters
The time spent in quiescence is correlated with the extent of inherited damage
This phenomenon is a major cause of heterogeneity in G1 and cell-cycle durations
Acknowledgments
We thank Mingyu Chung for pre-publication access to improved cell tracking scripts, Xuedong Liu, Daniel Durocher, and James Orth for comments on our manuscript, Galit Lahav and James Orth for reagents, and members of the Spencer Lab for general help, in particular Mingwei Min and Andrew Weekley. This work was supported primarily by an NIH K22 Early-Career Investigator Award to S.L.S (1K22CA188144-01), and in part by a Boettcher Webb-Waring Early-Career Investigator Award, a Kimmel Scholar Award, a Searle Scholar Award, and a Beckman Young Investigator Award to S.L.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
M.A. designed, conducted, and analyzed the experiments. H.P, A.B. and J.M. wrote MATLAB scripts used for data and image analysis; J.M. performed data analysis for Figures 4A and 4B. S.L.S conceived the project, suggested the experiments, interpreted the data, and wrote the manuscript with M.A.
References
- Al Mamun M, Albergante L, Moreno A, Carrington JT, Blow JJ, Newman TJ. Inevitability and containment of replication errors for eukaryotic genome lengths spanning megabase to gigabase. Proc Natl Acad Sci U S A. 2016;113:E5765–E5774. doi: 10.1073/pnas.1603241113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21:1719–1729. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G. p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci U S A. 2006;103:19842–19847. doi: 10.1073/pnas.0606343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novak B, Bakal C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun. 2017;8:14728. doi: 10.1038/ncomms14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol. 2001a;13:738–747. doi: 10.1016/s0955-0674(00)00280-5. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001b;490:117–122. doi: 10.1016/s0014-5793(01)02114-7. [DOI] [PubMed] [Google Scholar]
- Cappell SD, Chung M, Jaimovich A, Spencer SL, Meyer T. Irreversible APC(Cdh1) Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell. 2016;166:167–180. doi: 10.1016/j.cell.2016.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4:e83. doi: 10.1371/journal.pbio.0040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- El Achkar E, Gerbault-Seureau M, Muleris M, Dutrillaux B, Debatisse M. Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc Natl Acad Sci U S A. 2005;102:18069–18074. doi: 10.1073/pnas.0506497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Critical reviews in biochemistry and molecular biology. 2012;47:222–235. doi: 10.3109/10409238.2012.655374. [DOI] [PubMed] [Google Scholar]
- Fong CS, Mazo G, Das T, Goodman J, Kim M, O’Rourke BP, Izquierdo D, Tsou MF. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife. 2016;5 doi: 10.7554/eLife.16270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M, et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol. 2016;18:777–789. doi: 10.1038/ncb3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelot C, Magdalou I, Lopez BS. Replication stress in Mammalian cells and its consequences for mitosis. Genes (Basel) 2015;6:267–298. doi: 10.3390/genes6020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol. 2010;190:197–207. doi: 10.1083/jcb.200911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet. 1984;67:136–142. doi: 10.1007/BF00272988. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi M, Stacey DW. Cellular ras and cyclin D1 are required during different cell cycle periods in cycling NIH 3T3 cells. Mol Cell Biol. 1999;19:4623–4632. doi: 10.1128/mcb.19.7.4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RM, Petermann E. Replication fork dynamics and the DNA damage response. Biochem J. 2012;443:13–26. doi: 10.1042/BJ20112100. [DOI] [PubMed] [Google Scholar]
- Koundrioukoff S, Carignon S, Techer H, Letessier A, Brison O, Debatisse M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 2013;9:e1003643. doi: 10.1371/journal.pgen.1003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol. 2016;214:143–153. doi: 10.1083/jcb.201604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Clerici M, Lucchini G. The S-phase checkpoint and its regulation in Saccharomyces cerevisiae. Mutat Res. 2003;532:41–58. doi: 10.1016/j.mrfmmm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- Mankouri HW, Huttner D, Hickson ID. How unfinished business from S-phase affects mitosis and beyond. EMBO J. 2013;32:2661–2671. doi: 10.1038/emboj.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Torrejon MA, Porlan E, Banito A, Gomez-Ibarlucea E, Lopez-Contreras AJ, Fernandez-Capetillo O, Vidal A, Gil J, Torres J, Farinas I. Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell Stem Cell. 2013;12:88–100. doi: 10.1016/j.stem.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–6238. [PubMed] [Google Scholar]
- Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK, et al. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol. 2016;214:155–166. doi: 10.1083/jcb.201604081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015;528:286–290. doi: 10.1038/nature16139. [DOI] [PubMed] [Google Scholar]
- Moreno A, Carrington JT, Albergante L, Al Mamun M, Haagensen EJ, Komseli ES, Gorgoulis VG, Newman TJ, Blow JJ. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc Natl Acad Sci U S A. 2016;113:E5757–E5764. doi: 10.1073/pnas.1603252113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naetar N, Soundarapandian V, Litovchick L, Goguen KL, Sablina AA, Bowman-Colin C, Sicinski P, Hahn WC, DeCaprio JA, Livingston DM. PP2A–mediated regulation of Ras signaling in G2 is essential for stable quiescence and normal G1 length. Mol Cell. 2014;54:932–945. doi: 10.1016/j.molcel.2014.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A. 1974;71:1286–1290. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott DM. Regulation of cell reproduction. Cancer Res. 1968;28:1815–1820. [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153:613–620. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler O, Mizrahi SP, Weiss N, Agam O, Simon I, Balaban NQ. Lineage correlations of single cell division time as a probe of cell-cycle dynamics. Nature. 2015;519:468–471. doi: 10.1038/nature14318. [DOI] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltiel IA, Krenning L, Bruinsma W, Medema RH. The same, only different - DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci. 2015;128:607–620. doi: 10.1242/jcs.163766. [DOI] [PubMed] [Google Scholar]
- Shen KC, Heng H, Wang Y, Lu S, Liu G, Deng CX, Brooks SC, Wang YA. ATM and p21 cooperate to suppress aneuploidy and subsequent tumor development. Cancer Res. 2005;65:8747–8753. doi: 10.1158/0008-5472.CAN-05-1471. [DOI] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155:369–383. doi: 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol. 2010;20:1666–1671. doi: 10.1016/j.cub.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg A, Larsson O. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc Natl Acad Sci U S A. 1985;82:5365–5369. doi: 10.1073/pnas.82.16.5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MCF10A cells expressing the CDK2 sensor (DHB-Venus) were imaged every 12 min for 60 hr with a 10X 0.45NA objective. Cells were imaged for 12 hr in full-growth media and then for an additional 48 hr in full-growth media containing DMSO. Scale bar is 50µm (first frame).
MCF10A cells expressing the CDK2 sensor (DHB-Venus) were imaged every 12 min for 60 hr with a 10X 0.45NA objective. Cells were imaged for 12 hr in full-growth media and then for an additional 48 hr in full-growth media containing 0.4µM Aphidicolin. Scale bar is 50µm (first frame).