

Summary
The most common, and usually the only, endocrine disturbance in patients with hypothalamic hamartoma (HH) and epilepsy is central precocious puberty (CPP). The mechanism for CPP associated with HH may relate to ectopic generation and pulsatile release of gonadotropin-releasing hormone (GnRH) from the HH, but this remains an unproven hypothesis. Possible regulators of GnRH release that are intrinsic to HH tissue include the following: (1) glial factors (such as transforming growth factor α[TGFα) and (2) γ-aminobutyric acid (GABA)–mediated excitation. Both are known to be present in surgically-resected HH tissue, but are present in patients with and without a history of CPP, suggesting the possibility that symptoms related to HH are directly associated with the region of anatomic attachment of the HH to the hypothalamus, which determines functional network connections, rather than to differences in HH tissue expression or pathophysiology. CPP associated with HH presents with isosexual development prior to the age of 8 years in girls and 9 years in boys. It is not uncommon for CPP with HH to present in children at an earlier age in comparison to other causes of CPP, including in infancy. Surgical resection of the HH can be effective for treating CPP, but is reserved for patients with intractable epilepsy, since GnRH agonists are widely available and effective treatment. Other endocrine disturbances with HH are rare, but can include growth hormone deficiency, hypothyroidism, and adrenal insufficiency. Diabetes insipidus is commonly encountered postoperatively, but is not observed with HH prior to surgical intervention.
Keywords: Precocious puberty, Gelastic seizure, Hypothalamus, Pituitary, Gonadotropin-releasing hormone
Hypothalamic hamartoma (HH) are rare, congenital, benign mass lesions, located in the ventral hypothalamus. For the neurologist, they are highly associated with gelastic seizures and treatment-resistant epilepsy. An alternative clinical syndrome associated with HH is central precocious puberty (CPP), which can coexist in approximately 40% of HH patients with epilepsy. Other disturbances of endocrine function can also be observed in children with HH, particularly after surgical treatment. Herein, we review our current understanding of this complex neuroendocrine disease with a focus on hormonal abnormalities prior to any surgical intervention. We also identify important unanswered questions for future research.
Hypothalamic Hamartoma: Two Clinical Phenotypes
Two classical clinical phenotypes are associated with HH, consisting of the development of CPP or the presence of gelastic (laughing) seizures. As might be expected, patients with CPP and HH are referred to endocrinologists for evaluation and management, whereas children with epilepsy and HH are typically referred to pediatric neurologists. Textbook discussions usually reinforce this dichotomy, as they tend to be written by one specialist or the other, reflecting the ascertainment bias of the author. However, CPP and epilepsy frequently coexist in up to 45% of patients undergoing surgical treatment for epilepsy.1,2 The expertise of both clinical fields is required for multidisciplinary care of these rare and complex patients. This paper attempts to bridge that gap.
HH are highly diverse with respect to their anatomy and clinical expression. However, the anatomy of the HH as revealed by magnetic resonance imaging (MRI) is predictive of the clinical syndrome and it is appropriate to consider two recognized clinicopathologic subtypes. HH lesions associated with CPP usually have a base of attachment to the inferior surface of the hypothalamus, often with a thin stalk that arises from the region of the tuber cinereum. This result is a horizontal plane of attachment below the floor of the third ventricle (see Fig. 1D). These lesions are referred to as “parahypothalamic” or “pedunculated,” depending on the author.3–8 Conversely, HH lesions associated with gelastic seizures and treatment-resistant epilepsy usually have their base of attachment within the third ventricle, resulting in a vertical plane of attachment above the floor of the third ventricle (see Fig. 1A). These lesions are referred to as “intrahypothalamic” or “sessile.”3–8
Figure 1.
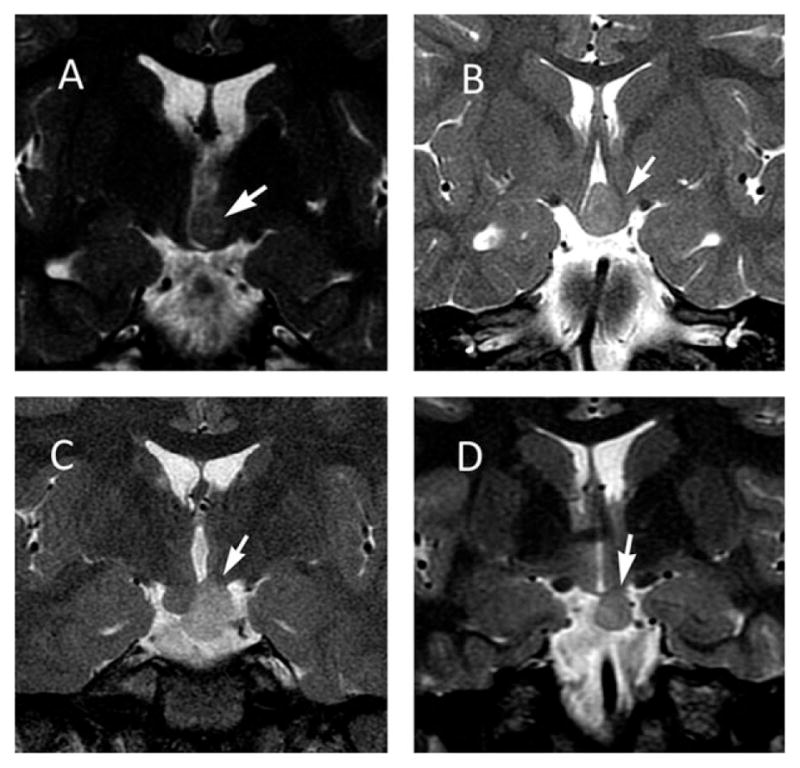
Series of four MRI slices in the coronal plan utilizing T2-weighted fast spin echo (FSE) sequences. Each is acquired from a different patient, all of whom had a history of gelastic seizures and treatment-resistant epilepsy requiring surgical resection. (A) Intrahypothalamic HH lesion (arrow) is attached vertically to the left lateral wall of the third ventricle, completely above the floor of the third ventricle. (B) Intermediate anatomy. (C) Intermediate anatomy. (D) Parahypothalamic HH lesion (arrow) attached horizontally to the underside of the hypothalamus on the right side, completely below the floor of the third ventricle. This lesion is pedunculated, but with a broad base of attachment that includes the region of the tuber cinereum. This patient also had history of central precocious puberty. (Copyright of Barrow Neurological Institute, used with permission.)
However, HH anatomy that is intermediate between these two prototypical subtypes is common. The HH lesions of individual patients occur on a smooth continuum between the poles of the parahypothalamic and intrahypothalamic subtypes (see Fig. 1). In our series of HH patients undergoing surgical treatment for epilepsy (n = 193), 40% have intermediate anatomic forms (see Figure 1). This continuum is recognized by the available classification systems for HH as proposed by Delalande and others.9 The Delalande classification system is shown in Figure 2. Type I lesions are parahypothalamic, with attachment completely below the normal plane of the floor of the third ventricle and type II are intrahypothalamic, with attachment completely above the floor of the third ventricle, whereas types II and IV have intermediate anatomy with attachment both above and below the floor of the third ventricle. Type III and IV lesions are often larger and are more likely to have bilateral, rather than unilateral, attachment to the hypothalamus (type IV are defined as “giant” lesions).9
Figure 2.

Classification system for HH, proposed by Delalande and Fohlen.9 Type I lesions have a horizontal base of attachment below the normal position of the floor of the third ventricle (also see Fig. 1D). These are most commonly attached in the region of the tuber cinereum and cause central precocious puberty. However, large type I HH lesions that attach both anteriorly and posteriorly may have precocious puberty and gelastic seizures. Type II lesions have a vertical plane of attachment to the wall of the third ventricle, completely above the normal position of the floor of the third ventricle. Type III lesions may be unilateral or bilateral, and have a plane of attachment that extends both above and below the normal position of the floor of the third ventricle. Consequently, these lesions have both vertical and horizontal planes of attachment. Type IV lesions were termed “giant” by Delalande and Fohlen. Our group uses a lesion volume of 8 cm3 as our criteria for using the type IV classification utilizing the formula for the volume of an ellipsoid derived from measuring the three primary axes. (Figure Copyright Barrow Neurological Institute, used with permission.)
The clinical symptoms of HH correlate with the region of attachment to the hypothalamus. Recent evidence suggests that the location of the HH on the anterior to posterior axis of the hypothalamus (that is, when viewed with the midline sagittal sequence on MRI) is the most important determining factor. HHs that occur in the anterior hypothalamus with attachment to the tuber cinereum and pituitary stalk are strongly correlated with CPP. Chan et al.10 evaluated a cohort of HH patients with epilepsy by comparing those with and without a prior history of CPP. Expression profiling of surgically resected HH tissue failed to reveal predictive findings (more on this below), but anatomic features on pre-operative MRI were predictive, such that all lesions associated with a prior history of CPP were located in the anterior hypothalamus with attachment or direct contact with the tuber cinereum or infundibulum (7 of 7; 100%), whereas most patients without a prior history of CPP had lesions that did not contact or attach to these structures (2 of 11; 18%; p = 0.002).10
Conversely, HH lesions associated with gelastic seizures and epilepsy (along with associated comorbidity including cognitive impairment and psychiatric symptoms) are in the posterior hypothalamus. Parvizi et al.11 have evaluated the MR imaging of 100 patients with HH and treatment-resistant epilepsy, and have found that all subjects (100%) had anatomic attachment of the HH lesions at the level of the mammillary bodies, as determined by high-resolution coronal imaging sequences. In an earlier report evaluating MRI in 72 patients with HH and epilepsy, Freeman et al.8 observed that 100% of cases included localization in the mammillary region of the hypothalamus when viewed on sagittal sequences and noted the intimate relationship between HH and the columns of the fornix, mammillary bodies, and mammillothalamic tracts that contribute to the limbic circuit.
HH lesions that attach to both the anterior and posterior regions of the hypothalamus (which in turn is highly associated with larger lesion volume of the HH) correlate with both gelastic seizures and CPP. In cohorts of HH cases with epilepsy, there is a significant association between larger lesion size and proximity to the tuber cinereum and pituitary stalk in those cases with a history of CPP in comparison to those who do not.8,10,11
Epidemiology
HH is a relatively rare abnormality. A population-based study in Sweden suggested that the prevalence of HH with epilepsy is one in 200,000 children and adolescents,12 whereas a similar study in Israel suggested a prevalence of HH with gelastic seizures to be one in 625,000 children.13 There are no recognized ethnic or geographical risk factors, although several studies have shown a slightly higher incidence in males.7,14 We are not aware of any population-based studies addressing the prevalence of HH and CPP, although male predominance is also recognized for these patients.15
There appears to be an approximate balance between the number of HH patients presenting with CPP or epilepsy. Based on a comprehensive review of the literature in 2003, consisting of 277 sporadic HH cases, Nguyen et al.16 reported that 104 (38%) had CPP only, 100 (36%) had epilepsy only, and 70 patients (25%) had both CPP and epilepsy. In a series originating from a single center in the People’s Republic of China, where surgical resection was the treatment of choice for both epilepsy and CPP (therefore minimizing referral bias), Li et al.14 reported a series of 214 patients, in which the initial symptoms consisted of CPP in 94 (44%) and gelastic seizures or other seizure types in 106 (50%), while 14 (7%) were deemed asymptomatic.
For HH patients undergoing surgery for treatment-resistant epilepsy, a prior history of CPP is present in approximately 40%.1,2,17 This likelihood correlates with the Delalande type, as discussed earlier. For patients with CCP secondary to HH followed in an endocrinology clinic at a single referral center, Cukier et al.18 reported comorbidity of at least one unprovoked seizure in 5 (33%) of 15 patients.
Clinical Features
Neurologic features of HH, consisting of gelastic seizures and often the later development of additional seizures types in association with cognitive and psychiatric comorbidity, are discussed separately in this issue and elsewhere.16,19–21 Prior to addressing the endocrine symptoms associated with HH, which (at least prior to any kind of surgical intervention) consist almost exclusively of CPP, we will briefly review the normal clinical course and physiology of puberty with an eye toward exploring basic mechanisms.
Normal Puberty
The neuroendocrine-gonadal axis matures between 20 and 24 weeks of gestation.22 Fetal levels of gonadotropin-releasing hormone (GnRH), luteinizing hormone (LH), and follicle-stimulating hormone (FSH), which are critical for developing sex-specific structures in utero, are elevated until just after birth and peak again between 1 and 4 months of life.23 Central inhibition of GnRH secretion then dominates until onset of puberty.24 LH and FSH are responsible for gonadal growth and sex steroid production.
Adrenarche is characterized by increased production of androgens by the adrenal zona reticularis, leading to pilosebaceous unit maturation, adult sweat gland function, and pubic hair growth. In females, ovarian growth leads to increased estrogen synthesis which is first evidenced by thelarche, occurring at an average age of 11 years. Other estrogen effects include increased thickness and secretory activity of the vaginal mucosa, and uterine and endometrial proliferation that culminates in menarche, on average just prior to 13 years of age. Pubic hair becomes more pronounced a few months following thelarche, and peak growth velocity is attained between thelarche and menarche.25 In males, the first sign of puberty is testicular enlargement occurring on average at 11.5 years of age, followed by genital and body hair growth. Peak growth velocity occurs on average at 13.5 years of age and increased muscle mass follows.
Current consensus is that puberty does not normally begin before 8 years of age in females and 9 years in males.26 The mechanisms by which normal puberty is achieved are not fully understood, but include a final common pathway consisting of pulsatile release of GnRH by specialized GnRH-secreting neurons in the hypothalamus, which in turn stimulates gonadotrophs in the anterior pituitary to release LH and FSH.27 These hormones act on the gonads, causing gonadal hypertrophy and production of gender-specific sex steroids, which are responsible for secondary sexual characteristics.
GnRH is a neuropeptide comprised of 10 amino acids, produced by neurons in the medial basal region of the hypothalamus in humans. These neurons are dispersed (without forming a highly discrete nucleus) and relatively few in number (1,000–3,000 total neurons).27 With onset of puberty, GnRH is enzymatically processed and packaged into secretory granules within neuronal processes which project to the median eminence and then are released to enter the hypophyseal portal system in pulses that occur every 60–120 min.28 This pulsatile pattern is required to successfully stimulate production of LH and FSH from the gonadotroph cells in the anterior pituitary gland. The release of LH and FSH from the anterior pituitary is also pulsatile.
Although the physiology of this portion of the hypothalamic–pituitary–gonadal axis is well-established, the mechanism by which the hypothalamus initiates pulsatile release of GnRH is not. The basic mechanisms by which puberty is triggered are complex, integrating multiple pathways and ligands, and are incompletely understood.29 Many genes that contribute to the development and function of hypothalamic GnRH-expressing neurons have been identified. Mutations within these genes in humans and animal models help to dissect these networks, suggesting that no single gene mutation is sufficient to explain the initiation and maintenance of the process.30 Some of the pathways that contribute to the initiation of puberty will be briefly highlighted.
G-protein–coupled receptor 54 (GPR54) and its peptide ligand kisspeptin are candidates for key regulators of pubertal onset.31,32 GPR54 is expressed on the cell surface of GnRH-secreting neurons, and kisspeptin binding directly activates the secretion of GnRH. Expression levels of messenger RNA (mRNA) for kisspeptin and GPR54 increase dramatically at the time of puberty.33 Loss of function mutations in the GPR54 gene were determined to be responsible for idiopathic hypogonadotropic hypogonadism in affected families.34,35 Exogenous administration of kisspeptin to juvenile primates results in marked gonadotropin release.36
Growth factors derived from glia also have a contributing role for the onset of puberty.30,37 Glia produce signaling molecules that stimulate GnRH release, including growth factors that act through tyrosine kinase receptor activation on target cells, such as transforming growth factor-α (TGFα), although many such ligands have been identified.38 TGFα binds to erbB1 receptors on local astrocytes and tanycytes (specialized supporting cells that attach to the ependyma of the third ventricle and penetrate into the hypothalamus with a functional role that includes transmitting neuroendocrine signaling compounds). Activation of erbB1 receptors in turn generates chemical messengers such as prostaglandin E2 (PGE2) which binds directly to GnRH-secreting neurons and enhances GnRH section.39 Although mutations within the TGFα pathway have not been identified in humans, overexpression of human TGFα in a transgenic mouse model facilitates precocious sexual development.39,40
Trans-synaptic neuron-to-neuron projections influence the functional activity of GnRH-secreting neurons. In the fully mature (post-pubertal state), glutamate mediates excitation and promotes GnRH release, whereas GABA exerts its customary role as an inhibitory neurotransmitter and inhibits GnRH secretion in some neurons.41,42 However, there is compelling evidence that GABA exerts an excitatory role and promotes the release of GnRH in the juvenile state and after puberty.43–46 This paradoxical excitatory influence of GABA is likely due to reversal of the transmembrane chloride gradient, as described over the last decade in multiple neuronal networks, including hypothalamic hamartoma.29,46 See Figure 3 for representation of the multiple factors that influence GnRH secretion. We will return to these factors when we discuss the pathophysiology of CPP associated with HH.
Figure 3.
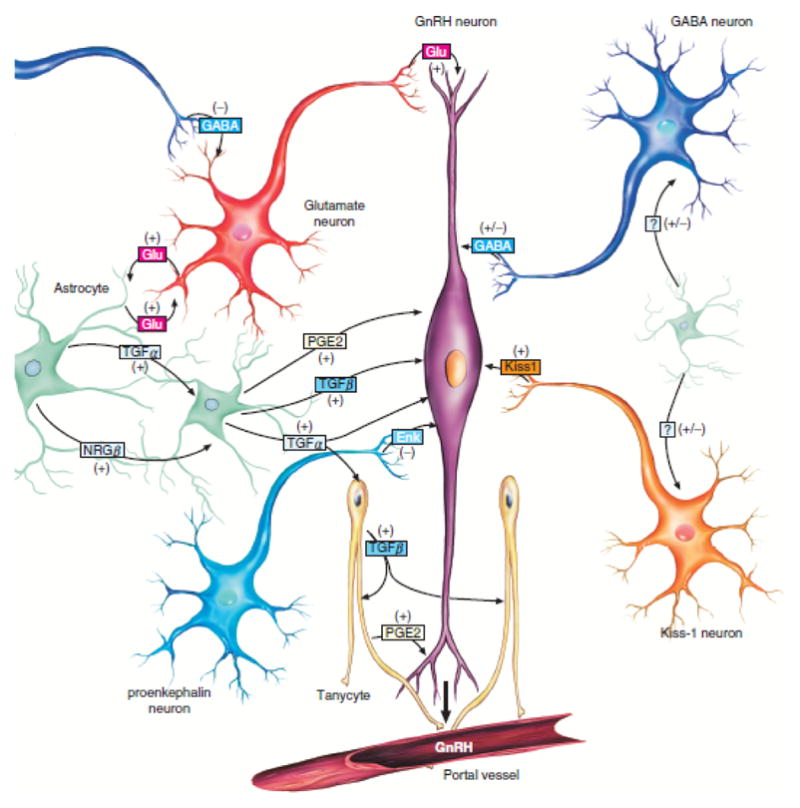
Cellular and molecular mechanisms contributing to GnRH release by normal human hypothalamus. Pulsatile release of GnRH from the GnRH neurons (purple) located in the medial basal hypothalamus is the final common pathway. Excitatory (glutamatergic) and inhibitory (GABAergic) neurons project directly onto the GnRH neuron. At least a subset of GnRH neurons demonstrates paradoxical excitation with GABA input, which is also observed in large HH neurons. Glia-generated influences are also present, with excitation mediated by transforming growth factor α (TGFα) and downstream factors. Kisspeptin-expressing neurons also project directly onto GnRH neurons. HH tissue is universally positive for TGFα but negative for kisspeptin expression.10 The exact molecular mechanisms responsible for premature pulsatile release of GnRH in association with HH are not understood, but ectopic release from the HH lesion is a viable hypothesis. (From Lomniczi A, Matagne V, Ojeda SR. Neuroendocrinology of puberty. In: Squire LR (Ed). Encyclopedia of Neuroscience. Elsevier, London, 2009. Used with permission.)
Premature Thelarche, Adrenarche and Pubarche
In girls, premature thelarche, when isolated, is usually a benign and self-limited condition without elevation of LH and estradiol to pubertal levels. However, premature thelarche is also encountered as the first sign of abnormal isosexual development with CPP with the distinguishing feature being pubertal levels of LH and estradiol (basal levels or levels obtained by GnRH stimulation testing). For girls with premature thelarche and otherwise normal endocrinologic evaluation at baseline, 14% will progress to CPP in time.47
In boys, premature testicular enlargement prior to 9 years is abnormal and further evaluation is indicated. Premature adrenarche may occur in isolation without testicular enlargement to pubertal size, but this requires evaluation to rule out causes of peripheral precocious puberty. The complete differential diagnosis for early changes of puberty is complex and beyond the scope of this article.
The main endocrine finding associated with HH is isosexual CPP. The average age of presentation for females is 2.5 years and for males is 3.7 years (see Table 1). In contrast, idiopathic CPP has an average age of onset of 5 years. A young child presenting with CPP has up to a 30% chance of having HH, as this is the most common non-idiopathic or secondary cause.48 It is likely that males are diagnosed on average a year later, because of more subtle findings. Females are noted to have breast development and pubic hair followed by menarche if untreated. In males, the development of pubic hair and penile enlargement usually leads to the diagnosis of CPP since testicular enlargement may be overlooked for some time.
Table 1.
Clinical features: central precocious puberty secondary to hypothalamic hamartoma
| Male mean [range] | Female mean [range] | |
|---|---|---|
| First physical sign of puberty (age in years) | 3.7 [0.5–9.4] | 2.5 [0.1–8.0] |
| First signs of thelarche (age in years) | NA | 1.7 [0.5–4.0] |
| First signs of pubarche (age in years) | 4.1 [0.6–10.3] | 3.8 [1.0–9.3] |
| First menses (menarche) (age in years) | NA | 1.3 [0.5–2.2] |
| First gelastic seizures (age in years) | 2.6 | 3.1 |
| Combined (male and female) mean [range] | ||
|
|
||
| Bone age minus chronological age (years) | 3.3 [0.4–10.5] | |
| Bone age elevation (SD above age-related normal) | 5.5 [1.6–7.8] | |
| Height (SD above age-related normal) | 2.8 [0–5.5] | |
| Comorbidity CPP and gelastic seizures together (%) | 36% | |
NA, not applicable; SD, standard deviation; CPP, central precocious puberty.
If left untreated, HH-induced CPP will progress to include all the physical changes associated with normal puberty, including changes in skin and hair (increased tendency to acne and hirsutism), increased muscle mass and bone growth, genital enlargement, behavior changes, and potentially even reproductive capacity. Although height is abnormally advanced at time of the precocious puberty, final adult height potential is drastically reduced due to early closure of bony growth plates.49 The diagnosis of CPP is confirmed by elevated serum levels of LH and FSH. GnRH stimulation testing may be performed if the diagnosis of CPP is unclear, identifying excessive elevation of LH and estradiol relative to age-matched normal values. A reasonable diagnostic evaluation for a child presenting with CPP includes a pelvic ultrasound and brain MRI.49
Treatment
CPP is treated effectively with GnRH agonist therapy. Exogenous administration in a nonpulsatile manner inhibits release of LH and FSH by the anterior pituitary with suppression of secondary sexual development and improvement in mature adult height.50 The success of GnRH agonist therapy is high and is discontinued when puberty is desired.51 Depot formulations given every 1–3 months or yearly implants of various GnRH agonists are commercially available. Monitoring pubertal progression is often all that is necessary, but random or stimulated LH and sex steroid levels can also be monitored in the laboratory to confirm efficacy. Pubic hair does not always regress as adrenarche can continue regardless of hypothalamic-pituitary-gonadal axis suppression.
GnRH agonist therapy is usually safe. Side effects include adverse skin reactions (sometimes requiring desensitization) in the absence of immunoglobulin G (IgG) antibodies toward the drug.52 An uncontrolled study has suggested that slipped capital femoral epiphysis can occur in children during therapy with GnRH-agonists, a series that included one girl with HH.53 The efficacy of GnRH agonists to suppress puberty associated with HH is high.54 Failures are most often caused by medication intolerance, usually local injection site reactions, which have been reported to occur in 1–8% of patients.
Pulsatile GnRH causes increased expression and release of LH and FSH, but continuous GnRH agonist therapy causes a decrease in gonadotroph GnRH-receptor expression as the likely mechanism of action.55 Therapy has been shown to cause regression of testicular or ovarian growth back to normal prepubertal volumes. Boys treated with GnRH agonists for CPP resulting from HH have normal physical examination and physiologic function of the pituitary-gonadal axis (as determined by GnRH stimulation testing) with mean follow-up of almost 9 years (range 4–13 years).56 Likewise, long-term outcome for girls treated with GnRH agonist therapy appears favorable.57,58 Menarche ensues between 9 and 18 months after discontinuation of therapy, and fertility is preserved.59
Surgical treatment (utilizing one of the multiple modalities and approaches now available) is usually required for the treatment of epilepsy associated with HH. As noted earlier, CPP associated with HH is usually treated medically with GnRH-agonist therapy. However, surgery is known to be effective for treating CPP.15,60,61 Surgical resection continues to be the treatment of choice for CPP associated with HH in communities where the cost of GnRH-agonist therapy is prohibitive or otherwise not available.14,62 Failure of surgical resection likely relates to incomplete resection of the HH lesion.63 Surgical resection is associated with possible complications, the details of which are beyond the scope of this review, but may include pituitary endocrinopathies and hypothalamic obesity syndrome.
Molecular and Cellular Pathogenesis
Although the location (the region of physical attachment to the hypothalamus) of HH lesions that cause CPP is firmly established, the molecular and cellular pathophysiology leading to premature initiation of puberty is not.64 The need for pulsatile release of GnRH as a final common pathway for initiation of puberty has been mentioned. Long-standing hypotheses for CPP associated with HH include premature initiation of pulsatile GnRH release by adjacent normal hypothalamus due to physical pressure or neurosecretory influences of the nearby HH. Alternatively, the HH itself may be an independent generator of pulsatile GnRH which in turn directly stimulates the anterior pituitary.15,64
Immunohistochemical studies have consistently demonstrated expression of GnRH protein (also referred to as luteinizing hormone–releasing hormone [LHRH]) in HH tissue.10,65–67 Ultrastructural examination of HH tissue demonstrates dense-core vesicles consistent with neuropeptide-containing neurosecretory granules.65,66,68 Although these findings are consistent with the hypothesis that HH tissue acts as an ectopic generator of pulsatile GnRH, it is important to note that there is no direct experimental evidence supporting physiologic release of GnRH from HH lesions. Moreover, immunoreactivity for GnRH in HH tissue has not been universally observed. Jung et al.69 report negative immunoreactivity for GnRH in two HH cases with prior history of CPP.
Surgically resected HH tissue has been investigated for the expression of pathways that are known to regulate GnRH release in normal hypothalamus, as potential candidates for regulating GnRH release from HH tissue, or alternatively, influencing nearby normal GnRH-secreting neurons in the medial basal hypothalamus. Expression of TGFα has been observed with positive immunoreactivity in HH cases with a prior history of CPP (n = 9).10,69 However, Chan et al.10 also report positive TGFα expression in the control group of HH cases without a prior history of CPP (n = 6). That is, all surgically resected HH specimens thus far reported have shown positive immunoreactivity for TGFα. The differentiating characteristic between those with and without a prior history of CPP was the region of attachment of the HH to the hypothalamus, as discussed previously.10
Conversely, it appears that most surgically resected HH tissue specimens, either with or without a prior history of CPP, do not express kisspeptin, as evaluated with reverse transcription polymerase chain reaction (RT-PCR) for KISS1 mRNA. Likewise, expression of mRNA for GPR54 (the kisspeptin receptor) was negative in HH tissue, regardless of the prior history for CPP.10
Our hypothesis is that all HH lesions (with or without a prior history of CPP) have similar expression patterns and cellular physiology, and that the clinical phenotype associated with HH relates directly to the region of the hypothalamus to which they anatomically (and functionally) connect. HHs that are functionally connected to the region of the pituitary stalk and tuber cinereum can cause CPP, whereas those functionally connected to the region of the mammillary bodies and limbic circuit result in epilepsy. We predict that transcription or proteomic profiling of HH tissue cohorts with and without CPP would show relatively limited differences. (Another prediction based on this hypothesis is that all HH lesions can generate intrinsic seizure activity, but gelastic seizures are clinically apparent only when there is functional integration into the neuronal networks present in the posterior hypothalamus.)
Additional links between the physiology of normal puberty and our current understanding of HH deserve speculative comment. Normal GnRH-secreting neurons in the medial basal region of the hypothalamus receive abundant local GABAergic innervation.70 GABAergic afferents to GnRH-secreting neurons integrate the influence of other physiologic effectors, including neuropeptide Y and leptin, which in turn reflect the appropriate energy balance required for puberty.71 GnRH neurosecretory neurons are paradoxically excited with depolarization to GABA throughout much of development.43,44,72
Large projection-type neurons in HH tissue are highly innervated by abundant surrounding GABAergic interneurons, which have been shown to have intrinsic pacemaker-like firing behavior.73 GnRH immunoreactivity in surgically resected HH tissue is observed in both large and small HH neurons (Kerrigan JF, Coons S unpublished observation). Large HH neurons have the immature property of depolarizing and firing in response to GABA ligands.74 This raises a hypothesis as to whether pulsatile release of GnRH from HH tissue could relate to the highly synchronized firing activity that is present in HH lesions.75 In vivo firing activity of GnRH-expressing neurons in normal mouse hypothalamus resulting in physiologic GnRH release (minimum of 10 Hz firing for 2 min duration in this experimental system) is well within the realm of paroxysmal network firing activity in human HH tissue.76 Study of perfused freshly resected HH tissue slices with electrophysiologic and pharmacologic methods would be valuable in addressing these unresolved questions.
Other Endocrine Manifestations of HH
The hallmark endocrine feature of HH is central precocious puberty. CPP usually occurs in isolation without other endocrine disturbances. This observation alone tends to suggest that HH lesions do not affect hypothalamic function by non-specific infiltration or mass effect, but rather give rise to symptoms based on specific interactions with normal neuronal and endocrine networks.
Taylor et al.77 retrospectively examined their experience with hypothalamic and pituitary disorders in their single-center endocrinology referral program. Of 176 patients, 22 (12.5%) had HH: one was completely asymptomatic, 4 had exclusively neurologic symptoms without endocrinopathy, and 17 had an endocrine disturbance. Of these 17 patients, all were diagnosed with CPP, and no other endocrine disturbance was identified (although 8 of 22 cases were identified as having obesity).77
Diabetes insipidus is commonly encountered as an acute and usually transient post-operative complication in 15–55% of HH patients undergoing transcallosal resection,1,2 but has not been reported prior to surgical intervention. We have observed that increased appetite and risk of obesity is the single most common long-term postoperative complication after HH resection, occurring in up to 20% of patients.2 We have not observed that HH patients with epilepsy have problems with obesity above and beyond that of other patients with treatment-resistant epilepsy prior to surgical intervention, but this has not been subjected to systematic investigation.
Martin et al.78 report a single case with growth-hormone deficiency and hypogonadotropic hypogonadism in a 17-year-old male adolescent with HH, deafness, short stature and delayed puberty. Nakhaeimoghadam et al.79 also report a single case of delayed onset of puberty in association with HH in a 14-year-old male adolescent with an earlier history for microphallus and testosterone therapy. We have not encountered delayed puberty in our HH case series to date (n = 193).
Voyadzis et al.80 report a single case with excessive corticotropin-releasing hormone (CRH) secretion in a 2-year-old boy with HH characterized clinically by excessive behavioral lability and paroxysms of rage. There were no physical signs of endocrinopathy, but CRH and adrenocorticotropic hormone (ACTH) levels were elevated and did not suppress with dexamethasone administration. Upon HH resection, behavior improved and CRH and ACTH levels normalized. Immunohistochemistry on the resected HH tissue showed positive immunoreactivity for CRH. These authors concluded that the HH lesion was CRH-secreting.80 We have not encountered a similar case, but CRH and ACTH levels (and their possible association with behavioral symptoms) have not been the subject of systematic investigation.
Rousseau-Nepton et al. report a 14-year-old girl with prior history of CPP and treatment with GnRH agonist therapy. There was no history for seizures. At age 14 years she had short stature, obesity and amenorrhea. Evaluation led to brain imaging and the diagnosis of HH at that time. Growth hormone stimulation testing confirmed growth hormone deficiency when evaluated at age 15 and 18 years.81
In previously published reports from our center (Barrow Neurological Institute) describing surgical intervention and outcome for HH patients with treatment-resistant epilepsy (N = 123 patients), we have observed the following endocrine abnormalities prior to any surgical treatment: CPP 40 patients (32.5%), hypothyroidism 3 (2.4%), growth hormone deficiency 1 (0.8%), hypothyroidism and glucocorticoid deficiency 1 (0.8%), and panhypopituitarism 1 (0.8%).2,17,82–84
Key Points.
Forty percent of children with hypothalamic hamartoma (HH) and epilepsy have central precocious puberty as a comorbid symptom
Central precocious puberty is more likely to occur with large HH lesions with attachment to the tuber cinereum
Central precocious puberty can be successfully treated with medical or surgical intervention
Other endocrinopathies (prior to surgical treatment) are uncommon in HH patients with epilepsy
Acknowledgments
Funding
Barrow Neurological Foundation and Arizona Biomedical Research Commission (JFK).
Biography
Victor S. Harrison is a Fellow, Division of Endocrinology, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, U.S.A.
Footnotes
Disclosure
The authors declare no conflicts of interest. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Freeman JL, Zacharin M, Rosenfeld JV, et al. The endocrinology of hypothalamic hamartoma surgery for intractable epilepsy. Epileptic Disord. 2003;5:239–247. [PubMed] [Google Scholar]
- 2.Ng YT, Rekate HL, Prenger EC, et al. Transcallosal resection of hypothalamic hamartoma for intractable epilepsy. Epilepsia. 2006;47:1192–1202. doi: 10.1111/j.1528-1167.2006.00516.x. [DOI] [PubMed] [Google Scholar]
- 3.Boyko OB, Curnes JT, Oakes WJ, et al. Hamartomas of the tuber cinereum: CT, MR, and pathologic findings. AJNR Am J Neuroradiol. 1991;12:309–314. [PMC free article] [PubMed] [Google Scholar]
- 4.Valdueza JM, Cristante L, Dammann O, et al. Hypothalamic hamartomas: with special reference to gelastic epilepsy and surgery. Neurosurgery. 1994;34:949–958. doi: 10.1227/00006123-199406000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Arita K, Ikawa F, Kurisu K, et al. The relationship between magnetic resonance imaging findings and clinical manifestations of hypothalamic hamartoma. J Neurosurg. 1999;91:212–220. doi: 10.3171/jns.1999.91.2.0212. [DOI] [PubMed] [Google Scholar]
- 6.Debeneix C, Bourgeois M, Trivin C, et al. Hypothalamic hamartoma: comparison of clinical presentation and magnetic resonance images. Horm Res. 2001;56:12–18. doi: 10.1159/000048084. [DOI] [PubMed] [Google Scholar]
- 7.Jung H, Probst EN, Hauffa BP, et al. Association of morphological characteristics with precocious puberty and/or gelastic seizures in hypothalamic hamartoma. J Clin Endocrinol Metab. 2003;88:4590–4595. doi: 10.1210/jc.2002-022018. [DOI] [PubMed] [Google Scholar]
- 8.Freeman JL, Coleman LT, Wellard RM, et al. MR imaging and spectroscopic study of epileptogenic hypothalamic hamartomas: analysis of 72 cases. AJNR Am J Neuroradiol. 2004;25:450–462. [PMC free article] [PubMed] [Google Scholar]
- 9.Delalande O, Fohlen M. Disconnecting surgical treatment of hypothalamic hamartoma in children and adults with refractory epilepsy and proposal of a new classification. Neurol Med Chir (Tokyo) 2003;43:61–68. doi: 10.2176/nmc.43.61. [DOI] [PubMed] [Google Scholar]
- 10.Chan YM, Fenoglio KA, Paraschos S, et al. Precocious puberty associated with hypothalamic hamartomas correlates with anatomic features but not with expression of GnRH, TGFα, or KISS1. Horm Res Paediatr. 2010;73:312–319. doi: 10.1159/000308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parvizi J, Le S, Foster B, et al. Gelastic epilepsy and hypothalamic hamartomas: neuroanatomical analysis of brain lesions in 100 patients. Brain. 2011;134:2960–2968. doi: 10.1093/brain/awr235. [DOI] [PubMed] [Google Scholar]
- 12.Brandberg G, Raininko R, Eeg-Olofsson O. Hypothalamic hamartoma with gelastic seizures in Swedish children and adolescents. Eu J Pediatr Neurol. 2004;8:35–44. doi: 10.1016/j.ejpn.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Shahar E, Kramer U, Mahajnah M, et al. Pediatric-onset gelastic seizures: clinical data and outcome. Pediatr Neurol. 2007;37:29–34. doi: 10.1016/j.pediatrneurol.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Li CD, Luo SQ, Tang J, et al. Classification of hypothalamic hamartoma and prognostic factors for surgical outcome. Acta Neurol Scand. 2014;130:18–26. doi: 10.1111/ane.12209. [DOI] [PubMed] [Google Scholar]
- 15.Zuniga OF, Tanner SM, Wild WO, et al. Hamartoma of CNS associated with precocious puberty. Am J Dis Child. 1983;137:127–133. doi: 10.1001/archpedi.1983.02140280025007. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen D, Singh S, Zaatreh M, et al. Hypothalamic hamartomas: seven cases and review of the literature. Epilepsy Behav. 2003;4:246–258. doi: 10.1016/s1525-5050(03)00086-6. [DOI] [PubMed] [Google Scholar]
- 17.Ng YT, Rekate HL, Prenger EC, et al. Endoscopic resection of hypothalamic hamartomas for refractory symptomatic epilepsy. Neurology. 2008;70:1543–1548. doi: 10.1212/01.wnl.0000310644.40767.aa. [DOI] [PubMed] [Google Scholar]
- 18.Cukier P, Castro LHM, Banaskiwitz N, et al. The benign spectrum of hypothalamic hamartomas: infrequent epilepsy and normal cognition in patients presenting with central precocious puberty. Seizure. 2013;22:28–32. doi: 10.1016/j.seizure.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Berkovic SF, Andermann F, Melanson D, et al. Hypothalamic hamartomas and ictal laughter: evolution of a characteristic epileptic syndrome and diagnostic value of magnetic resonance imaging. Ann Neurol. 1988;23:429–439. doi: 10.1002/ana.410230502. [DOI] [PubMed] [Google Scholar]
- 20.Kerrigan JF, Ng Y-T, Chung SS, et al. The hypothalamic hamartoma: a model of subcortical epileptogenesis and encephalopathy. Semin Pediatr Neurol. 2005;12:119–131. doi: 10.1016/j.spen.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Mittal S, Mittal M, Montes JL, et al. Hypothalamic hamartomas. Part 1. Clinical, neuroimaging, and neurophysiological characteristics. Neurosurg Focus. 2013;34:E6. doi: 10.3171/2013.3.FOCUS1355. [DOI] [PubMed] [Google Scholar]
- 22.Clements JA, Reyes FI, Winter JS, et al. Ontogenesis of gonadotropin-releasing hormone in the human fetal hypothalamus. Proc Soc Exp Biol Med. 1980;163:437–444. doi: 10.3181/00379727-163-40793. [DOI] [PubMed] [Google Scholar]
- 23.Winter JS, Faiman C, Hobson WC, et al. Pituitary-gonadal relations in infancy: I. Patterns of serum gonadotropin concentrations from birth to four years of age in man and chimpanzee. J Clin Endocrinol Metab. 1975;40:545–551. doi: 10.1210/jcem-40-4-545. [DOI] [PubMed] [Google Scholar]
- 24.Krsmanovic LZ, Stojilkovic SS, Catt KJ. Pulsatile gonadotropin-releasing hormone release and its regulation. Trends Endocrinol Metab. 1996;7:56–59. doi: 10.1016/1043-2760(96)00007-0. [DOI] [PubMed] [Google Scholar]
- 25.Tanner JM, Davies PS. Clinical longitudinal standards for height and height velocity for North American children. J Pediatr. 1985;107:317–329. doi: 10.1016/s0022-3476(85)80501-1. [DOI] [PubMed] [Google Scholar]
- 26.Parent AS, Teilmann G, Juul A, et al. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev. 2003;24:668–693. doi: 10.1210/er.2002-0019. [DOI] [PubMed] [Google Scholar]
- 27.Sisk CL, Foster DL. The neural basis of puberty and adolescence. Nat Neurosci. 2004;7:1040–1047. doi: 10.1038/nn1326. [DOI] [PubMed] [Google Scholar]
- 28.Herde MK, Iremonger KJ, Constantin S, et al. GnRH neurons elaborate a long-range projection with shared axonal and dendritic functions. J Neurosci. 2013;33:12689–12697. doi: 10.1523/JNEUROSCI.0579-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terasawa E, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev. 2001;22:111–151. doi: 10.1210/edrv.22.1.0418. [DOI] [PubMed] [Google Scholar]
- 30.Ojeda SR, Lomniczi A, Mastronardi C, et al. The neuroendocrine regulation of puberty: is the time ripe for a systems biology approach? Endocrinology. 2006;147:1166–1174. doi: 10.1210/en.2005-1136. [DOI] [PubMed] [Google Scholar]
- 31.Dungan HM, Clifton DK, Steiner RA. Kisspeptin neurons as central processors in the regulation of gonadotropin-releasing hormone secretion. Endocrinology. 2006;147:1154–1158. doi: 10.1210/en.2005-1282. [DOI] [PubMed] [Google Scholar]
- 32.Terasawa E, Guerriero KA, Plant TM. Kisspeptin and puberty in mammals. Adv Exp Med Biol. 2013;784:253–273. doi: 10.1007/978-1-4614-6199-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navarro VM, Castellano JM, Fernandez-Fernandez R, et al. Developmental and hormonally regulated messenger ribonucleic acid expression of KiSS-1 and its putative receptor, GPR54, in rat hypothalamus and potent luteinizing hormone-releasing activity of KiSS-1 peptide. Endocrinology. 2004;145:4565–4574. doi: 10.1210/en.2004-0413. [DOI] [PubMed] [Google Scholar]
- 34.Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 35.de Roux N, Genin E, Carel JC, et al. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shahab M, Mastronardi C, Seminara SB, et al. Increased hypothalamic GPR54 signaling: a potential mechanism for initiation of puberty in primates. Proc Natl Acad Sci USA. 2005;102:2129–2134. doi: 10.1073/pnas.0409822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharif A, Baroncini M, Prevot V. Role of glia in the regulation of gonodatropin-releasing hormone neuronal activity and secretion. Neuroendocrinology. 2013;98:1–15. doi: 10.1159/000351867. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Segura LM, McCarthy MM. Role of glia in neuroendocrine function. Endocrinology. 2004;145:1082–1086. doi: 10.1210/en.2003-1383. [DOI] [PubMed] [Google Scholar]
- 39.Ma YJ, Berg-von der Emde K, Rage F, et al. Hypothalamic astrocytes respond to transforming growth factor α with secretion of neuroactive substances that stimulate the release of luteinizing hormone-releasing hormone. Endocrinology. 1997;138:19–25. doi: 10.1210/endo.138.1.4863. [DOI] [PubMed] [Google Scholar]
- 40.Ma YJ, Dissen GA, Merlino G, et al. Overexpression of a human transforming growth factor-α (TGFα) transgene reveals a dual antagonistic role of TGFα in female sexual development. Endocrinology. 1994;135:1392–1400. doi: 10.1210/endo.135.4.7925101. [DOI] [PubMed] [Google Scholar]
- 41.Ottem EN, Godwin JG, Petersen SL. Glutamatergic signaling through the N-methyl-D-aspartate receptor directly activates medial subpopulations of luteinizing hormone-releasing hormone (LHRH) neurons, but does not appear to mediate the effects of estradiol on LHRH gene expression. Endocrinology. 2002;143:4837–4845. doi: 10.1210/en.2002-220707. [DOI] [PubMed] [Google Scholar]
- 42.Han SK, Todman MG, Herbison AE. Endogenous GABA release inhibits the firing of adult gonadotropin-releasing neurons. Endocrinology. 2004;145:495–499. doi: 10.1210/en.2003-1333. [DOI] [PubMed] [Google Scholar]
- 43.DeFazio RA, Heger S, Ojeda SR, et al. Activation of A-type γ-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16:2872–2891. doi: 10.1210/me.2002-0163. [DOI] [PubMed] [Google Scholar]
- 44.Moenter SM, DeFazio RA. Endogenous γ-aminobutyric acid can excite gonadotropin-releasing hormone neurons. Endocrinology. 2005;146:5374–5379. doi: 10.1210/en.2005-0788. [DOI] [PubMed] [Google Scholar]
- 45.Constantin S, Iremonger KJ, Herbison AE. In vivo recordings of GnRH neuron firing reveal heterogeneity and dependence upon GABAA receptor signaling. J Neurosci. 2013;33:9394–9401. doi: 10.1523/JNEUROSCI.0533-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watanabe M, Fukuda A, Nabekura J. The role of GABA in the regulation of GnRH neurons. Front Neurosci. 2014;8:387. doi: 10.3389/fnins.2014.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pasquino AM, Pucarelli I, Passeri F, et al. Progression of premature thelarche to central precocious puberty. J Pediatr. 1995;126:11–14. doi: 10.1016/s0022-3476(95)70492-2. [DOI] [PubMed] [Google Scholar]
- 48.Brito VN, Latronico AC, Arnhold IJ, et al. Update on the etiology, diagnosis and therapeutic management of sexual precocity. Arq Bras Endocrinol Metabol. 2008;52:18–31. doi: 10.1590/s0004-27302008000100005. [DOI] [PubMed] [Google Scholar]
- 49.Bajpai A, Sharma J, Kabra M, et al. Precocious puberty: clinical and endocrine profile and factors indicating neurogenic precocity in Indian children. J Pediatr Endocrinol Metab. 2002;15:1173–1181. doi: 10.1515/jpem.2002.15.8.1173. [DOI] [PubMed] [Google Scholar]
- 50.Boepple PA, Mansfield MJ, Wierman ME, et al. Use of a potent, long acting agonist of gonadotropin-releasing hormone in the treatment of precocious puberty. Endocr Rev. 1986;7:24–33. doi: 10.1210/edrv-7-1-24. [DOI] [PubMed] [Google Scholar]
- 51.Mahachoklertwattana P, Kaplan SL, Grumbach MM. The luteinizing hormone-releasing hormone-secreting hypothalamic hamartoma is a congenital malformation: natural history. J Clin Endocrinol Metab. 1993;77:118–124. doi: 10.1210/jcem.77.1.8325933. [DOI] [PubMed] [Google Scholar]
- 52.Styne DM, Harris DA, Egli CA, et al. Treatment of true precocious puberty with a potent luteinizing hormone-releasing factor agonist: effect on growth, sexual maturation, pelvic sonography, and the hypothalamic-pituitary-gonadal axis. J Clin Endocrinol Metab. 1985;61:142–151. doi: 10.1210/jcem-61-1-142. [DOI] [PubMed] [Google Scholar]
- 53.Inman M, Hursh BE, Mokashi A, et al. Occurrence of slipped capital femoral epiphysis in children undergoing gonadotropin-releasing hormone agonist therapy for the treatment of central precocious puberty. Horm Res Paediatr. 2013;80:64–68. doi: 10.1159/000351028. [DOI] [PubMed] [Google Scholar]
- 54.de Brito VN, Latronico AC, Arnhold IJ, et al. Treatment of gonadotropin dependent precocious puberty due to hypothalamic hamartoma with gonadotropin releasing hormone agonist depot. Arch Dis Child. 1999;80:231–234. doi: 10.1136/adc.80.3.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loumaye E, Catt KJ. Agonist-induced regulation of pituitary receptors for gonadotropin-releasing hormone. Dissociation of receptor recruitment from hormone release in cultured gonadotrophs. J Biol Chem. 1983;258:12002–12009. [PubMed] [Google Scholar]
- 56.Feuillan PP, Jones JV, Barnes KM, et al. Boys with precocious puberty due to hypothalamic hamartoma: reproductive axis after discontinuation of gonadotropin-releasing hormone analog therapy. J Clin Endocrinol Metab. 2000;85:4036–4038. doi: 10.1210/jcem.85.11.6951. [DOI] [PubMed] [Google Scholar]
- 57.Feuillan PP, Jones JV, Barnes K, et al. Reproductive axis after discontinuation of gonadotropin-releasing hormone analog treatment of girls with precocious puberty: long term follow-up comparing girls with hypothalamic hamartoma to those with idiopathic precocious puberty. J Clin Endocrinol Metab. 1999;84:44–49. doi: 10.1210/jcem.84.1.5409. [DOI] [PubMed] [Google Scholar]
- 58.Heger S, Muller M, Ranke M, et al. Long-term GnRH agonist treatment for female central precocious puberty does not impair reproductive function. Mol Cell Endocrinol. 2006;254–255:217–220. doi: 10.1016/j.mce.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 59.Pasquino AM, Pucarelli I, Accardo F, et al. Long-term observation of 87 girls with idiopathic central precocious puberty treated with gonadotropin-releasing hormone analogs: impact on adult height, body mass index, bone mineral content, and reproductive function. J Clin Endocrinol Metab. 2008;93:190–195. doi: 10.1210/jc.2007-1216. [DOI] [PubMed] [Google Scholar]
- 60.Starceski PJ, Lee PA, Albright AL, et al. Hypothalamic hamartomas and sexual precocity: evaluation of treatment options. Am J Dis Child. 1990;144:225–228. doi: 10.1001/archpedi.1990.02150260105040. [DOI] [PubMed] [Google Scholar]
- 61.Albright AL, Lee PA. Hypothalamic hamartomas and sexual precocity. Pediatr Neurosurg. 1992;18:315–319. doi: 10.1159/000120682. [DOI] [PubMed] [Google Scholar]
- 62.Luo S, Li C, Ma Z, et al. Microsurgical treatment for hypothalamic hamartoma in children with precocious puberty. Surg Neurol. 2002;57:356–362. doi: 10.1016/s0090-3019(02)00694-8. [DOI] [PubMed] [Google Scholar]
- 63.Stephen MD, Zage PE, Waguespack SG. Gonadotropin-dependent precocious puberty: neoplastic causes and endocrine considerations. Int J Pediatr Endocrinol. 2011;2011:184502. doi: 10.1155/2011/184502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jung H, Parent A-S, Ojeda SR. Hypothalamic hamartoma: a paradigm/model for studying the onset of puberty. Endocr Dev. 2005;8:81–93. doi: 10.1159/000084095. [DOI] [PubMed] [Google Scholar]
- 65.Judge DM, Kulin HE, Page R, et al. Hypothalamic hamartoma: a source of luteinizing-hormone-releasing factor in precocious puberty. N Engl J Med. 1977;296:7–10. doi: 10.1056/NEJM197701062960102. [DOI] [PubMed] [Google Scholar]
- 66.Hochman HI, Judge DM, Reichlin S. Precocious puberty and hypothalamic hamartoma. Pediatrics. 1981;67:236–244. [PubMed] [Google Scholar]
- 67.Culler FL, James HE, Simon ML, et al. Identification of gonadotropin-releasing hormone in neurons of hypothalamic hamartoma in a boy with precocious puberty. Neurosurgery. 1985;17:408–412. doi: 10.1097/00006123-198509000-00003. [DOI] [PubMed] [Google Scholar]
- 68.Beggs J, Nakada S, Fenoglio K, et al. Hypothalamic hamartomas associated with epilepsy: ultrastructural features. J Neuropathol Exp Neurol. 2008;67:657–668. doi: 10.1097/NEN.0b013e31817d8085. [DOI] [PubMed] [Google Scholar]
- 69.Jung H, Carmel P, Schwartz MS, et al. Some hypothalamic hamartomas contain transforming growth factor alpha, a puberty-inducing growth factor, but not luteinizing hormone-releasing hormone neurons. J Clin Endocrinol Metab. 1999;84:4695–4701. doi: 10.1210/jcem.84.12.6185. [DOI] [PubMed] [Google Scholar]
- 70.Clarkson J, Herbison AE. Development of GABA and glutamate signaling at the GnRH neuron in relation to puberty. Mol Cell Endocrinol. 2006;254–255:32–38. doi: 10.1016/j.mce.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 71.Sullivan SD, Moenter SM. γ-Aminobutyric acid neurons integrate and rapidly transmit permissive and inhibitory metabolic cues to gonadotropin-releasing hormone neurons. Endocrinology. 2004;145:1194–1202. doi: 10.1210/en.2003-1374. [DOI] [PubMed] [Google Scholar]
- 72.Han SK, Abraham IM, Herbison AE. Effect of GABA on GnRH neurons switches from depolarization to hyperpolarization at puberty in the female mouse. Endocrinology. 2002;143:1459–1466. doi: 10.1210/endo.143.4.8724. [DOI] [PubMed] [Google Scholar]
- 73.Wu J, Xu L, Kim DY, et al. Electrophysiological properties of human hypothalamic hamartomas. Ann Neurol. 2005;58:371–382. doi: 10.1002/ana.20580. [DOI] [PubMed] [Google Scholar]
- 74.Wu J, Gao M, Shen JX, et al. Mechanisms of intrinsic epileptogenesis in human gelastic seizures with hypothalamic hamartoma. CNS Neurosci Ther. 2015;21:104–111. doi: 10.1111/cns.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steinmetz PN, Wait SD, Lekovic GP, et al. Firing behavior and network activity of single neurons in human epileptic hypothalamic hamartoma. Front Neurol. 2013;4:210. doi: 10.3389/fneur.2013.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Campos P, Herbison AE. Optogenetic activation of GnRH neurons reveals minimal requirements for pulsatile luteinizing hormone secretion. Proc Natl Acad Sci USA. 2014;111:18387–18392. doi: 10.1073/pnas.1415226112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taylor M, Couto-Silva AC, Adan L, et al. Hypothalamic-pituitary lesions in pediatric patients: endocrine symptoms often precede neuro-ophthalmic presenting symptoms. J Pediatr. 2012;161:855–863. doi: 10.1016/j.jpeds.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 78.Martin DD, Seeger U, Ranke M, et al. MR imaging and spectroscopy of a tuber cinereum hamartoma in a patient with growth hormone deficiency and hypogonadotropic hypogonadism. AJNR Am J Neuro-radiol. 2003;24:1177–1180. [PMC free article] [PubMed] [Google Scholar]
- 79.Nakhaeimoghadam M, Rostami P, Zare-Shahabadi A, et al. Hypothalamic hamartoma in an unusual case with delayed puberty. Acta Med Iran. 2013;51:819–821. [PubMed] [Google Scholar]
- 80.Voyadzis JM, Guttman-Bauman I, Santi M, et al. Hypothalamic hamartoma secreting corticotrophin-releasing hormone. J Neurosurg. 2004;100:212–216. doi: 10.3171/ped.2004.100.2.0212. [DOI] [PubMed] [Google Scholar]
- 81.Rousseau-Nepton I, Kaduri S, Garfield N, et al. Hypothalamic hamartoma associated with central precocious puberty and growth hormone deficiency. J Pediatr Endocrinol Metab. 2014;27:117–121. doi: 10.1515/jpem-2013-0108. [DOI] [PubMed] [Google Scholar]
- 82.Abla AA, Shetter AG, Chang SW, et al. Gamma knife surgery for hypothalamic hamartomas and epilepsy: patient selection and outcomes. J Neurosurg. 2010;113:207–214. doi: 10.3171/2010.8.GKS101027. [DOI] [PubMed] [Google Scholar]
- 83.Abla AA, Rekate HL, Wilson DA, et al. Orbitozygomatic resection for hypothalamic hamartoma and epilepsy: patient selection and outcome. Childs Nerv Syst. 2011;27:265–277. doi: 10.1007/s00381-010-1250-7. [DOI] [PubMed] [Google Scholar]
- 84.Drees C, Chapman K, Prenger E, et al. Seizure outcome and complications following hypothalamic hamartoma treatment in adults: endoscopic, open, and Gamma Knife procedures. J Neurosurg. 2012;117:255–261. doi: 10.3171/2012.5.JNS112256. [DOI] [PubMed] [Google Scholar]
- 85.Albright AL, Lee PA. Neurosurgical treatment of hypothalamic hamartomas causing precocious puberty. J Neurosurg. 1993;78:77–82. doi: 10.3171/jns.1993.78.1.0077. [DOI] [PubMed] [Google Scholar]
- 86.Beningfield SJ, Bonnici F, Cremin BJ. Magnetic resonance imaging of hypothalamic hamartomas. Br J Radiol. 1988;61:1177–1180. doi: 10.1259/0007-1285-61-732-1177. [DOI] [PubMed] [Google Scholar]
- 87.Cascino GD, Andermann F, Berkovic SF, et al. Gelastic seizures and hypothalamic hamartomas: evaluation of patients undergoing chronic intracranial EEG monitoring and outcome of surgical treatment. Neurology. 1993;43:747–750. doi: 10.1212/wnl.43.4.747. [DOI] [PubMed] [Google Scholar]
- 88.Castano De La Mota C, Martin Del Valle F, Perez Villena A, et al. Hypothalamic hamartoma in paediatric patients: clinical characteristics, outcomes and review of the literature. Neurologia. 2012;27:268–276. doi: 10.1016/j.nrl.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 89.Chamouilli JM, Razafimahefa B, Rierron H. Precocious puberty and hypothalamic hamartoma: treatment with triptorelin during eight years. Arch Pediatr. 1995;2:438–441. doi: 10.1016/0929-693x(96)81178-x. [French] [DOI] [PubMed] [Google Scholar]
- 90.Commentz JC, Helmke K. Precocious puberty and decreased melatonin secretion due to a hypothalamic hamartoma. Horm Res. 1995;44:271–275. doi: 10.1159/000184640. [DOI] [PubMed] [Google Scholar]
- 91.Hahn FJ, Leibrock LG, Huseman CA, et al. The MR appearance of hypothalamic hamartoma. Neuroradiology. 1988;30:65–68. doi: 10.1007/BF00341946. [DOI] [PubMed] [Google Scholar]
- 92.Hamilton RL. Case of the month. July 1996-precocious puberty. Brain Pathol. 1997;7:711–712. doi: 10.1111/j.1750-3639.1997.tb01085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsiao PH, Tsai WY, Lee JS, et al. Hypothalamic hamartoma and precocious puberty: report of a case. J Formos Med Assoc. 1992;91:1017–1020. [PubMed] [Google Scholar]
- 94.Katayama H, Miyao M, Kobayashi S, et al. A case of hypothalamic hamartoma with gelastic seizures, precocious puberty, and poly-syndactyly. No To Hattatsu. 1993;25:341–346. [Japanese] [PubMed] [Google Scholar]
- 95.Koelfen W, Wentz J. Precocious puberty and laugh attacks. Monatss-chr Kinderheilkd. 1991;139:479–481. [German] [PubMed] [Google Scholar]
- 96.Kuzniecky R, Guthrie B, Mountz J, et al. Intrinsic epileptogenesis of hypothalamic hamartomas in gelastic epilepsy. Ann Neurol. 1997;42:60–67. doi: 10.1002/ana.410420111. [DOI] [PubMed] [Google Scholar]
- 97.Lona Soto A, Takahashi M, Yamashita Y, et al. MRI findings of hypothalamic hamartoma: report of five cases and review of the literature. Comput Med Imaging Graph. 1991;15:415–421. doi: 10.1016/0895-6111(91)90169-v. [DOI] [PubMed] [Google Scholar]
- 98.Marliani AF, Tampieri D, Melancon D, et al. Magnetic resonance imaging of hypothalamic hamartomas causing gelastic epilepsy. Can Assoc Radiol J. 1991;42:335–359. [PubMed] [Google Scholar]
- 99.Nishio S, Fujiwara S, Aiko Y, et al. Hypothalamic hamartoma. Report of two cases. J Neurosurg. 1989;70:640–645. doi: 10.3171/jns.1989.70.4.0640. [DOI] [PubMed] [Google Scholar]
- 100.Rappaport R, Brauner R. Lack of adrenarche in two children with precocious puberty secondary to hypothalamic hamartoma. Horm Res. 1989;31:226–229. doi: 10.1159/000181121. [DOI] [PubMed] [Google Scholar]
- 101.Robben SG, Tanghe HL, Drop SL. Hypothalamic hamartoma. J Belge Radiol. 1994;77:221. [PubMed] [Google Scholar]
- 102.Romner B, Trumpy JH, Marhaug G, et al. Hypothalamic hamartoma causing precocious puberty treated by surgery: case report. Surg Neurol. 1994;41:306–309. doi: 10.1016/0090-3019(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 103.Shenoy SN, Raja A. Hypothalamic hamartoma with precocious puberty. Pediatr Neurosurg. 2004;40:249–252. doi: 10.1159/000082302. [DOI] [PubMed] [Google Scholar]
- 104.Wentz KU, Kolfen W, Suchalla R. Precocious puberty and gelastic epilepsy in hamartoma of the tuber cinereum. Rofo. 1993;158:280–282. doi: 10.1055/s-2008-1032650. [German] [DOI] [PubMed] [Google Scholar]
- 105.Zaatreh M, Tennison M, Greenwood RS. Successful treatment of hypothalamic seizures and precocious puberty with GnRH analogue. Neurology. 2000;55:1908–1910. doi: 10.1212/wnl.55.12.1908. [DOI] [PubMed] [Google Scholar]
- 106.Zucchini S, di Natale B, Ambrosetto P, et al. Role of magnetic resonance imaging in hypothalamic-pituitary disorders. Horm Res. 1995;44(Suppl 3):8–14. doi: 10.1159/000184666. [DOI] [PubMed] [Google Scholar]