

Abstract
Objective
Vascular precursor cells with angiogenic potentials are important for tissue repair, which is impaired in diabetes mellitus. MicroRNAs are recently discovered key regulators of gene expression, but their role in vascular precursor cell–mediated angiogenesis in diabetes mellitus is unknown. We tested the hypothesis that the microRNA miR-27b rescues impaired bone marrow–derived angiogenic cell (BMAC) function in vitro and in vivo in type 2 diabetic mice.
Approach and Results
BMACs from adult male type 2 diabetic db/db and from normal littermate db/+ mice were used. miR-27b expression was decreased in db/db BMACs. miR-27b mimic improved db/db BMAC function, including proliferation, adhesion, tube formation, and delayed apoptosis, but it did not affect migration. Elevated thrombospondin-1 (TSP-1) protein in db/db BMACs was suppressed on miR-27b mimic transfection. Inhibition of miR-27b in db/+ BMACs reduced angiogenesis, which was reversed by TSP-1 small interfering RNA (siRNA). miR-27b suppressed the pro-oxidant protein p66shc and mitochondrial oxidative stress, contributing to its protection of BMAC function. miR-27b also suppressed semaphorin 6A to improve BMAC function in diabetes mellitus. Luciferase binding assay suggested that miR-27b directly targeted TSP-1, TSP-2, p66shc, and semaphorin 6A. miR-27b improved topical cell therapy of diabetic BMACs on diabetic skin wound closure, with a concomitant augmentation of wound perfusion and capillary formation. Normal BMAC therapy with miR-27b inhibition demonstrated reduced efficacy in wound closure, perfusion, and capillary formation. Local miR-27b delivery partly improved wound healing in diabetic mice.
Conclusions
miR-27b rescues impaired BMAC angiogenesis via TSP-1 suppression, semaphorin 6A expression, and p66shc-dependent mitochondrial oxidative stress and improves BMAC therapy in wound healing in type 2 diabetic mice.
Keywords: microRNAs, diabetes mellitus, type 2, wound healing
Diabetic foot ulcers are estimated to occur in 15% of all patients with diabetes mellitus and in 84% of all diabetes mellitus–related lower extremity amputations.1 Augmentation of wound perfusion by correcting vascular defects may be a key strategy in developing therapeutic protocols for diabetic wounds. Circulating endothelial progenitor cells (EPCs) are endothelial precursors that are essentially involved in angiogenesis.2 Recent studies suggest that some early EPCs from ex vivo expansion were a heterogeneous cell population with angiogenic potentials3 and are better defined as bone marrow–derived circulating angiogenic cells. Despite this controversy, various kinds of circulating EPCs have been reported to contribute to vascular and tissue regeneration in humans.4 However, EPCs are found to be dysfunctional in patients with diabetes mellitus,5,6 and the underlying mechanisms and resulting impact on tissue repair are not fully understood.
MicroRNAs (miRNAs) are small, noncoding RNAs that regulate cellular activities by repressing gene expression.7–9 In angiogenesis, miRNA studies were mostly performed using mature cells.10,11 We previously investigated the contribution of miR-34a to EPC dysfunction in aging.12 In diabetes mellitus, in which EPC dysfunction has been demonstrated, miRNA regulation has never been reported. Inhibiting 1 miRNA, miR-27b, has been reported to decrease endothelial cell sprouting and to regulate the development of adipocytes.13,14 However, the role of miR-27b in controlling angiogenic cell activities in diabetes mellitus and its possible regulation in wound healing have not yet been reported.
miRNAs interferes with mRNA translation by binding to the 3′-untranslated region (UTR) of target mRNAs. Thrombospondin-1 (TSP-1) and thrombospondin-2 (TSP-2) are extracellular glycoproteins that modulate cell-to-cell interactions.15 High glucose levels have been reported to be a cell-type–specific posttranscriptional regulating effect on TSP-1.16 TSP-2 is reported to be upregulated by oxidative stress in human endothelial cells.17 Putative miR-27b binding sites have been found in TSP-1 mRNA (http://cbio.mskcc.org/mirnaviewer/)18 and TSP-2 mRNA (microRNA.org). It is also reported that miR-27b directly targets semaphorin 6A (Sema6A) and Sprouty 2, both of which display antiangiogenic functions by impairing endothelial sprouting.19,20 However, it is unknown whether these antiangiogenic molecules are suppressed by miR-27b in angiogenic cells. Of particular note, miR-27b also has binding sites on mRNA coding p66shc (microRNA.org), a pro-oxidant protein that increases mitochondrial reactive oxygen species (ROS) level. A recent study indicates that p66shc deletion reduces oxidative stress induced by high glucose, suggesting p66shc as a potential therapeutic target in diabetic vasculopathy.21 To date, no information is available on the regulation of miR-27b on these molecules in diabetes mellitus–related angiogenesis impairment.
The aim of this study was to reveal novel insights into how miRNAs improve stem/progenitor cell angiogenesis and wound healing in diabetes mellitus. Specifically, we tested the hypothesis that miR-27b is critical to bone marrow–derived angiogenic cell (BMAC) function and wound healing in type 2 diabetic mice. We determined how miR-27b promoted diabetic BMAC functions by investigating several target molecules in vitro. Furthermore, the efficacy of BMAC therapy combined with miR-27b mimic/inhibitor or the direct effects of miR-27b mimic/inhibitor were evaluated in a type 2 diabetic mouse excisional wound model. We think that stem cell therapy miRNA manipulation studies will provide useful information on future development of a clinical regimen for diabetic-induced vascular complications.
Results
Decreased miR-27b Contributed to BMAC Dysfunction in Diabetes Mellitus
Human circulating angiogenic cells and mouse BMACs were cultured and characterized using our established, previously described methods (Figure I in the online-only Data Supplement). Quantitative real-time polymerase chain reaction demonstrated that miR-27b was significantly decreased in circulating angiogenic cells from patients with newly diagnosed type 2 diabetes mellitus (Figure 1A) and in type 2 diabetic db/db BMACs (Figure 1B). miR-27b mimic/inhibitor was used to manipulate miR-27b levels (Figure 1B). db/db BMACs with miR-27b mimic transfection displayed significantly increased proliferation (Figure 1C), decreased apoptosis (Figure 1D), improved tube formation (Figure 1E), and adhesion (Figure 1F). In contrast, db/+ BMACs with miR-27b inhibitor showed significantly impaired proliferation, tube formation, and adhesion and increased apoptosis (Figure 1C–1F). Angiogenic mediators, such as vascular endothelial growth factor and stromal cell–derived factor 1α, were upregulated by miR-27b mimic (Figure 1G), whereas angiopoietin-1 did not change.
Figure 1.

Decreased miR-27b contributed to bone marrow–derived angiogenic cell (BMAC) dysfunction in diabetes mellitus. A, miR-27b expression in BMACs from healthy subjects (n=5) vs patients with diabetes mellitus (n=6). *P<0.05 vs healthy BMACs. B, miR-27b expression in BMACs with miR-27b mimic/inhibitor. n=4, *P<0.05 vs db/+ scramble. C, BMAC proliferation with miR-27b mimic/inhibitor transfection (n=5). D, Apoptotic BMACs with miR-27b mimic/inhibitor transfection (n=5). E, BMAC tube formation with miR-27b mimic/inhibitor transfection (n=6). Representative pictures of tube network are shown on the right. F, BMAC adhesion with miR-27b mimic/inhibitor transfection (n=5). In all bar graphs, *P<0.05 vs db/+ scramble. #P<0.05 vs db/db scramble. G, miR-27b increased stromal cell–derived factor 1α (SDF-1α) and vascular endothelial growth factor A (VEGFA), but not aniogiopoietin-1 (n=5). P<0.05 vs scramble.
MiR-27b Augmented BMAC Function Via TSP-1 Suppression
A luciferase reporter system was used to test the direct bindings of miR-27b to human TSP-1 (gene symbol THBS1) mRNA or human TSP-2 (gene symbol THBS2) mRNA 3′UTR. The relative luciferase activities of TSP-1 and p66shc 3′UTR reporter were diminished significantly after miR-27b cotransfection, as compared with mutant controls (Figure 2A), suggesting that miR-27b interferes with TSP-1 and TSP-2 mRNA. But the repression of miR-27b on TSP-2 was weaker than that on TSP-1. TSP-1 protein levels were increased in db/db BMACs versus db/+ BMACs (Figure 2B), which can be reduced by miR-27b mimic (Figure 2C). In db/+ BMACs, miR-27b inhibition significantly increased TSP-1 protein levels (Figure 2D). TSP-2 protein expression was not suppressed by miR-27b mimic in db/db BMACs (Figure 2E). miR-27b inhibitor–induced impairment in tube formation was partly rescued by TSP-1 siRNA (Figure 2F), suggesting that miR-27b regulates BMAC function partly through targeting TSP-1. TSP-1 protein levels were still reduced after TSP-1 siRNA and miR-27b inhibitor transfection (Figure II in the online-only Data Supplement). To determine whether CD36 or CD47 is the major receptor in mediating the effects of TSP-1 on BMACs, CD36 and CD47 expressions were detected by flow cytometry. Both CD36 and CD47 expressions were elevated in db/db BMACs, which were reduced by TSP-1 siRNA (Figure 2G). Furthermore, we treated CD36−/− or CD47−/− BMACs with recombinant mouse TSP-1 protein using an established protocol. Compared with CD36−/− BMACs, CD47−/− BMACs showed much better tube formation and less reduction when stimulated by recombinant mouse TSP-1 (Figure 2H). The activation of the Akt/endothelial nitric oxide (NO) synthase pathway represents an important protective mechanism in the vascular system.22,23 Western blot analysis suggested that CD47−/− BMACs possessed higher Akt/endothelial NO synthase pathway activation compared with wild-type BMACs and CD36−/− BMACs (Figure 2I and 2J), suggesting that CD47 was the major receptor for TSP-1 in BMACs in diabetes mellitus.
Figure 2.

miR-27b rescued diabetic bone marrow–derived angiogenic cell (BMAC) angiogenic function via thrombospondin (TSP)-1 suppression. A, Luciferase activity of TSP-1 and TSP-2 mRNA 3′-untranslated region (UTR) reporter were suppressed by miR-27b (n=5). B, Secreted TSP-1 protein from db/+ and db/db BMACs (n=4). *P<0.05 vs db/+ BMACs. C, Secreted TSP-1 from db/db BMACs was suppressed by miR-27b (n=4). *P<0.05 vs scramble. D, Secreted TSP-1 from db/+ BMACs was increased by miR-27b inhibitor. n=4,*P<0.05 vs scramble. E, Secreted TSP-2 protein on miR-27b mimic transfection in db/db BMACs (n=5, no statistical significance among 3 groups). F, Tube formation in db/+ BMACs with miR-27b inhibitor and TSP-1 small interfering RNA (siRNA) (n=4). *P<0.05 vs db/+ BMACs with scramble. #P<0.05 vs db/+ BMACs with miR-27b inhibitor. G, Flow cytometry analysis of CD36 and CD47 expression (n=5). *P<0.05 vs the other 2 groups. H, Tube networks of wild-type (WT), CD47−/−, and CD36−/− BMACs treated with recombinant mouse TSP-1 (rTSP-1). n=4, *P<0.05 vs WT plus vehicle; #P<0.05 vs WT plus rTSP-1. Representative pictures are shown on the right. I, Phosphorylation of Akt (ser473) of WT, CD47−/−, and CD36−/− BMACs treated with rTSP-1 (n=4). *P<0.05 vs WT plus vehicle; #P<0.05 vs WT+rTSP-1. J, Phosphorylation of endothelial nitric oxide synthase (eNOS; s1177) of WT, CD47−/−, and CD36−/− BMACs treated with rTSP-1 (n=4). *P<0.05 vs WT plus vehicle; #P<0.05 vs WT plus rTSP-1. THBS1 indicates human TSP-1; and THBS2, human TSP-2.
MiR-27b Rescues BMAC Angiogenesis Via Suppression of Mitochondrial ROS and p66shc Expression
The db/db BMACs demonstrated increased mitochondrial ROS and p66shc protein, which were both significantly suppressed by miR-27b mimic (Figure 3A and 3B). In db/+ BMACs, mitochondrial ROS was elevated by miR-27b inhibitor (Figure 3A). Luciferase reporter assay indicated that miR-27b directly bound to the 3′UTR of p66shc (Src homology 2 domain containing transforming protein 1) mRNA (Figure 3C). To address whether miR-27b regulates BMAC angiogenesis via suppression of p66shc, p66shc was knocked down by adenovirus-mediated p66shc small silencing RNA transfection before miR-27b inhibitor transfection. The elevation of mitochondrial ROS induced by miR-27b inhibition was reversed by knocking down p66shc (Figure 3D). Proliferation (Figure 3E) and tube formation assays (Figure 3F) suggested that the impairment induced by miR-27b inhibitor was partially reversed by adenovirus-mediated p66shc small silencing RNA. Furthermore, in db/db BMACs, antioxidant enzyme manganese superoxide dismutase was increased by miR-27b mimic transfection, whereas copper/zinc superoxide dismutase and catalase were not affected (Figure 3G).
Figure 3.

miR-27b suppressed mitochondrial oxidative stress. A, MitoSOX fluorescence in bone marrow–derived angiogenic cells (BMACs) on miR-27b mimic/inhibitor transfection (n=5). *P<0.05 vs db/+ scramble; #P<0.05 vs db/db scramble. B, miR-27b mimic suppressed p66shc protein expression in diabetic BMACs (n=4). *P<0.05 vs the other 2 groups. C, Luciferase activity of p66shc (SHC1) mRNA 3′-untranslated region (UTR) reporter was suppressed by miR-27b (n=6). P<0.05 vs mutant sequence. D, MitoSOX fluorescence in BMACs on adenovirus-mediated p66shc small silencing RNA (Ad-p66shcRNAi) and miR-27b inhibitor transfection (n=4). *P<0.05 vs the other 2 groups. E, Proliferation on transfection with Ad-p66shcRNAi and miR-27b inhibitor (n=4). *P<0.05 vs db/+ scramble; #P<0.05 vs miR-27b inhibitor plus adenovirus-β-galactosidase (Ad-β-gal). F, BMAC tube formation on transfection with Ad-p66shcRNAi and miR-27b inhibitor (n=4). *P<0.05 vs db/+ scramble; #P<0.05 vs miR-27b inhibitor plus Ad-β-gal. G, Manganese superoxide dismutase (MnSOD), copper/zinc superoxide dismutase (Cu/ZnSOD), and catalase protein expression in db/db BMACs on miR-27b mimic transfection (n=4 per group). *P<0.05 vs scramble.
miR-27b Augmented Diabetic BMAC Function Via Suppression of Sema6A But Not Sprouty 2
Sema6A and Sprouty 2 were 2 tested targets of miR-27b in endothelial cells, but not in BMACs.19,20 The protein expression of Sema6A was elevated in db/db BMACs, which was suppressed by miR-27b mimic, whereas miR-27b inhibitor increased Sema6A protein expression in db/+ BMACs (Figure 4A). However, miR-27b did not suppress Sprouty 2 protein expression in BMACs (Figure 4B). Impaired db/db BMAC tube formation (Figure 4C and 4D) and adhesion (Figure 4F) were improved by Sema6A siRNA. Furthermore, knocking down Sema6A reversed the detrimental effects of miR-27b inhibition on db/+ BMAC tube formation (Figure 4C and 4E) and adhesion (Figure 4G). Sprouty 2 siRNA improved db/db tube formation, but it did not reverse the effects of miR-27b on BMAC function. The protein expressions of Sema6A and Sprouty 2 after oligo transfections are shown in Figure III in the online-only Data Supplement. The 3′UTR vector binding assay suggested that both Sema6A and Sprouty 2 were direct targets of miR-27b (Figure 4H).
Figure 4.

miR-27b suppresses semaphorin 6A (Sema6A) in diabetic bone marrow–derived angiogenic cells (BMACs). A, Sema6A protein expression in BMACs was suppressed by miR-27b (n=6). *P<0.05 vs db/+ with scramble; #P<0.05 vs db/db with scramble. B, Sprouty 2 protein expression was not suppressed by miR-27b (n=6). C, Representative pictures of tube network formed by BMACs with oligo transfections. D, Tube formation of db/db BMACs with scramble, Sema6A small interfering RNA (siRNA), or Sprouty 2 siRNA using db/+ with scramble as controls (n=5). E, Tube formation of db/+ BMACs with miR-27b inhibitor plus Sema6A siRNA or Sprouty 2 siRNA (n=5). F, Adhesion of db/db BMACs with scramble, Sema6A siRNA, or Sprouty 2 siRNA (n=5). There was no significant statistical difference between db/db scramble and db/db Sprouty 2 siRNA. G, Adhesion of db/+ BMACs with miR-27b inhibitor plus Sema6A siRNA or Sprouty 2 siRNA (n=5). D–G, *P<0.05 vs db/+ with scramble; #P<0.05 vs db/db with scramble. H, Luciferase activity of Sema6A and Sprouty 2 mRNA 3′-untranslated region reporters were suppressed by miR-27b (n=4). *P<0.05 vs its mutant sequence.
BMAC Cell Therapy With miR-27b Manipulation–Regulated Diabetic Wound Closure
The in vivo therapeutic potentials of BMACs were tested on 6-mm excisional skin wounds. Wound closure in db/db mice was significantly delayed compared with that in db/+ mice (Figure 5A and 5B). Next, either 1×106 db/+ BMACs or db/db BMACs were topically transplanted onto db/db wounds immediately after wounding. The db/+ BMACs significantly accelerated wound closure starting on day 2, whereas db/db BMACs did not show significant acceleration until day 6.
Figure 5.

miR-27b improved bone marrow–derived angiogenic cell (BMAC) therapy on wound healing in type 2 diabetic mice. A, Representative pictures of cutaneous wounds taken on days 0, 4, 8, 12, and 16 after wounding. B, Wound closure in db/db and db/+ mice with or without BMAC therapy (n=5 per group). C, Wound closure demonstrates the effect of miR-27b mimic on db/db BMAC therapy (n=5 per group). D, Wound closure rates demonstrates the effect of miR-27b inhibition on db/+ BMAC therapy (n=5 per group). E, Representative pictures of wound perfusion on day 6 as measured by laser Doppler. F, Wound perfusion index in db/db and db/+ mice with or without BMAC therapy (n=4 per group). G, Wound perfusion index demonstrating the effect of miR-27b inhibitor on db/+ BMAC therapy (n=4 per group). H, Wound perfusion index demonstrating the effect of miR-27b mimic on db/db BMAC therapy (n=5). In all graphs, *P<0.05 vs db/db wounds plus phosphate-buffered saline (PBS). #P<0.05 vs db/+ wounds plus PBS. ΔP<0.05 vs db/db BMAC therapy. $P<0.05 vs db/+ BMAC therapy. siRNA indicates small interfering RNA; and TSP, thrombospondin.
More importantly, miR-27b overexpression significantly accelerated wound closure compared with db/db BMAC therapy starting on day 2 (Figure 5C). There was no difference between db/db miR-27bmimic-BMAC therapy and db/+ BMAC therapy. In contrast, wounds with db/+ miR-27binhibitor-BMACs had significantly slower closure rates compared with wounds with db/+ BMACs (Figure 5D). To specify the role of TSP-1 in miR-27b–regulated BMAC function, TSP-1 expression was knocked down by siRNA in db/+ miR-27binhibitor-BMACs (termed “db/+ miR-27binhibitorTSP-1siRNA-BMACs”). The db/+ miR-27binhibitorTSP-1siRNA-BMACs partly rescued their poor healing rates (P<0.05 versus db/+ miR-27binhibitor-BMAC therapy; Figure 5D). The miR-27b and TSP-1 expressions after transfections are shown in Figure IV in the online-only Data Supplement.
BMAC Cell Therapy With miR-27b Manipulation–Regulated Diabetic Wound Perfusion
Representative pictures of wound area perfusion taken by laser Doppler were selected on day 6 (Figure 5E). The db/+ wounds had better perfusion than db/db wounds throughout the whole healing process, whereas db/db wounds only had a mild increase in perfusion (Figure 5F). The db/+ BMAC therapy partly increased wound blood flow (Figure 5F), whereas db/db BMAC therapy did not increase wound blood flow.
The db/db wounds with db/db miR-27bmimic-BMACs displayed better perfusion than wounds with db/db BMACs starting on day 4 after wounding (Figure 5H). There was no difference between db/db miR-27bmimic-BMAC therapy and db/+ BMAC therapy. Conversely, miR-27b inhibition in db/+ BMAC transplantation significantly decreased their efficacy in prompting wound blood flow (Figure 5G), but the overall perfusion of this group was still higher than that of db/db wounds treated with phosphate-buffered saline (P<0.05). Furthermore, db/+ miR-27binhibitor TSP-1siRNA-BMAC therapy partly increased wound blood flow compared with db/+ miR-27binhibitor-BMACs (Figure 5G).
Direct miR-27b Manipulation Altered Wound Closure Rates and Perfusion
The db/db skin had decreased expression of miR-27b at basal level and on wounding (Figure 6A and 6B). miR-27b mimic or inhibitor was used to manipulate local miR-27b expressions during wound closure. Our results demonstrated that miR-27b mimic accelerated wound closure (Figure 6C and 6D) and increased wound blood flow (Figure 6E and 6F). miR-27b inhibitor significantly delayed normal wound closure (Figure 6C and 6D) and decreased blood flow (Figure 6E and 6F). These data indicated that miR-27b inhibition leads to disturbed tissue repair and regeneration, as well as impaired wound angiogenesis.
Figure 6.

Direct miR-27b manipulation on db/db and db/+ wound closure and perfusion. A, miR-27b expression in unwounded skin from db/db and db/+ mice (n=5). *P<0.05 vs db/+. B, miR-27b expression on day 6 wound samples from db/db and db/+ mice with or without miR-27b manipulation (n=5). *P<0.05 vs db/+ scramble; #P<0.05 vs db/db scramble. C, Representative pictures of cutaneous wounds transfected with miR-27b mimic/inhibitor or scramble, on days 0, 4, 8, 12, and 16 after wounding. D, Wound closure of wounds transfected with miR-27b mimic/inhibitor or respective scramble (n=5). *P<0.05 vs db/+ scramble; #P<0.05 vs db/db scramble. E, Representative pictures of wound perfusion on day 6 as measured by laser Doppler. F, Wound perfusion index on wounds transfected with miR-27b mimic/inhibitor or respective scramble (n=5). *P<0.05 vs db/+ scramble; #P<0.05 vs db/db scramble.
miR-27b Improved Diabetic Wound BMAC Therapy to Augment In Vivo Angiogenesis
Wounds and surrounding skin tissues were recovered on day 6 after wounding and capillaries were stained for the endothelial cell-specific marker CD31 (representative pictures are shown in Figure V in the online-only Data Supplement). Capillary formation was much more active in db/+ mice than in db/db mice (Figure 7A). The db/db mice treated with db/+ BMACs demonstrated more capillary formation than nontreated db/db mice. However, db/db BMACs could not achieve the same efficacy as db/+ BMACs. miR-27b overexpression in db/db BMACs significantly enhanced capillary formation compared with db/db BMAC controls. Conversely, the effect of db/+ BMAC on capillary formation was abolished by miR-27b inhibition (Figure 7A). Direct transfection of miR-27b mimic on db/db wounds partly increased capillaries, whereas direct transfection of miR-27b inhibitor decreased wound capillaries (Figure 7B).
Figure 7.

Wound capillary formation and incorporation of bone marrow–derived angiogenic cells (BMACs). A, Capillary formation in wounds with different BMAC therapies. *P<0.05 vs db/+ mice with phosphate-buffered saline (PBS). #P<0.05 vs db/db mice with PBS. $P<0.05 vs db/+ BMAC therapy. ΔP<0.05 vs db/db BMAC therapy. B, Capillary formation in wounds with direct miR-27b mimic/inhibitor transfection. *P<0.05 vs db/+ mice with scramble. #P<0.05 vs db/db mice with scramble. C, BMACs were integrated into the vasculature in wounds harvested on day 6 after wounding. Cell nuclei were stained in blue, CD31 was stained in green, and bromodeoxyuridine (BrdU)-labeled BMACs were stained in red. Representative picture showed that BrdU-labeled EPCs integrated into vascular-like structures and the dermis.
To see whether transplanted BMACs integrate into the cutaneous vasculature and surrounding tissue, db/+ BMACs labeled with bromodeoxyuridine (BrdU) were transplanted onto wounds of db/db mice at the time of wounding. On day 6 of wound healing, the wound and surrounding skin tissues were recovered and stained for BrdU and CD31. The data showed that BrdU-labeled EPCs integrated into vascular-like structures and the dermis (Figure 7C).
Discussion
In this study, we tested the hypothesis that miR-27b rescues impaired BMAC angiogenesis and improves topical BMAC therapy on wound healing in type 2 diabetic mice. Our major findings are the following: decreased miR-27b expression is responsible for the functional loss of BMAC in diabetes mellitus; miR-27b protects BMAC function through ≥3 pathways, via suppression of the antiangiogenic molecules TSP-1 and Sema6A, and via suppression of p66shc, thus suppressing mitochondrial oxidative stress; miR-27b improves the efficacy of diabetic BMAC therapy on wound closure, wound perfusion, and capillary formation; and direct miR-27b delivery partly determines wound closure and wound perfusion pattern.
In diabetic wounds, natural wound-healing processes are inadequate to prevent tissue ischemia and necrosis. One important reason is impaired angiogenesis.24 Stem/progenitor cell therapy may serve as a potential therapeutic approach for such a problem.25 However, outcomes from clinical investigations reveal limited efficacy of patient-derived EPC therapy in treating wounds, partly because of the concern that EPCs are reduced in number and impaired in function.26 In our studies, diabetic BMAC therapy was less efficient in accelerating wound closure and promoting wound perfusion compared with normal BMAC therapy, suggesting that diabetic BMAC therapies per se may not fully compensate for impaired wound regeneration in diabetes mellitus. Therefore, intrinsic BMAC dysfunction needs to be corrected before BMACs are used for autologous therapy in treating diabetic complications.
Posttranscriptional miRNA gene regulation plays an essential role in angiogenesis. In this study, we found that miR-27b was decreased in angiogenic cells from patients with type 2 diabetes mellitus and from type 2 diabetic mice. Upregulation of miR-27b rescued diabetic BMAC tube formation, adhesion, and proliferation and delayed apoptosis, suggesting a pivotal role of miR-27b in BMAC function in diabetes mellitus. Furthermore, our data showed that 2 angiogenic factors, vascular endothelial growth factor and stromal cell–derived factor 1α, were upregulated on miR-27b mimic transfection in BMACs, indicating that miR-27b also enhanced BMAC paracrine functions. miRNA regulation is a complex network. miR-27 belongs to the miR-23/27/24 cluster. This cluster has been shown to be involved in angiogenesis and endothelial apoptosis in cardiac ischemia and retinal vascular development.20,27 We detected miR-23a/b, miR-24, and other angiogenesis-related miRNAs in diabetic BMACs, such as let-7f, miR-221, and miR-222 (Figure VI in the online-only Data Supplement). miR-24 was elevated in diabetic BMACs, whereas other miRNAs showed no differences. miR-24 induces endothelial apoptosis and impairs angiogenesis in myocardial infarction.28 It is possible that multiple miRNAs participate in 1 aspect of cell activity and that other miRNAs, such as miR-24, are involved in revascularization in diabetes mellitus.
In addition, we examined some major causal factors of the development of diabetic complications: high glucose and oxidative.29,30 Normal BMACs were treated with high glucose (25 μmol/L for 7 days starting from day 0 of culture, using mannitol as controls)29 or LY83583 (a ROS generator, 12 μmol/L for 24 hours starting from day 7 of culture)30 using established protocols. miR-27b levels were determined by real-time polymerase chain reaction after the treatment. Our results demonstrated that high glucose or LY83583 did not significantly alter mir-27b expression in BMACs (data not shown). Because these are artificial interventions and may not represent in vivo situations, future in vivo studies in human and animal models will be needed to verify what aspects of diabetes mellitus causes the downregulation of miR-27b.
miRNAs regulate cell activities by repressing their downstream targets. Our data indicated that miR-27b directly interacted with TSP-1 and TSP-2 mRNA, but the impact of miR-27b on TSP-1 expression was more drastic than that on TSP-2 expression. TSP-1 is a potent, endogenous, and detrimental factor to angiogenesis.31 Increased TSP-1 levels have been reported in hyperglycemia and diabetes mellitus.32 In diabetes mellitus, increased TSP-1 essentially suppresses the angiogenic responses of multiple cell types to injury and ischemia.33 Our results showed that diabetic BMACs expressed both enhanced mRNA (Figure VII in the online-only Data Supplement) and protein expression of TSP-1, which was significantly suppressed by miR-27b overexpression, indicating that miR-27b suppressed TSP-1 gene expression. Recent research revealed critical roles of the major receptor of TSP, CD47, in acute regulation of cardiovascular dynamics.34 The TSP1-CD47 ligand receptor axis controls several important cellular processes, inhibiting survival factors such as NO, cGMP, cAMP, and vascular endothelial growth factor.35 CD47−/− BMACs showed much better tube formation and less reduction when stimulated by recombinant mouse TSP-1 compared with CD36−/− BMACs. Western blot analysis suggested that CD47−/− BMACs possessed higher activation of the Akt/endothelial NO synthase pathway compared with wild-type BMACs and CD36−/− BMACs, suggesting that CD47 was the major receptor for TSP-1 in db/db BMACs.
Oxidative stress has been implicated in diabetes mellitus–related vascular damage.36 All main molecular mechanisms seem to reflect the overproduction of superoxide by the mitochondrial electron-transport chain.37 In our studies, miR-27b mimic suppressed mitochondrial ROS in diabetic BMACs, whereas miR-27b inhibitor increased mitochondrial ROS in normal BMACs, strongly suggesting that miR-27b is crucial for BMAC resistance to oxidative stress. p66shc is a mammalian adaptor protein from the ShcA family. p66shc−/− mice exhibit longer life spans compared with their wild-type littermates, with reduced intracellular ROS level and mitochondrial DNA alterations, indicating that p66shc is a key component of the intracellular redox state.38,39 In our study, diabetic BMACs possessed higher levels of p66shc protein than normal BMACs, which were suppressed by miR-27b. p66shc knockdown before miR-27b inhibition partly rescued BMAC proliferation and tube formation as mitochondrial ROS remained low. These data suggest that p66shc serves as an important target of miR-27b in its regulation of redox status in BMACs, which is critical for BMAC angiogenesis. In addition, we found that manganese superoxide dismutase, but not copper/zinc superoxide dismutase or catalase, was increased on miR-27b mimic transfection, further supporting the regulatory role of miR-27b in mitochondrial redox homeostasis.
Sema6A and Sprouty 2 were reported as direct targets of miR-27.19,20 Sema6A is transmembrane axonal guidance protein with many unexplained functional aspects.40 We observed that Sema6A, but not Sprouty 2, was elevated in diabetes mellitus and suppressed by miR-27b, although miR-27b was bound with both of their mRNA 3′UTRs. Knocking down Sema6A significantly reversed the detrimental effects of miR-27b inhibition on BMAC function, suggesting that Sema6A is one of the miR-27b target molecules in diabetes mellitus. In addition to these 2 molecules, miR-27b has been reported to target peroxisome proliferator-activated receptor-γ to regulate adipogenesis and target Sema6A in ECs.14 miR-27b was also shown to target the Notch ligand Delta-like ligand 4 and Sprouty 2, and to promote venous differentiation in zebrafish.41 miR-27b may have targeted other molecules involved in angiogenesis and in other cell activities. Specifying each potential target and downstream pathway is interesting and requires future work.
To test the impact of miR-27b on BMAC function in vivo, we used these miR-27b–manipulated BMACs as cell therapy for tissue repair. Our data demonstrated that miR-27b mimic significantly improved the efficacy of db/db BMAC therapy on wound closure and perfusion. CD31 staining indicated an increase in capillary formation on db/db miR-27bmimic–treated BMACs. In contrast, db/+ miR-27binhibitor-BMAC therapy demonstrated poor efficacy in improving diabetic wounds and decreased wound capillary formation compared with db/+ BMAC therapy. These results suggest that miR-27b is essential for BMACs to function in tissue regeneration. Histological analysis of wounds that received BrdU-labeled BMACs suggested continued existence of donor cells in the wound area. The majority of cells were incorporated into the wound vasculature. In vivo wound healing is a situation in which multiple cellular and molecular components participate in skin structure regeneration. It becomes very difficult to trace the effects of BMAC-derived molecules. However, our in vivo findings are exciting because if modifying cell function by manipulating a single miRNA before transplantation would help to improve wound healing, then autologous cell transplantation in patients with diabetes mellitus may possibly gain favorable efficacy via manipulating ≥2 proangiogenic miRNAs.
We also questioned whether direct delivery of miR-27b can achieve better or at least equal efficacy in diabetic wounds. First, we found that basal miR-27b expression was reduced in diabetic mice and that it was not elevated as in normal mice on wounding. miR-27b overexpression partly accelerated wound healing and increased wound blood flow, whereas miR-27b inhibition significantly delayed normal wound healing. Direct miRNA transfection affects all cell types within the wound area, including keratinocytes. Hildebrand et al42 observed that miR-27b is 1 of 10 miRNAs associated with human keratinocytes in vitro and in vivo. miR-27b was upregulated in neonatal epidermal keratinocytes and in terminal skin keratinocytes. We tested the effects of miR-27b on keratinocyte functions, including migration, proliferation, and apoptosis (Figure VIII in the online-only Data Supplement). Our results demonstrated that miR-27b improved keratinoctye migration and that miR-27b inhibition increased keratinocyte apoptosis and mitochondrial ROS, whereas miR-27b overexpression decreased mitochondrial ROS. Based on these observations, it is thought that miR-27b also directly benefits keratinocyte functions and regulates keratinocyte oxidative stress.
In conclusion, our study suggests that miR-27b rescued impaired BMAC angiogenic function in type 2 diabetic mice, partly via repressing the antiangiogenic molecules TSP-1 and Sema6A, as well as pro-oxidant protein p66shc (Figure 8). miR-27b gene therapy enhances the efficacy of diabetic angiogenic cells for wound angiogenesis and wound repair in diabetic mice. Understanding the mechanisms by which genes and pathways are regulated by angiogenic cell–expressed miRNAs is a great challenge, but it provides important mechanistic insights into complex regulatory networks underlying biological processes in angiogenesis.
Figure 8.
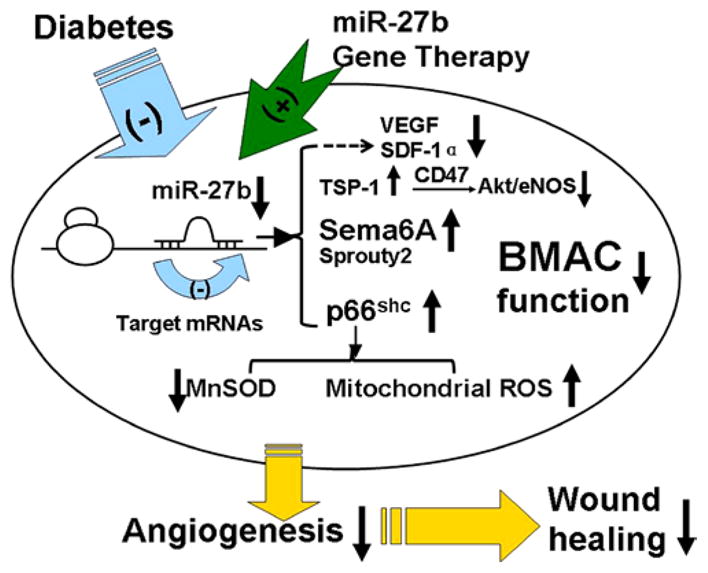
Schema of hypothesis. Physically, miR-27b in bone marrow–derived angiogenic cells (BMACs) suppresses the expression of antiangiogenic molecules semaphorin 6A (Sema6A), p66shc, and thrombospondin (TSP)-1, protecting BMAC angiogenesis, repressing mitochondrial reactive oxygen species, and improving wound healing. In diabetes mellitus, miR-27b expression in BMACs was decreased, which harms BMAC angiogenesis. Gene therapy miR-27b rescues BMAC functions and improves BMAC therapy on diabetic wounds, accelerating wound closure and increasing wound perfusion. eNOS indicates endothelial nitric oxide synthase; MnSOD, manganese superoxide dismutase; SDF-1α, stromal cell–derived factor 1α; and VEGF, vascular endothelial growth factor.
Materials and Methods
Animals
The db/db (BKS.Cg-m+/+Leprdb/J) mouse is a well-established diabetic animal model, demonstrating multiple vascular dysfunction and delayed wound healing similar to the condition found in patients with type 2 diabetes.1 Male diabetic mice (db/db, age=10–12 weeks, plasma glucose 389.24±45.67 mg/dL) and their age- and gender-matched non-diabetic healthy littermates (db/+, BKS.Cg-m−/− Lepdb/− lean, plasma glucose 168.36±25.89 mg/dL) were purchased from The Jackson Laboratory (Bar Harbor, ME). CD47 global knockout (CD47−/−,C57BL/6 background) and wildtype (C57BL/6) were from Jackson Laboratory. CD36 knockout (CD36−/−, C57BL/6 background) mice were in-house colony in Department of Surgery at University of Pittsburgh. The animals were maintained under controlled environmental condition (12h: 12h light/dark cycle, temperature approximately 25°C), and provided with standard laboratory food and water ad libitum. All animal procedures were performed according to University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) guidelines.
Human subjects
Six Type 2 diabetic patients with newly confirmed diagnosis (male=3, female=3, age=56±6.3 years, fasting glucose=9.4±1.8mmol/l) and five healthy age-matched volunteers (male=1, female=4, age=54±1.8 years, fasting glucose=5.1±0.5mmol/l) were recruited based on published criteria.2 All the participants have acquired the nature of this study and given their written informed consent forms for the study, which was approved by the Ethics Committee of our hospital. Mononuclear cells were isolated from peripheral blood (20mL) by density gradient centrifugation and cultured using the same method described in online supplement 3, 4. All the processes were performed by a single operator who was unaware of the subjects’ state. Circulating angiogenic cells were cultured for 7 days and harvested for miR-27b detection.
BMAC in vitro culture and profiles
In vitro expansion of BMACs was performed as described in our recent reports.3, 4 Bone marrow mononuclear cells from the tibias and femurs of mice were plated on a culture flask coated with rat plasma vitronectin (Sigma-Aldrich) and maintained in Endothelial Growth Media (EGM-2, Lonza) in 37°C, 5% CO2 for seven days. On day 4 in culture, non-adherent cells were washed away. New medium was applied. After seven days in culture, to test the BMAC phenotype, attached cells were labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine perchlorate-labeled acetylated LDL (Dil-ac-LDL, 1μg/ml, Sigma) and FITC-labeled Ulex europeus agglutinin (Ulex-Lectin, 1μg/ml; Sigma) for one hour. After nuclei staining by Hoechst 33258 (5μg/ml, Invitrogen), the samples were viewed with an inverted fluorescent microscope (Nikon). Pictures were taken in 200× high power fields. Cells demonstrating double-positive fluorescence of Dil-ac-LDL and Ulex-Lectin were identified as differentiating BMACs. Endothelial functional molecules including von Willebrand Factor (vWF), Vascular Endothelium Cadherin (VE-Cadherin) and endothelial nitric oxide synthase (eNOS) were detected using Western blot (please see “Western Blot Analysis” for details). In addition, the expression of stem cell markers such as Sca-1, CD34 and endothelial lineage markers such as Flk-1 and VE-Cadherin (CD144) were analyzed by flow cytometry and compared with freshly isolated bone marrow mononuclear cells. Adherent cells were gently detached using 5 mM EDTA/PBS at 37ºC, washed, and incubated for one hour on ice in PBS/0.5% (w/v) BSA together with FITC-labeled mouse antibodies (Abs) against Sca-1(1μg/106 cells, BD Bioscience), PE-labeled mouse Abs against Flk-1 (0.5μg/106 cells, BD Bioscience), FITC-labeled mouse Abs against CD34 (1μg/106 cells, BD Bioscience), PE-labeled mouse Abs against CD11b (0.5μg/106 cells, BD Bioscience), or their corresponding isotypic control Abs, respectively. To detect VE-Cadherin, cells were incubated with goat anti-mouse VE-cadherin Abs (2.5 μg/106 cells; R&D Systems) on ice for 20 minutes and then labeled with rabbit anti-goat Alexa 488 Abs (0.625 μg/106 cells; R&D Systems) on ice for 20 minutes. As staining control, 0.5% goat serum was used, followed by identical secondary antibody staining. Flow cytometry was performed using FACScan system and analyzed with Cell Quest software (BD Bioscience). Each analysis included at least 10,000 events.
Small interfering RNA and Mimic/Inhibitor Transfection
In vitro expansion of BMACs was performed as described in our recent reports.3, 4 For small interfering RNA (siRNA)-mediated gene knockdown, BMACs after 7 days in culture were replanted at a density of 2.5×104 to 3.5×104/cm2. SiRNA duplexes were transfected into BMACs with DharmaFECT transfection reagent I (all from Dharmacon), according to the protocol of the manufacturer. For over-expression or inhibition of miR-27b, BMACs were transfected with its specific mimic or inhibitor, 2′-O-Methyl oligoribonucleotides against miR-27b synthesized by Dharmacon (100nM), with DharmaFECT transfection reagent I (Dharmacon) according to the protocol of the manufacturer. In each experiment, non-related scramble oligo was used as a negative control. After 60 hours of silencing RNA and mimic/inhibitor transfection, the cells were harvested for the in vitro and in vivo experiments. In some experiments, db/+ BMACs were transfected with miR-27b inhibitor and/or siRNAs. First, transfection of siRNA or non-target siRNA (scramble, 100nM) was performed for 48–60 hours. Medium was changed. Second, transfection of miR-27b inhibitor or scramble oligo (100nM) was performed for 48–60 hours. The protein expression of target molecule was determined by western blot.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA from BMACs were isolated by mirVanaTM miRNA Isolation Kit (Ambion) and collected as large RNA (for TSP-1 mRNA detection) and small RNA (for miR-27b detection). 3 For mRNA expressions, primers were as follows: TSP-1 forward ACT GGT GAA GGG CCA AGA TCT, reverse: 5′-GGA TCA GGT TGG CAT TCT CAA-3′; 18s (as internal control) forward: 5′-CGG GTC GGG AGT GGG T-3′; reverse: 5′-GAA ACG GCT ACC ACA TCC AAG-3′. Quantitative real-time PCR was performed using cDNA generated from 200ng of large RNA. For miR-27b detection, cDNA was generated from 10 ng of small RNA using specific reverse transcription primers for miR-27b synthesized by Ambion, Inc. Quantitative real-time PCR was performed using its specific real-time PCR primers synthesized by Amion, Inc. Amplification and detection of specific products were performed with the ABI PRISM 7500 Sequence Detection System with the cycle profile according to the protocol of mirVana qRT-PCR miRNA Detection Kit (Ambion, Inc), using sno55 as an internal control. Fluorescent signals were normalized to an internal reference, and the threshold cycle (Ct) was set within the exponential phase of the PCR. The relative gene expression was calculated by comparing cycle times for each target PCR. The miR-27b PCR Ct value was normalized by subtracting the sno55 Ct value, which gave the ΔCt value. The relative expression levels of each target between groups were then calculated using the following equation: relative gene expression = 2-(ΔCt_treatments-ΔCt_controls). 3
Western Blot Analysis
Western blot analysis was performed by SDS/PAGE as we described previously.5 For intracellular protein measurement, BMACs were lysed using Cell Lytic MT lysis buffer (Sigma) with Protease Inhibitor Cocktail (1:100 v/v, Sigma) for 20 minutes on ice. After centrifugation for 15 minutes at 12,000 g (4°C), the protein content of the samples was determined by BCA assay (Bio-Rad). For secreted TSP-1 or TSP-2 protein measurement, the culture medium was replaced with 2 ml of serum-free medium and the cells were incubated for 4 hours.6 The conditioned medium of each sample was collected, concentrated with a Centricon-10 (Amicon, Danvers, MA), and protein concentrations were determined with the BCA assay. At the same time, all the BMACs of each sample were lysed to extract cytoplasmic protein for the detection of β-actin. The TSP-1 or TSP-2 levels in culture medium were normalized by β-actin. Equal amounts of protein (30μg) were loaded onto SDS/PAGE and blotted onto nitrocellulose membranes (Bio-rad). Immunobloting was performed by using antibodies directed against each target molecule: Akt (rabbit anti-mouse Akt, 1:1000; Cell signaling); phosphorylated-Aktser473 (rabbit anti-mouse Akt, 1:1000; Cell signaling); eNOS (rabbit anti-mouse eNOS, 1:1000; Cell signaling), phosphorylated-eNOSs1177 (rabbit anti-mouse eNOS, 1:1000; Cell signaling), TSP-1 (mouse monoclonal anti-TSP-1, 1:400 Abcam), TSP-2 (mouse monoclonal anti-TSP-2, 1:400; Abcam), p66shc (mouse anti-SHC, 1:1000; BD Biosciences), Sema6A (goat anti-mouse Semaphorin 6A, 1:500, R&D); Sprouty2 (rabbit anti-mouse Sprouty2, 1:1000, Abcam); MnSOD (mouse monoclonal anti-MnSOD,1:500, Abcam), Cu/ZnSOD (rabbit anti-mouse Cu/ZnSOD, 1:500, Abcam) and catalase (rabbit anti-mouse catalase,1:200, Santa Cruz), VEGF (rabbit anti-mouse VEGF,1:1000, Millipore), Angiopoietin-1 (rabbit anti-mouse Angiopoietin-1,1:500, Abcam), SDF-1α (rabbit anti-mouse SDF-1α,1:250, Santa Cruz), β-actin (mouse monoclonal anti-β-actin, 1:10000, Sigma, served as loading control). Secondary antibodies included IRDye 800–conjugated rat anti-mouse antibody (1:4000, Rockland), Alexa Fluor 680 goat anti-rabbit IgG antibody (1:2500, Rockland) and Alexa Fluor 800 donkey anti-goat IgG antibody (1:2500, Rockland). The blot was read with an Odyssey imager (Li-Cor). Molecular band intensity was determined with Odyssey 2.1 software (Li-Cor).
BMAC Function Assays (Proliferation, Tube Formation, Migration, Adhesion, Apoptosis)
In tube formation assay, BMACs in EBM-2 plus 5% FBS were plated in a 48-well cell culture plate (5×104 cells per well) pre-coated with 150μl of growth factor-reduced Matrigel-Matrix (BD Biosciences) as described previously. 7 After 24-hour incubation, images of tube morphology were taken by inverted microscope (Nikon) at the magnification ×40 and tube lengths were measured at 5 random fields per well. In adhesion assay, BMACs were plated in a 96-well plates (2.5×104 cells per well) pre-coated with 2.5 g/ml vitronectin. After 1-hour incubation, non-adherent cells were washed away and adherent cells were fixed with 2% Paraformaldehyde. Nuclei were stained with Hoechst (5μg/L, Sigma) for 20 minutes. The number of adherent cells was counted at the magnification ×100, and the mean value of 4 wells was determined for each sample.8 The migration assay was performed using a modified Boyden’s chamber assay as described previously.6 Around 5×104 BMACs were placed into upper Boyden’s chamber with EBM-2 plus 5% FBS. The lower chamber was loaded with EBM-2 plus 5% FBS and VEGF (50ng/ml). BMACs were allowed to migrate for 24 hours. The cells remaining on the upper chamber were mechanically removed. Cells on the lower side of the membrane were fixed and stained with Hoechst, counted at magnification ×100. The mean value of 5 different fields was determined for each sample. Cell proliferation was evaluated by MTS assay using CellTiter 96® Aqueous One Solution Cell Proliferation Assay Kit (Promega). BMACs in EGM-2 were plated in a 96-well culture plate (1×104 cells per well) overnight before different treatments. After the treatments, cell were washed with pre-warmed E’BM-2 and100μl EGM-2 with 20μl assay aliquots were added. BMACs were cultured for 4 hours in 5% CO2,37ºC. The absorbance was read at 490 nm in 100 μL of soluble formazan medium with a microplate spectrophotometer. Cell number was then calculated from a standard cure and expressed as folds of the controls. Cell apoptosis was evaluated with Annexin V-FITC/PI Apoptosis Detection Kit (Invitrogen). The cells were stained with Annexin-V-FITC and propidium iodide in 1 × binding buffer for 15 min at room temperature. Flow-cytometric analyses were performed on FACScan system and analyzed with Cell Quest software (BD Bioscience). Each analysis included at least 10,000 events.
Luciferase Reporter Assays of miR-27b to 3′UTR of Target mRNAs
The luciferase target assay was performed as previously described 9–11. Synthetic oligonucleotides as indicated by NCBI reference sequence bearing either human TSP-1 (THBS1) mRNA 3′UTR (NM_003246), human TSP-2 (THBS2) mRNA 3′UTR (NM_003247.2), human p66shc (SHC1) mRNA 3′UTR(NM_183001.4), human Semaphorin 6A (Sema6A) mRNA 3′UTR (NM_020796), or Sprouty2 (SPRY2) mRNA 3′UTR (NM_005842) was cloned into pMirTarget plasmid after the stop codon of luciferase, respectively. The co-transfection of mutant sequence of each mRNA 3′UTR and miR-27b mimic served as controls. In this vector system, a red fluorescent protein (RFP) under a CMV promoter is used to monitor the transfection and normalizing transfection efficiency. For examples, HEK 293 cells (50% confluence in each well of 6-well plate) were co-transfected with 100ng of TSP-1 (THBS1) 3′UTR plasmid and 0.1nmol of miR-27b mimic (Dharmacon), all combined with Turbofect 8.0 (Origene) according to manufacture’s protocol. As control, similar oligonucleotides to TSP-1 mRNA 3′UTR with mutant miR-27b binding site was cloned into pMirTarget plasmids and co-transfected with miR-27b mimic (see below for mutant sequences). The same protocol was used to co-transfect other 3′UTR plasmid and miR-27b mimic. After 48 h, cells were washed and lysed with Reporter Lysis Buffer (Promega), and their luciferase activity was measured using the GloMax® 96 Microplate Luminometer (Promega). The relative reporter activity was obtained by normalization to each rmutant plasmid co-transfection. A reduced firefly luciferase activity indicated the direct binding of miR-27b to the cloned target sequence.

MitoSOX staining
The mitochondrial reactive oxygen species were detected using MitoSOX™ Red mitochondrial superoxide indicator (Invitrogen) as we previously described.12 MitoSOX™ is a novel fluorogenic dye for highly selective detection of superoxide in the mitochondria of live cells. Once in the mitochondria, MitoSOX™ Red reagent is oxidized by superoxide and exhibits red fluorescence. BMACs were harvested after miR-27b mimic/inhibitor transfection and incubated in HBSS containing 5μM MitoSOX™ for 10 minutes. Immediately after the incubation, flow cytometry was performed using FACScan system and analyzed with Cell Quest software (BD Bioscience). Each analysis included at least 10,000 events.
Ad-p66shcRNAi transfection
For p66shc down-regulation, BMACs were transfected by recombinant adenovirus Ad-p66shcRNAi at the titer of 10MOI, as established previously.13 Dr. Kaikobad Irani kindly donated the virus. This adenovirus encodes a short hairpin loop RNA with a 19-mer sequence corresponding to bases 45–63 of the cDNA of p66shc and is unique to mRNA of p66shc.14 Ad-β-gal was also used for internal control. Adenovirus transfection was performed in EGM-2 supplemented with 2% FBS for 24h, followed by change of EGM-2 with 5% FBS. Then miR-27b inhibitor transfection was performed for 60h, as described above.
Topical BMAC therapy on wound healing in vivo
Wounds were created on the dorsal surface of the mouse as previously described.15 Full-thickness skins were removed using a 6-mm punch biopsy without hurting the underlying muscle. In mice with BMAC therapy, 1×106 BMACs with different gene manipulation in 30 μl PBS were topically transplanted onto the wound area immediately after punch. The grouping was as follows: 1) db/+ wound with PBS; 2) db/db wound with PBS; 3) db/db wound with db/+ BMACs; 4) db/db wound with db/db BMACs; 5) db/db wound with db/db miR-27bmimic-BMACs; 6) db/db wound with db/+ miR-27binhibitor-BMACs; 7) db/db wound with db/+ miR-27binhibitorTSP-1siRNA-BMACs.
Wounds were covered with transparent oxygen-permeable wound dressing (Bioclusive, Johnson & Johnson). The dressings were changed every other day. Wound closure rates were measured by tracing the wound area onto acetate paper every other day until day 10. The tracings were digitized, and the areas were calculated with a computerized algorithm and converted to percent wound closure (Image J). Wound closure rates were calculated as Percentage Closed (y%) =[(Area on Day0–Open Area on Dayx )/Area on Day0]×100, as described previously .16
Laser Doppler imaging (LDI) using PeriScan PIM3 system (Perimed AB, Sweden) was performed to measure the perfusion of the wound areas immediately after wounding and every other day until day 16 as described previously.17, 18 Low-intensity (0.20) laser beams (630nm) were used to scan across wound surface with fixed distance (20cm) from each wound site under standard room illumination. The scan area included the entire wound site and margins. In the following measurements, the same position, detection area, and distances were referred to the first detection profile gained from the same mouse. Blood flow measurements of the microvasculature were calculated with a 214 × 214 pixel resolution, with each pixel being an actual measurement of flux or blood flow (speed of blood cells × blood volume). Color-coded images representing the microvascular blood flow distribution were recorded. Low perfusion was displayed in dark blue and highest perfusion areas were displayed in red. Color images were analyzed by self-contained software Perimed LDPIwin 3.0. A circle was drawn to capture the original size of wound area and the mean perfusion data within this circle were expressed as perfusion index.
Local delivery of miR-27b inhibitor on normal wounds
The effect of topical miR-27b mimic/inhibitor transfection on normal wound healing was measured. An established model of local oligonucleotide delivery via F-127 pluronic gel was applied as previously described.19 Excisional full-thickness cutaneous wounds were created in db/+ mice as described above. Briefly, 0.25 nmol of miR-27b inhibitor (equals to ~3.5 μg) was preloaded into the 10μl, 30% F-127 pluronic gel (Sigma) at 4°C. DharmaFECT transfection reagent I (Dharmacon) was added at a final concentration of 1% in F-127 pluronic gel loaded with miR-27b inhibitor. The gel was applied locally to the surface of wound area and absorbed within 2 hours. The procedure was repeated every other day from day 0 to day 8. The control wounds were treated with 1% F-127 pluronic gel loaded with 0.25 nmol of scramble oligo and 1% of DharmaFECT transfection reagent I. Wound closure rates and wound blood flow were monitored as described above. On day 6, some of wound samples were harvested for miR-27b expression using real-time PCR.
Wound capillary formation
Wounds were recovered from mice on Day 6. Capillary density in the healing wounds was quantified by histological analysis. Wound samples were fixed with zinc chloride fixative (BD) for 24 hours, then embedded in paraffin, and sectioned at 4-μm intervals. Slides were deparaffinized and hydrated, then placed in Tris-Buffered Saline (pH 7.5) for 5 minutes for pH adjustment. Endogenous peroxidase was blocked by 3% Hydrogen Peroxide/Methanol bath for 20 minutes, followed by distilled H2O rinses. Slides were blocked with normal rabbit serum (Vector Laboratories) for 30 minutes, then incubated for 60 minutes at room temperature with an anti-CD31 antibody (1:50; Santa Cruz), and further incubated with Vectastain Elite ABC Reagent (Vector Laboratories) for 30 minutes and Nova Red (Vector Laboratories) for 15 minutes. Slides were counterstained with Gill (Lerner) 2 Hematoxylin (VWR Scientific) for 10 seconds, differentiated in 1% aqueous glacial acetic acid, and rinsed in running tap water. Ten random microscopic fields (×200 magnification) were counted to determine the number of capillaries per wound. Pictures were taken under a Zeiss Axioskop microscope using Image-Pro Plus sofeware (Media Cybernetics).
BMAC Integration
BMACs were isolated as described above and cultured in EGM-2. BrdU staining was performed on BMACs. On day 5 of culture EPCs were labeled with 5-bromo-2′-deoxyuridine and 5-fluro-2′deoxyuridine (BrdU labeling reagent, Invitorgen). BrdU labeling reagent was diluted 1:100 in EGM-2, filtered through a 0.2 :m filter, and warmed to 37°C. 1 mL of BrdU/EGM-2 was added to cells in a 6 well plate and incubated overnight. The wells were washed 3 times with PBS followed by trypsinization to re-suspend the cells. a number of 1 × 106 BMACs were then transplanted to a db/db wound (6 mm punch biopsy) as described above. After 6 days of wound healing, the mouse was euthanized, and the wound and surrounding skin was recovered and fixed in 10% formalin for 24 hours. Slides were double-stained with an anti-CD31 antibody followed by BrdU antibody (1:50; Santa Cruz Biotechnology Inc.) incubation. Pictures were taken under a Zeiss Axioskop microscope using Image-Pro Plus sofeware (Media Cybernetics).
miR-27b mimic/inhibitor transfection on human primary epidermal keratinocytes
Human primary epidermal keratinocytes were purchased from ATCC and cultured in dermal cell basal medium supplemented with keratinocyte growth kit (ATCC) according to the manufacturer’s protocol. For over-expression or inhibition of miR-27b, keratinocytes were transfected with its specific mimic or inhibitor, 2′-O-Methyl oligoribonucleotides against miR-27b synthesized by Dharmacon (100nM), with DharmaFECT transfection reagent I (Dharmacon) according to the protocol of the manufacturer. In each experiment, non-related scramble oligo was used as a negative control. After 60 hours of silencing RNA and mimic/inhibitor transfection, the cells were harvested for the in vitro experiments.
Migration, proliferation, and apoptosis of epidermal keratinocytes
Keratinocyte migration was tested in an in vitro scratch assay as previously described, with slight modification 20. A number of 3×105 keratinocytes were seeded in six-well plates. After reaching 100% confluence, cells were washed twice with PBS and scratched using a pipette-tip. The resulting scratch was investigated under the microscope and the area for photographs was marked using a pen on the bottom of the well. Immediately after scratching, the first photograph was taken (initial wound size) exactly at the marked area. After six hours and 12 hours, the same areas of the scratch wounds were photographed again and the reduction in wound width was measured using ImageJ 1.45 s software (National Institutes of Health, Bethesda, MD). Cell proliferation was evaluated by MTS assay using CellTiter 96® Aqueous One Solution Cell Proliferation Assay Kit (Promega). Keratinocytes in dermal cell basal medium were plated in a 96-well culture plate (1×104 cells per well) overnight before different treatments. After the treatments, cell were washed with pre-warmed E’BM-2 and 100μl EGM-2 with 20μl assay aliquots were added. Keratinocytes were cultured for four hours in 5% CO2,37ºC. The absorbance was read at 490 nm in 100 μL of soluble formazan medium with a microplate spectrophotometer. Cell number was then calculated from a standard cure and expressed as folds of the controls. Cell apoptosis was evaluated with Annexin V-FITC/PI Apoptosis Detection Kit (Invitrogen). The cells were stained with Annexin-V-FITC and PI in 1 × binding buffer for 15 minutes at room temperature. Flow-cytometric analyses were performed on FACScan system and analyzed with Cell Quest software (BD Bioscience). Each analysis included at least 10,000 events.
Data Analysis
All values were expressed as mean±SEM. The statistical significance of differences between the 2 groups was determined with Mann-Whitney U nonparametric test. When more than two treatment groups were compared, one-way ANOVA followed by LSD post hoc testing was used.21 For the in vivo wound closure data, two-way repeated measures ANOVA followed by Bonferroni post hot testing was used to compare both differences between treatments and time courses. For wound perfusion data, Receive Operating Characteristic (ROC) Curve Analysis was performed to compare the differences between treatments. In all tests, p<0.05 was considered statistically significant.
Supplementary Material
Significance.
This study provides mechanistic observations that miR-27b deficiency in bone marrow–derived angiogenic cell functions is responsible for poor angiogenesis in diabetes mellitus, and that targets of miR-27b, including semaphorin 6A, thrombospondin-1, and p66shc, have been identified in the regulation of angiogenesis. MicroRNAs are potential gene regulators that may compensate for the insufficient effects of single-molecule therapy because 1 microRNA seems to affect cell activities via targeting multiple genes. Although we have begun to appreciate the importance of microRNAs in the regulation of angiogenesis, much work remains to elucidate the potential microRNAs involved in this process and the target pathways affected.
Acknowledgments
Sources of Funding
This work was supported, in part, by the National Institutes of Health (NIH)/National Institute of General Medical Sciences R01 GM077352 (to A.F. Chen), Department of Veterans Affairs Rehabilitation Research and Development Merit Awards I01RX000244 and 1I01RX000652 (to A.F. Chen), American Diabetes Association Research Award 7-11-BS-23 (to A.F. Chen), the National Science Foundation of China Key Research Project 81130004 (to A.F. Chen), and NIH 1R21CA158650 (to Q.D. Wang). Dr Jie-Mei Wang was the awardee of China Overseas Scholarship 20070320, American Heart Association Postdoctoral Fellowship 0920110G, and American Heart Association Scientist Development Grant 13SDG16930098.
Nonstandard Abbreviations and Acronyms
- BMAC
bone marrow–derived angiogenic cell
- EPC
endothelial progenitor cell
- miRNA
microRNA
- ROS
reactive oxygen species
- Sema6A
semaphorin 6A
- TSP
thrombospondin
- UTR
untranslated region
Footnotes
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.113.302104/-/DC1.
Disclosures
None.
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 3.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1584–1595. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 5.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 6.Marrotte EJ, Chen DD, Hakim JS, Chen AF. Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J Clin Invest. 2010;120:4207–4219. doi: 10.1172/JCI36858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 8.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117:2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 10.Suárez Y, Sessa WC. MicroRNAs as novel regulators of angiogenesis. Circ Res. 2009;104:442–454. doi: 10.1161/CIRCRESAHA.108.191270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parmacek MS. MicroRNA-modulated targeting of vascular smooth muscle cells. J Clin Invest. 2009;119:2526–2528. doi: 10.1172/JCI40503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao T, Li J, Chen AF. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am J Physiol Endocrinol Metab. 2010;299:E110–E116. doi: 10.1152/ajpendo.00192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59–68. doi: 10.1161/CIRCRESAHA.107.153916. [DOI] [PubMed] [Google Scholar]
- 14.Karbiener M, Fischer C, Nowitsch S, Opriessnig P, Papak C, Ailhaud G, Dani C, Amri EZ, Scheideler M. microRNA miR-27b impairs human adipocyte differentiation and targets PPARgamma. Biochem Biophys Res Commun. 2009;390:247–251. doi: 10.1016/j.bbrc.2009.09.098. [DOI] [PubMed] [Google Scholar]
- 15.Roberts DD. Thrombospondins: from structure to therapeutics. Cell Mol Life Sci. 2008;65:669–671. doi: 10.1007/s00018-007-7483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharyya S, Marinic TE, Krukovets I, Hoppe G, Stenina OI. Cell type-specific post-transcriptional regulation of production of the potent antiangiogenic and proatherogenic protein thrombospondin-1 by high glucose. J Biol Chem. 2008;283:5699–5707. doi: 10.1074/jbc.M706435200. [DOI] [PubMed] [Google Scholar]
- 17.Lopes N, Gregg D, Vasudevan S, Hassanain H, Goldschmidt-Clermont P, Kovacic H. Thrombospondin 2 regulates cell proliferation induced by Rac1 redox-dependent signaling. Mol Cell Biol. 2003;23:5401–5408. doi: 10.1128/MCB.23.15.5401-5408.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urbich C, Kaluza D, Frömel T, Knau A, Bennewitz K, Boon RA, Bonauer A, Doebele C, Boeckel JN, Hergenreider E, Zeiher AM, Kroll J, Fleming I, Dimmeler S. MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood. 2012;119:1607–1616. doi: 10.1182/blood-2011-08-373886. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Q, Gallagher R, Ufret-Vincenty R, Li X, Olson EN, Wang S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23~27~24 clusters. Proc Natl Acad Sci U S A. 2011;108:8287–8292. doi: 10.1073/pnas.1105254108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Stefano V, Cencioni C, Zaccagnini G, Magenta A, Capogrossi MC, Martelli F. p66ShcA modulates oxidative stress and survival of endothelial progenitor cells in response to high glucose. Cardiovasc Res. 2009;82:421–429. doi: 10.1093/cvr/cvp082. [DOI] [PubMed] [Google Scholar]
- 22.Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS, Chung HT, Kwon YG, Kim YM. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am J Physiol Heart Circ Physiol. 2006;291:H2836–H2846. doi: 10.1152/ajpheart.00113.2006. [DOI] [PubMed] [Google Scholar]
- 23.Liang C, Ren Y, Tan H, He Z, Jiang Q, Wu J, Zhen Y, Fan M, Wu Z. Rosiglitazone via upregulation of Akt/eNOS pathways attenuates dysfunction of endothelial progenitor cells, induced by advanced glycation end products. Br J Pharmacol. 2009;158:1865–1873. doi: 10.1111/j.1476-5381.2009.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117:1219–1222. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Wu F, Xia WH, Zhang YY, Xu SY, Cheng F, Liu X, Zhang XY, Wang SM, Tao J. CXCR4 gene transfer contributes to in vivo reendothelialization capacity of endothelial progenitor cells. Cardiovasc Res. 2010;88:462–470. doi: 10.1093/cvr/cvq207. [DOI] [PubMed] [Google Scholar]
- 26.Caballero S, Sengupta N, Afzal A, Chang KH, Li Calzi S, Guberski DL, Kern TS, Grant MB. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–967. doi: 10.2337/db06-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bang C, Fiedler J, Thum T. Cardiovascular importance of the microRNA-23/27/24 family. Microcirculation. 2012;19:208–214. doi: 10.1111/j.1549-8719.2011.00153.x. [DOI] [PubMed] [Google Scholar]
- 28.Fiedler J, Jazbutyte V, Kirchmaier BC, et al. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation. 2011;124:720–730. doi: 10.1161/CIRCULATIONAHA.111.039008. [DOI] [PubMed] [Google Scholar]
- 29.Urbich C, Dernbach E, Rössig L, Zeiher AM, Dimmeler S. High glucose reduces cathepsin L activity and impairs invasion of circulating progenitor cells. J Mol Cell Cardiol. 2008;45:429–436. doi: 10.1016/j.yjmcc.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Sorrentino SA, Bahlmann FH, Besler C, Müller M, Schulz S, Kirchhoff N, Doerries C, Horváth T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser U. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2007;116:163–173. doi: 10.1161/CIRCULATIONAHA.106.684381. [DOI] [PubMed] [Google Scholar]
- 31.Isenberg JS, Martin-Manso G, Maxhimer JB, Roberts DD. Regulation of nitric oxide signalling by thrombospondin 1: implications for anti-angiogenic therapies. Nat Rev Cancer. 2009;9:182–194. doi: 10.1038/nrc2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ii M, Takenaka H, Asai J, Ibusuki K, Mizukami Y, Maruyama K, Yoon YS, Wecker A, Luedemann C, Eaton E, Silver M, Thorne T, Losordo DW. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res. 2006;98:697–704. doi: 10.1161/01.RES.0000209948.50943.ea. [DOI] [PubMed] [Google Scholar]
- 33.Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, Plow EF. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation. 2003;107:3209–3215. doi: 10.1161/01.CIR.0000074223.56882.97. [DOI] [PubMed] [Google Scholar]
- 34.Roberts DD, Miller TW, Rogers NM, Yao M, Isenberg JS. The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biol. 2012;31:162–169. doi: 10.1016/j.matbio.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isenberg JS, Frazier WA, Roberts DD. Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cell Mol Life Sci. 2008;65:728–742. doi: 10.1007/s00018-007-7488-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahangdale S, Yeh SY, Malhotra A, Veves A. Therapeutic interventions and oxidative stress in diabetes. Front Biosci (Landmark Ed) 2009;14:192–209. doi: 10.2741/3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 38.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 39.Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M, Petronilli V, Minucci S, Bernardi P, Lanfrancone L, Pelicci PG. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- 40.Janssen BJ, Robinson RA, Pérez-Brangulí F, Bell CH, Mitchell KJ, Siebold C, Jones EY. Structural basis of semaphorin-plexin signalling. Nature. 2010;467:1118–1122. doi: 10.1038/nature09468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abe S, Sakabe S, Hirata M, Kamuro H, Asahara N, Watanabe M. Study on optimal fat content in total parenteral nutrition in partially hepatectomized rats. J Nutr Sci Vitaminol (Tokyo) 1997;43:187–198. doi: 10.3177/jnsv.43.187. [DOI] [PubMed] [Google Scholar]
- 42.Hildebrand J, Rütze M, Walz N, Gallinat S, Wenck H, Deppert W, Grundhoff A, Knott A. A comprehensive analysis of microRNA expression during human keratinocyte differentiation in vitro and in vivo. J Invest Dermatol. 2011;131:20–29. doi: 10.1038/jid.2010.268. [DOI] [PubMed] [Google Scholar]
References
- 1.Emanueli C, Caporali A, Krankel N, Cristofaro B, Van Linthout S, Madeddu P. Type-2 diabetic Lepr(db/db) mice show a defective microvascular phenotype under basal conditions and an impaired response to angiogenesis gene therapy in the setting of limb ischemia. Front Biosci. 2007;12:2003–2012. doi: 10.2741/2205. [DOI] [PubMed] [Google Scholar]
- 2.Esposito K, Maiorino MI, Di Palo C, Gicchino M, Petrizzo M, Bellastella G, Saccomanno F, Giugliano D. Effects of pioglitazone versus metformin on circulating endothelial microparticles and progenitor cells in patients with newly diagnosed type 2 diabetes--a randomized controlled trial. Diabetes Obes Metab. 2010;13:439–445. doi: 10.1111/j.1463-1326.2011.01367.x. [DOI] [PubMed] [Google Scholar]
- 3.Zhao T, Li J, Chen AF. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. American journal of physiology. 299:E110–116. doi: 10.1152/ajpendo.00192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marrotte EJ, Chen DD, Hakim JS, Chen AF. Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J Clin Invest. 2010;120:4207–4219. doi: 10.1172/JCI36858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du YH, Guan YY, Alp NJ, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117:1045–1054. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]
- 6.Ii M, Takenaka H, Asai J, Ibusuki K, Mizukami Y, Maruyama K, Yoon YS, Wecker A, Luedemann C, Eaton E, Silver M, Thorne T, Losordo DW. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res. 2006;98:697–704. doi: 10.1161/01.RES.0000209948.50943.ea. [DOI] [PubMed] [Google Scholar]
- 7.Luo JD, Hu TP, Wang L, Chen MS, Liu SM, Chen AF. Sonic hedgehog improves delayed wound healing via enhancing cutaneous nitric oxide function in diabetes. Am J Physiol Endocrinol Metab. 2009;297:E525–531. doi: 10.1152/ajpendo.00308.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamada H, Kim MK, Iwakura A, et al. Estrogen receptors alpha and beta mediate contribution of bone marrow-derived endothelial progenitor cells to functional recovery after myocardial infarction. Circulation. 2006;114:2261–2270. doi: 10.1161/CIRCULATIONAHA.106.631465. [DOI] [PubMed] [Google Scholar]
- 9.Bonauer A, Carmona G, Iwasaki M, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 10.Felli N, Fontana L, Pelosi E, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18081–18086. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen CP, Bordeleau ME, Pelletier J, Sharp PA. Short RNAs repress translation after initiation in mammalian cells. Mol Cell. 2006;21:533–542. doi: 10.1016/j.molcel.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 12.Wang XR, Zhang MW, Chen DD, Zhang Y, Chen AF. AMP-activated protein kinase rescues the angiogenic functions of endothelial progenitor cells via manganese superoxide dismutase induction in type 1 diabetes. American journal of physiology. Endocrinology and metabolism. 2011;300:E1135–1145. doi: 10.1152/ajpendo.00001.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim CS, Jung SB, Naqvi A, Hoffman TA, DeRicco J, Yamamori T, Cole MP, Jeon BH, Irani K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circulation research. 2008;103:1441–1450. doi: 10.1161/CIRCRESAHA.108.181644. [DOI] [PubMed] [Google Scholar]
- 14.Yamamori T, White AR, Mattagajasingh I, Khanday FA, Haile A, Qi B, Jeon BH, Bugayenko A, Kasuno K, Berkowitz DE, Irani K. P66shc regulates endothelial NO production and endothelium-dependent vasorelaxation: implications for age-associated vascular dysfunction. J Mol Cell Cardiol. 2005;39:992–995. doi: 10.1016/j.yjmcc.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Gallagher KA, Liu ZJ, Xiao M, Chen H, Goldstein LJ, Buerk DG, Nedeau A, Thom SR, Velazquez OC. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–1259. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen SL, Myers CA, Charboneau A, Young DM, Boudreau N. HoxD3 accelerates wound healing in diabetic mice. Am J Pathol. 2003;163:2421–2431. doi: 10.1016/S0002-9440(10)63597-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nanney LB, Wamil BD, Whitsitt J, Cardwell NL, Davidson JM, Yan HP, Hellerqvist CG. CM101 stimulates cutaneous wound healing through an anti-angiogenic mechanism. Angiogenesis. 2001;4:61–70. doi: 10.1023/a:1016752925761. [DOI] [PubMed] [Google Scholar]
- 18.Ebrahimian TG, Pouzoulet F, Squiban C, Buard V, Andre M, Cousin B, Gourmelon P, Benderitter M, Casteilla L, Tamarat R. Cell therapy based on adipose tissue-derived stromal cells promotes physiological and pathological wound healing. Arterioscler Thromb Vasc Biol. 2009;29:503–510. doi: 10.1161/ATVBAHA.108.178962. [DOI] [PubMed] [Google Scholar]
- 19.Reynolds LE, Conti FJ, Silva R, Robinson SD, Iyer V, Rudling R, Cross B, Nye E, Hart IR, Dipersio CM, Hodivala-Dilke KM. alpha3beta1 integrin-controlled Smad7 regulates reepithelialization during wound healing in mice. J Clin Invest. 2008;118:965–974. doi: 10.1172/JCI33538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ross C, Alston M, Bickenbach JR, Aykin-Burns N. Oxygen tension changes the rate of migration of human skin keratinocytes in an age-related manner. Exp Dermatol. 2010;20:58–63. doi: 10.1111/j.1600-0625.2010.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder K, Kohnen A, Aicher A, Liehn EA, Buchse T, Stein S, Weber C, Dimmeler S, Brandes RP. NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ Res. 2009;105:537–544. doi: 10.1161/CIRCRESAHA.109.205138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.