

Abstract
X-linked agammaglobulinemia (XLA) is a rare genetic disorder, caused by mutations in BTK (Bruton’s Tyrosine Kinase) gene. Deep high-throughput RNA sequencing (RNA-Seq) approach was utilized to explore the possible differences in transcriptome profiles of primary monocytes in XLA patients compared with healthy subjects. Our analysis revealed the differences in expression of 1,827 protein-coding genes, 95 annotated long non-coding RNAs (lncRNAs) and 20 novel lincRNAs between XLA patients and healthy subjects. GO and KEGG pathway analysis of differentially expressed (DE) protein-coding genes showed downregulation of several innate immune-related genes and upregulation of oxidative phosphorylation and apoptosis-related genes in XLA patients compared to the healthy subjects. Moreover, the functional prediction analysis of DE lncRNAs revealed their potential role in regulating the monocytes cell cycle and apoptosis in XLA patients. Our results suggested that BTK mutations may contribute to the dysregulation of innate immune system and increase susceptibility to apoptosis in monocytes of XLA patients. This study provides significant finding on the regulation of BTK gene in monocytes and the potential for development of innovative biomarkers and therapeutic monitoring strategies to increase the quality of life in XLA patients.
Introduction
X-linked agammaglobulinemia (XLA) is one of the inherited forms of Primary Immunodeficiency Diseases (PIDs)1. It is caused by mutations in the BTK (Bruton’s Tyrosine Kinase) gene, which results in defective development and maturation of B cell within the bone marrow and a considerable decrease or complete absence of mature B cells in peripheral blood2. Due to the absence of mature B cells, XLA patients have significantly decreased levels of all major immunoglobulins in the serum and consequently, would be subjected to severe and chronic bacterial infections3. The BTK expression is not restricted to B cells, it is also expressed in myeloid cells such as neutrophils4, natural killer (NK) cells5, and monocytes6. The significance of BTK for macrophage function was first seen in X-linked immunodeficient (XID) mice infected with microfilaria7. The experiments showed a delayed microfilaria clearance together with low levels of IL-12A (Interleukin 12A), IL-1 (Interleukin 1) and TNF (Tumor Necrosis Factor) production as well as decrease in NO (Nitric oxide) production in XID mice7. Similarly, Schmidt and colleagues showed that in primary macrophages, BTK was activated by TLR4 (Toll-like Receptor 4) and is essential for normal TLR-induced IL-10 (Interleukin 10) production in various populations of macrophages8. Additionally, BTK also plays a crucial role in initiating TLR3 signaling in BTK deficient macrophages9. In the absence of BTK, TLR3-induced PI3K (Phosphoinositide 3-Kinase), AKT (V-Akt Murine Thymoma Viral Oncogene Homolog 1) and MAPK (MAP Kinase Phosphorylation) signaling as well as activation of NFκB (Nuclear Factor Kappa B), IRF3 (Interferon Regulatory Factor 3), and AP-1 transcription factors were defective9. Further investigations on the human monocytic THP1 cell line showed interactions of TLR8 and TLR9 with BTK, in which defective BTK leads to impaired TLR8 and TLR9 signaling and causes susceptibility of XLA patients to viral infections10. It has also been reported that BTK contributed in TLR4 signaling to NFκB, and may also involve in signaling by ligands for TLR2, TLR6, TLR8, and TLR9 and also with MYD88 (Myeloid Differentiation Primary Response 88), MAL (MyD88-Adapter-Like) and IRAK1 (Interleukin 1 Receptor Associated Kinase 1)11, 12. The decreased chemotaxis and defective FcγR (Fc-gamma Receptors), CR1 (Complement Receptor 1) and CR3 (Complement Receptor 3)-mediated phagocytosis has also been reported in monocytes from XLA patients compared to healthy subjects13.
In addition to the protein coding genes, long non-coding RNAs (lncRNAs) have also been shown to play important roles in immune cell development and processes such as anti-viral responses, NFκB signaling, and inflammatory responses14, 15. lncRNAs are the biggest class of non-coding RNAs in mammalians, having more than 200 nucleotides length and without coding potential16. The lncRNAs dysregulated expression has been also reported in many human disease, such as cancer17, 18, neurological disorders19, autoimmune disease20, 21, and microbial susceptibility22.
Monocytes are essential components of the innate immune system. They are produced from a common myeloid progenitor cells in the bone marrow and circulate in the blood vessels for short times. During inflammatory conditions, they move into peripheral tissues, differentiating into macrophages and dendritic cells. The effect of primary monocyte with BTK deficient in XLA patients is not well studied. There is no or limited data exist on the genome-wide transcriptome expression profile of primary monocytes in XLA patients. In addition, the molecular mechanisms underlying the functions of lncRNAs in primary monocytes of XLA have not been studied yet. We recently published a gene reference catalogue and lncRNAs landscape of human primary monocytes from healthy subjects23, 24. In this study, we performed deep high-throughput RNA sequencing (RNA-Seq) analysis on primary monocytes from XLA patients and healthy subjects to investigate the effect of BTK gene expression deficiency on innate immune function of XLA patients. We identified the set of protein-coding genes and lncRNAs which were differentially expressed (DE) between XLA patients and healthy subjects. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses predicted the functions of these DE protein-coding genes and lncRNAs in primary monocytes of XLA patients.
Results
Transcriptome profiling
We performed deep RNA-Seq of primary monocytes from healthy subjects and XLA patients. The patients were diagnosed to have XLA with absence or low circulating B cells, low serum immunoglobulin isotypes and deficient of BTK protein expression in their monocytes. Molecular genetic tests revealed BTK gene mutations in all patients. Novel BTK invariant splice site mutation was identified in one of the patients25. The history of the patient’s serum immunoglobulin levels before receiving intravenous human immunoglobulin (IVIG) therapy, and the nucleotide change that occurred in each patient and its consequences in the protein synthesis is shown in Table 1.
Table 1.
Clinical and immunological data of the XLA patients.
| Ig levels at diagnosis (mg/dL) | Mutations | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Age (years) | Age at onset (years)a | Age at diagnosis (years)b | Family historyc | IgG | IgM | IgA | CD19+ (%) | BTK expression | Nucleotide | Protein | Protein Domain |
| P1 | 12 | 1 | 4 | − | N/A | N/A | N/A | 1 (12−22) | 7.7% | c.1888A > T | p.M630L | Kinase |
| P2 | 13 | 1 | 6 | + | 41(550−1200) | <12(40−95) | 48 (60–170) | 0 (12−22) | 6% | IVS9 + 1 G > C | Skipping of exon 9 | SH3 & SH2 |
| P3 | 18 | 2 | 7 | − | 91.1 (550−1200) | 11.3 (40−95) | UD (60−170) | 0 (12−22) | 0.04% | g.34430_34447 delCAAAGTCATGATgtgagt | p.A446_N451 ins(28 amino acids) | Kinase |
N/A, not available. UD, undetectable. aAge at the which an individual acquires, develops, or first experiences a condition or symptoms of a disease. bAge at the start of intravenous immunoglobulin replacement. c“ + ”, indicates that family members [boy (s)] died at a young age because of infection. dNormal expression is >94%.
RNA was extracted from classical monocytes (CD14++CD16−) isolated from peripheral blood mononuclear cells (PBMCs) using a negative selection method. The purified RNA was used to carry out poly-adenylated paired-end RNA sequencing using Illumina HiSeq 2000 platform. After discarding adaptor sequences and low-quality sequences, we obtained approximately 1.2 million reads from all samples. The trimmed reads were mapped to human reference genome (Ensembl GRCH38.79) and transcripts were assembled using aligned reads. The transcripts from each XLA patients and healthy subjects were then separately merged to form two sets of single non-redundant transcripts. The expression levels of transcripts were quantified across each dataset. By applying an FPKM > 0.1 threshold, we have identified the expression of total 11,777 protein-coding genes and 3,363 lncRNAs in XLA patients, and 11,644 protein-coding genes and 3,190 lncRNAs in healthy subjects. Using multi-step mapping and filtering criteria, the expression of 430 and 380 potential novel lincRNAs were also identified in XLA patients and healthy subjects, respectively.
Differentially expressed (DE) protein-coding genes and lncRNAs in XLA patients compared to healthy subjects
The differential gene expression analysis between XLA patients and healthy subjects was conducted using Cuffdiff and the genes with q-value ≤ 0.01 and log2 fold-change ≥ 1 or ≤ −1 were defined as differentially expressed (DE). Using these criteria, a total of 1,827 DE protein-coding genes, 95 DE annotated lncRNAs and 20 DE novel long intergenic non-coding RNAs (lincRNAs) were identified in XLA patients compared to the healthy subjects. Figure 1 shows the hierarchical clustering of the expression patterns of DE protein-coding genes and DE lncRNAs between XLA patients and healthy subjects.
Figure 1.
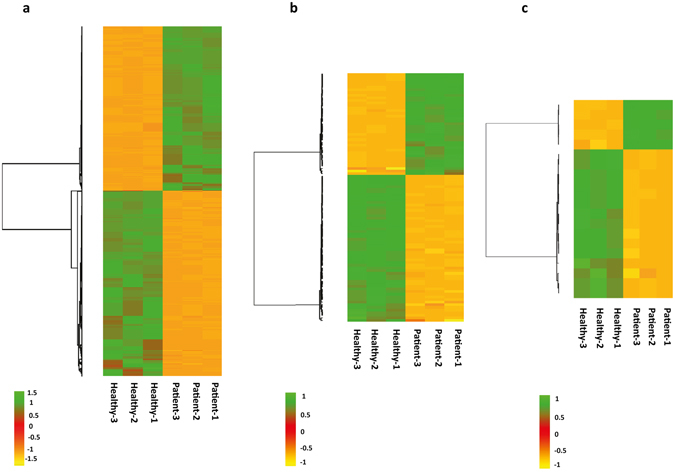
Hierarchical clustering of DE protein-coding genes and DE lncRNAs in primary monocytes of XLA patients compared to healthy subjects. (a) DE protein-coding genes (b) DE annotated lncRNAs and (c) DE novel lincRNAs. The green and orange shades indicate the expression above and below the relative expression, respectively, across all samples.
Differentially expressed (DE) protein-coding genes
Out of the 1,827 DE protein-coding genes, 859 genes were upregulated and 968 genes were downregulated in XLA patients compared to the healthy subjects (Supplementary Table S1). The expression of BTK was detected to be significantly downregulated (log2 fold-change < −7) in XLA patients compared to healthy subjects. The functional consequences of identified DE protein-coding genes in XLA patients compared to healthy subjects were characterized through GO enrichment and KEGG pathway analysis (Fig. 2, Supplementary Table S2). The significant GO biological process terms for upregulated genes were related to mitochondrial function and organization including: “oxidative phosphorylation”, mitochondrial ATP synthesis coupled to electron transport”, and “electron transport chain”, as well as “apoptotic” process and “response to oxidative stress” (Fig. 2a). The expression of several apoptosis-related genes such as BAX (BCL2 Associated X Protein) and BAD (BCL2 Associated Agonist of Cell Death) and oxidative stress response genes such as SOD1 (Superoxide Dismutase 1, Soluble), GPX1 (Glutathione Peroxidase 1), GPX4 (Glutathione Peroxidase 4), PRDX1 (Peroxiredoxin 1), PRDX5 (Peroxiredoxin 5) were observed to be significantly upregulated in XLA patients compared to healthy subjects. However, the GO biological process terms for downregulated genes were significantly related to monocytes immune system functions including: “intracellular signaling cascade”, “immune response” and “innate immune response” (Fig. 2b). Furthermore, the KEGG pathway analysis revealed that the upregulated genes were enriched in 9 pathways, most significantly in “Oxidative phosphorylation” (Fig. 2c). The oxidative phosphorylation system consists of five protein complexes namely; I (NDUF; NADH: Ubiquinone Oxidoreductase), II (SHD; Src Homology 2 Domain Containing Transforming Protein D), III (UQCR; Ubiquinol-Cytochrome C Reductase), IV (COX; Cytochrome C Oxidase), and V (ATP; Adenosine Triphosphate). In this study, the upregulation of the several components of complexes I, III, IV and V were observed in primary monocytes of XLA patients compared to the healthy subjects (Table 2). The GO analysis also revealed that the downregulated genes were enriched in 29 pathways, most significantly in several immune-related pathways such as “Fc gamma R-mediated phagocytosis”, “Chemokine signaling pathway”, “Toll-like receptor signaling pathway” and “MTOR signaling pathway” (Fig. 2d). The core downregulated genes contributing to the enrichment of the immune-related pathways in primary monocyte of XLA patients is presented in Table 3.
Figure 2.
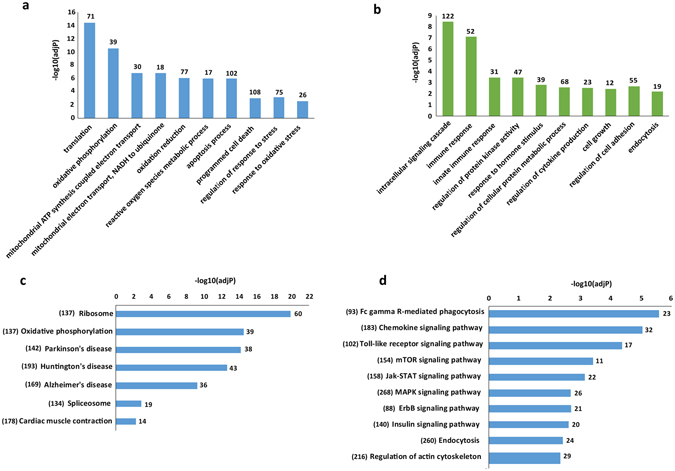
The GO and KEGG pathway analysis of DE protein-coding genes in primary monocytes of XLA patients compared to healthy subjects. The significant GO biological process terms (adj.P-value < 0.01) enriched for (a) upregulated genes, and (b) downregulated genes. The number of identified DE protein-coding genes enriched in each GO terms is depicted above bars of x-axis in the figure. The significant KEGG pathway terms enriched for (c) upregulated genes and (d) downregulated genes. Each statistical significance value (adj.p-value) was negative log-10 base transformed.The numbers in the brackets indicated the total number of genes available in the KEGG database for each pathway terms. The number of identified DE protein-coding genes enriched in each KEGG pathways is depicted above bars of x-axis in the figure.
Table 2.
The upregulated genes involved in Oxidative Phosphorylation pathway in primary monocytes of the XLA patients compared to healthy subjects.
| Oxidative Phosphorylation Pathway (P-value: 6.50E-18; Adj.P-value = 5.20E-16) | |
|---|---|
| Oxidative Phosphorylation system subunits | Gene Name |
| Complex I: NADH dehydrogenase | NDUFA1, NDUFA12, NDUFA2, NDUFA3, NDUFA4, NDUFA6, NDUFA8, NDUFAB1, NDUFB11, NDUFB4, NDUFB5, NDUFB7, NDUFB8, NDUFS4, NDUFS5, NDUFS6, NDUFS7 |
| Complex III: Cytochorom c reductase | UQCR10, UQCRB, UQCRFS1, UQCRH, UQCRHL |
| Complex IV: Cytochorom c oxidase | COX14, COX17, COX4I1, COX5A, COX5B, COX6B1, COX6C, COX7A2, COX7C |
| Complex V: ATPase | ATP1A3, ATP5D, ATP5E, ATP5G2, ATP5G3, ATP5H, ATP5I, ATP5J, ATP6V0B, ATP6V0E1, ATP6V1F, ATPIF1 |
Table 3.
The downregulated genes involved in immune-related pathways in primary monocytes of XLA patients compared to healthy subjects.
| Pathway Terms | P-value | Adj.P-value | Gene Name |
|---|---|---|---|
| Fc gamma R-mediated phagocytosis | 1.10E-07 | 2.60E-06 | FCGR2A, ASAP1, GAB2, DOCK2, DNM1L, INPP5D, MAPK1, MAP2K1, PAK1, PIKFYVE, PIK3CG, PIK3R5, PLCG1, PRKCB, PRKCE, PTPRC, SYK, LYN, VASP, VAV3, PIK3R1, RAF1, AKT2 |
| Chemokines signaling pathway | 4.70E-07 | 9.30E-06 | CXCL16, CXCR4, CXCR1, CXCR2, JAK2, ROCK2, ADCY6, ADCY7, ADCY9, ADRBK2, DOCK2, FOXO3, GRB2, GNB4, GNG12, GNG2, MAPK1, MAP2K1, NRAS, PAK1, PIK3CG, PIK3R1, PIK3R5, PRKCB, PRKX, STAT2, STAT5B, ROCK1, SOS1, SOS2, BRAF, VAV3 |
| Toll-like receptors signaling pathway | 3.10E-06 | 4.30E-05 | TLR1, TLR5, TLR2, TLR4, TLR6, TLR7, TBK1, IKBKE, JUN, MAPK1, MAP2K1, MAP2K4, MYD88, PIK3CG, PIK3R1, PIK3R5, FOS |
| MTOR signaling pathway | 5.20E-05 | 3.80E-04 | MTOR, RICTOR, HIF1A, MAPK1, PIK3CG, PIK3R1, PIK3R5, RPS6KA3, TSC1, ULK2, BRAF |
| Jak-STAT signaling pathway | 1.20E-04 | 7.10E-04 | CREBBP, CBL, EP300, JAK1, JAK2, CSF2RB, GRB2, IL10RA, IL13RA1, IL6R, IL6ST, PIK3CG, PIK3R1, PIK3R5, PRLR, STAT2, STAT5A, STAT5B, SOS1, SOS2, SOCS4, SOCS7 |
| MAPK signaling pathway | 4.30E-04 | 2.10E-03 | RAPGEF2, ATF2, DUSP1, DUSP6, FLNB, GRB2, GNA12, GNG12, HSPA1A, JUN, MAPK1, MAP2K1, MAP2K4, MAP3K1, MAP4K4, NRAS, PAK1, PRKCB, PRKX, RPS6KA3, SOS1, SOS2, TGFBR1, FOS, BRAF |
| ErbB signaling pathway | 5.10E-04 | 2.40E-03 | CBL, EREG, GRB2, JUN, MTOR, MAPK1, MAP2K1, MAP2K4, NRAS, PAK1, PIK3CG, PIK3R, PIK3CG, PIK3R, PIK3R5, PLCG1, PRKCB, STAT5A, STAT5B, SOS1, SOS2, ABL2, BRAF |
| Insulin signaling pathway | 8.30E-04 | 3.80E-03 | CBL, GRB2, INPP5D, IRS2, MTOR, MAPK1, MAP2K1, NRAS, PDE3B, PIK3CG, PIK3R1, PIK3R5, PRKCI, PRKX, PTPRF, SOS1, SOS2, SOCS4, TSC1, BRAF |
| Endocytosis | 1.00E-03 | 4.50E-03 | ARAP2, ASAP1, ACAP2, CBL, DNAJC6, EHD3, IQSEC1, RAB11, FIP1, ADRBK2, CXCR4, CLTCL1, DNM1L, EPN2, HSPA1A, CXCR1, CXCR2, LDLR, NEDD4, PIKFYVE, PRKCI, RNF41, SMAP2, TGFBR1, VPS36 |
| Regulation of actin cytoskeleton | 2.20E-03 | 9.60E-03 | IQGAP1, IQGAP2, ARHGEF12, ROCK2, ACTN1, CYFIP1, FGFR1, FN1, GNA12, GNA13, GNG12, ITGA4, ITGAV, ITGB1, MAPK1, MAP2K1, NRAS, PAK1, PIKFYVE, PIK3CG, PIK3R1, PIK3R5, PPP1R12A, ROCK1, SSH2, SOS1, SOS2, BRAF, VAV3 |
Gene interaction network of DE protein-coding genes
To explore the dysregulated gene interactions in primary monocytes of XLA patients, the interaction networks were generated for DE protein-coding genes which were significantly enriched in upregulated and downregulated KEGG pathways in XLA patients compared to healthy subjects (Fig. 3). The network contained 1,601 interactions between 78 upregulated and 103 downregulated genes.
Figure 3.
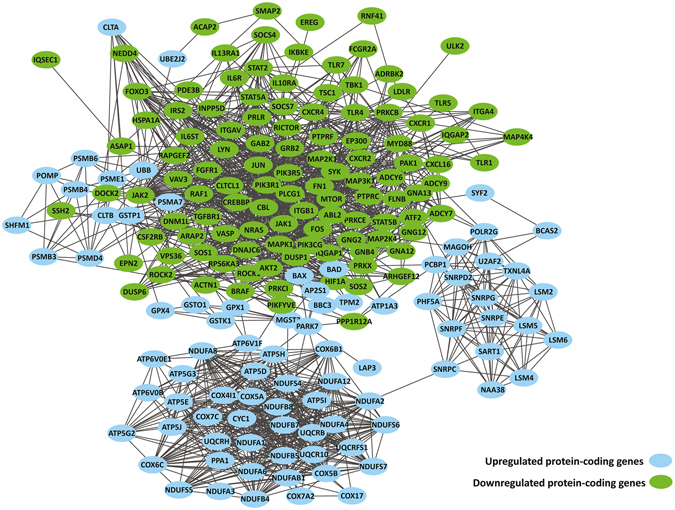
Interaction network analysis of DE protein-coding genes in primary monocytes of XLA patients compared to healthy subjects. The DE protein-coding genes were connected in a network based on the protein-protein interactions.
Differentially expressed (DE) long non-coding RNAs (lncRNAs)
The total of 95 DE annotated lncRNAs were detected in primary monocytes of XLA patients compared to healthy subjects in which 56 and 39 lncRNAs were upregulated and downregulated, respectively (Supplementary Table S3). Several lncRNAs which were known to be involved in the regulation of gene expression, cell cycle and apoptosis were detected among DE lncRNAs in primary monocytes of XLA patients. These lncRNAs include: HOTAIRM1 (HOXA Transcript Antisense RNA, Myeloid-Specific 1)26, DANCR (Differentiation Antagonizing Non-Protein Coding RNA)27, GAS5 (Growth Arrest Specific 5)28, LINC-PINT (Long Intergenic Non-Protein Coding RNA, P53 Induced Transcript)29, HEIH (Hepatocellular Carcinoma Associated Transcript)30 and RMRP (RNA Component Of Mitochondrial RNA Processing Endoribonuclease)31 which were upregulated and, TUG1 (Taurine Upregulated 1)32, which was downregulated in XLA patients compared to the healthy subjects. In addition, expression of the 20 DE novel lincRNAs were identified between XLA patients and healthy subjects. Among the 20 DE novel lincRNAs, 5 lincRNAs were upregulated and 15 lincRNAs were downregulated in XLA patients compared to healthy subjects, respectively (Supplementary Table S4).
DE lncRNAs co-located with protein-coding genes
lncRNAs have been reported to coordinate the regulation of neighboring protein-coding genes (co-located genes). To identify the potential function of the identified DE lncRNAs, the protein-coding genes which genomic locations within ~5 kb upstream and ~1 kb downstream of the DE lncRNAs and which may extend to 1000 kb in both directions were searched. The analysis revealed that out of 95 DE annotated lncRNAs, 85 lncRNAs were corresponded to 144 protein-coding genes. Moreover, among the 20 DE novel lincRNAs, 18 novel lincRNAs were linked to 28 protein-coding genes (Supplementary Table S5). Majority of DE lncRNAs region-gene associations were found to be distal binding events while approximately only 9% regions were within 5 kb of transcription start sites (TSS) (Supplementary Fig. S1).
Next, the function of DE lncRNAs was examined based on GO enrichment and KEGG pathway analysis of their co-located genes (Fig. 4, Supplementary Table S2). The GO enrichment analysis based on biological process demonstrated that the DE annotated lncRNAs co-located genes were mainly involved in “reproductive process”, “positive regulation of cell proliferation” and “induction of apoptosis by extracellular signals” (Fig. 4a). On the other hands, the DE novel lincRNAs co-located genes were related to “regulation of cell differentiation” and “regulation of cell activation” (Fig. 4b). The KEGG pathway analysis revealed that the DE annotated lncRNAs co-located genes were significantly involved in “Metabolic pathways”, and “Cytokine- cytokine receptor interaction” (Fig. 4c). While the novel lincRNAs co-located genes were related to “Focal adhesion” and “Regulation of actin cytoskeleton” (Fig. 4d).
Figure 4.
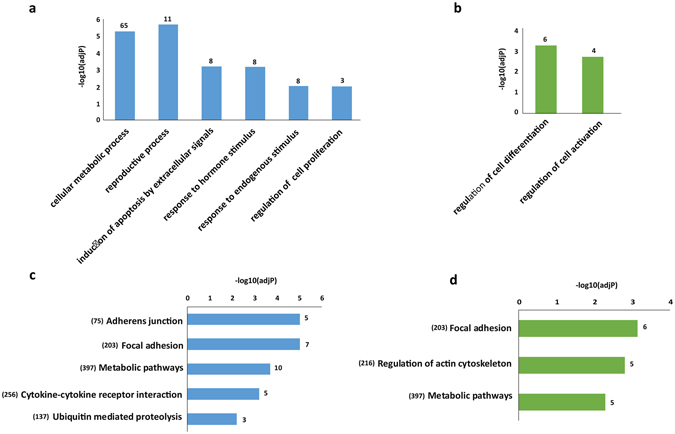
The GO and KEGG pathway analysis of DE lncRNAs co-located genes in primary monocytes of XLA patients compared to healthy subjects. The significant GO biological process terms enriched for (a) DE annotated lncRNAs co-located genes, and (b) DE novel lincRNAs co-located genes. The number of DE lncRNAs co-located genes enriched in each GO terms is depicted above the bars in the figure. The significant KEGG pathway terms enriched for (c) DE annotated lncRNAs co-located genes, and (d) DE novel lincRNAs co-located genes. Each statistical significance value (adj.p-value) was negative log-10 base transformed. The numbers in the brackets indicate the total numbers of genes available in the KEGG database for each pathway terms. The number of identified DE lncRNAs co-located genes enriched in each KEGG pathways is depicted above the bars in the figure.
Besides that, we examined whether any DE lncRNAs co-located genes was differentially expressed in XLA patients compared to healthy subjects. The comparison of the DE annotated and novel lncRNAs co-located genes with DE protein-coding genes led to identification of 23 genomically co-located DE protein-coding genes (Supplementary Table S6). For instance, DE annotated lncRNAs; HOTAIRM1, DANCR and GAS5 were co-located with DE protein-coding genes: HOXA1, USP46, ZBTB48 respectively, which are involved in regulating the gene expression, morphogenesis and differentiation, ubiquitin protease activity, and MHC II promoter compotes. The results also indicated that DE novel lincRNAs TCONS_00030433, TCONS_00041961 and TCONS_00295657 were co-located with DE protein-coding genes VAV3 (Vav Guanine Nucleotide Exchange Factor 3) - involve in phagocytosis, DOCK1 (Dedicator of cytokinesis) - involve in kinase activity and DUSP22 (Dual Specificity Phosphatase 22) - involve in ubiquitin protease activity.
Gene interaction network of DE lncRNAs with their co-located DE protein-coding genes
To unravel the interaction between DE annotated lncRNAs and DE novel lincRNAs and their co-located DE protein-coding genes, putative interactive networks were constructed using Cytoscape. The network contained 80 interactions between 21 DE annotated and DE novel lincRNAs with 23 co-located DE protein-coding genes (Fig. 5).
Figure 5.
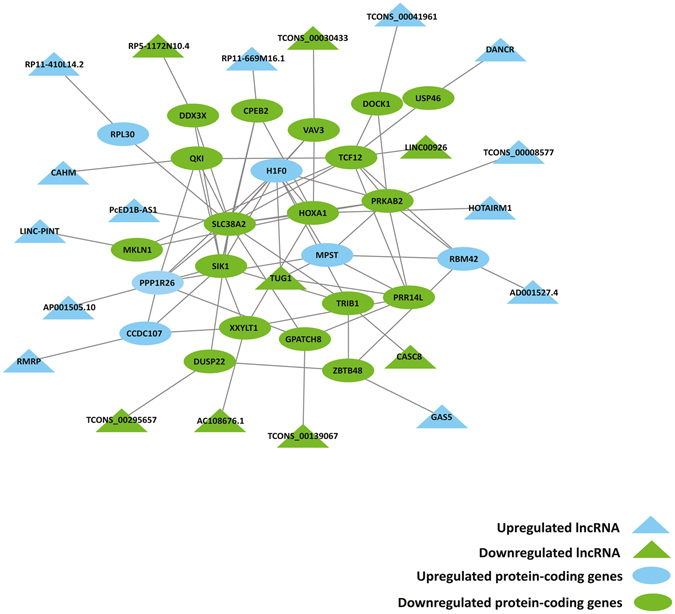
The interaction network of DE lncRNAs with their co-located DE protein-coding genes in primary monocytes of XLA patients compared to healthy subjects. The DE protein-coding genes were connected in a network based on the protein-protein interactions. The edges for lncRNAs that were close in genomic space to the DE protein-coding genes were automatically added into the network.
qRT-PCR validation
To further confirm our findings as described above, expression levels of selected DE protein-coding genes and DE annotated and novel lincRNAs were measured by Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) analysis. The candidate genes were 10 DE protein-coding genes: FCGR2A, CXCR2, TLR1, TLR5, ATP5D, NDUFA1, UQCRB, SOD1, MTOR and BAX which were enriched in several significant upregulated and downregulated KEGG pathways. Also included were 7 DE annotated lncRNAs; HOTAIRM1, DANCR, GAS5, LINC-PINT, RMRP, HEIH, TUG1 and 3 DE novel lincRNAs; TCONS_00041961, TCONS_00295657, TCONS_00298577, which co-located with DE protein-coding genes in XLA patients compared to the healthy subjects. The qRT-PCR validation results showed that in total, 90% of the selected genes reached significance (P-value ≤ 0.05) (Fig. 6). Comparison between RNA-Seq and qRT-PCR log2 fold-change ratio revealed similar expression trends for selected genes with a Pearson correlation (r) value of 0.83 (P-value < 0.001), demonstrating the reliability of our RNA-Seq data analysis (Fig. 6).
Figure 6.
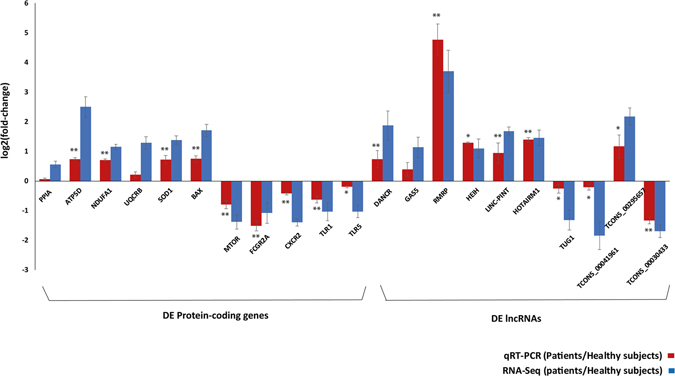
The qRT-PCR validation of DE protein-coding genes and DE lncRNAs in primary monocytes of XLA patients compared to the healthy subjects. The comparison of log2 fold-change of DE protein-coding genes and DE lncRNAs were determined by RNA-Seq analysis (blue) and qRT-PCR validation (red). PPIA was used as endogenous control for normalizing the expression levels. x-axis shows genes; y-axis shows the log2 ratio of expression in XLA patients compared to healthy subjects. Statistical significance was calculated using paired Student’s T-test. The asterisks above the bars denote statistically significant differences from healthy subjects obtained by qRT-PCR, *P-value < 0.05, **P-value < 0.01.
Discussion
In this study, we investigated the effect of BTK gene expression deficiency on primary monocyte’s immune function in XLA patients using deep RNA-Seq analysis. The BTK gene is situated at band Xq21.3 to Xq22, long arm of the X chromosome, spanning 37.5 kb that contains 19 exons. The BTK protein has 5 different functional domains including PH, TH, SH3, SH2, and TK. Over 800 mutations have been identified in BTK 33. These mutations varied in types and scattered throughout all domains of the BTK protein, which all resulting in overall dysfunction of BTK protein. Recently, Xia-Fang and colleagues examined the genetic background and clinical features of 174 patients with XLA and reported that there was no relationship between BTK mutations and clinical symptoms in XLA patients34.
We compared transcriptome profiles of primary monocytes of healthy subjects and XLA patients, who shared similar clinical symptoms despites having different type of mutations in BTK gene. Our analysis showed a total of 1,827 DE protein-coding genes, of which 859 genes were upregulated and 968 genes were downregulated in XLA patients compared to the healthy subjects (Supplementary Table S1). Based on the GO and KEGG pathways analysis, detailed information on the biological functions and potential mechanisms of actions of DE protein-coding genes were identified. The GO enrichment analysis showed that downregulated genes were mainly involved in the regulation of immune response. Pathway enrichment analysis also revealed that downregulated genes were mainly enriched in several pathways belonged to the innate immune system such as: “Fc gamma R-mediated phagocytosis”, “Chemokine signaling pathways”, “Toll like receptors signaling pathway” and “MTOR signaling pathway”, reflects the deficiencies of innate immune function in primary monocytes of the XLA patients.
The expression of FCGR2A (also known as FcγRIIA or CD32, involved in “Fc gamma R-mediated phagocytosis”) was significantly decreased in primary monocytes of the XLA patients which is consistent with the findings about decreased expression of FCGR2A in monocytes from XLA patients due to BTK deficiency13. In addition to FCGR2A, our study identified the downregulation of 22 core enrichment genes involved in “Fc gamma R-mediated phagocytosis” pathway in XLA patients compared to healthy subjects (Table 3). Majority of these genes encode for kinases in the early signaling events [such as LYN (LYN Proto-Oncogene, Src Family Tyrosine Kinase) and SYK (Spleen Tyrosine Kinase) Kinases] as well as the genes encode for proteins involved in cytoskeleton rearrangement. Decreased expression of 32 core genes involved in “chemokines signaling pathway” including 4 chemokine receptors (CXCL16, CXCR1, CXCR2, CXCR4) were also observed in primary monocytes of the XLA patients (Table 3). A direct role for BTK in signaling by CXCR4 and in chemokine-controlled adhesion and migration in B cells of XLA patients has been shown previously35. Similar observations regarding the regulatory role of BTK on CXCR4 as well as CXCL16, CXCR1, and CXCR2 were found in primary monocytes of XLA patients in which the BTK deficiency may lead to downregulation of these chemokine receptors.
Another significant finding of this study is the overall downregulation of “Toll-like Receptor (TLR) signaling pathway” in primary monocytes of the XLA patients compared to healthy subjects (Table 3). The BTK has been shown to be involved in TLR signaling, where it interacted with TLR2, TLR4, TLR6, TLR7, TLR8 and TLR9 and facilitated their transduction of downstream signals and phosphorylation10–12. In addition to these findings, our study demonstrated for the first time, significant decreased in expression of TLR1 and TLR5 in primary monocytes of the XLA patients compared to healthy subjects. This suggested that the BTK may also associates with TLR1 and TLR5 expression in primary monocyte of XLA patients, in which mutants of BTK may inhibit their signaling. Similar observation of TLR1 signaling deficiency due to the BTK mutant was reported in mice36.
Our analysis also revealed the downregulation of the MTOR (Mechanistic Target Of Rapamycin) genes along with the other 10 core genes of the “MTOR signaling pathway” in the XLA patients compared to healthy subjects (Table 3). Recently, Ezell and colleagues reported similar observation regarding the possible regulatory role of BTK on MTOR signaling in activated Diffuse Large B Cell Lymphoma (DLBCL)37. MTOR plays an important role in cell differentiation and growth, cellular metabolism and cancer metabolism. It can sense the growth factors, nutrients, insulin, energy and environmental changes, then transmit signals to downstream targets to activate the metabolic and cellular reactions38. In addition, MTOR was recently found to be associated with the regulation of both the innate39 and adaptive immune response40. Low expression of MTOR was observed in cells that are more dependent on mitochondrial oxidative phosphorylation for energy supply41. Overexpression of mitochondrial components due to great energy production demand has been reported in several disease states such as cancers42, Acquired Immune Deficiency Syndrome (AIDs)43 and Alzheimer’s disease (AD)44. We also detected low expression of MTOR and overexpression of multiple components of mitochondrial complexes I (NDUF; NADH: Ubiquinone Oxidoreductase), III (UQCR; Ubiquinol-Cytochrome C Reductase), IV (COX; Cytochrome C Oxidase) and V (ATP; Adenosine Triphosphate) in XLA patients compared to the healthy subjects (Table 2). This indicate great energy demand in primary monocytes of the XLA patients compared to healthy subjects. Furthermore, the upregulated genes involved in the production of reactive oxygen species (ROS), oxidative stress response and apoptotic process were observed in the XLA patients compared to healthy subjects. It is well known that during the oxidative phosphorylation, mitochondria consume most of the cellular oxygen and produce the majority of ROS. High concentration of ROS in the cell would lead to state termed oxidative stress, in which the excess ROS induces oxidative damage on cellular components and activate apoptosis pathways and cell death45. This may possibly explain the upregulation of several genes involved in the oxidative phosphorylation, ROS production, response to oxidative stress and apoptosis in monocytes of XLA patients.
In addition to the protein-coding genes, lncRNAs can also act as key regulators of various biological processes in the immune system16, 17. Our analysis of the lncRNAs expression between the XLA patients and healthy subjects showed a total of 95 DE annotated lncRNAs (56 upregulated and 39 downregulated) and 20 DE novel lincRNAs (5 upregulated and 15 downregulated) (Supplementary Tables S3 and S4). Several DE lncRNAs were identified in the XLA patients, which were known to contribute to regulation of gene expressions and cell cycle. Overexpression of these lncRNAs have been reported to suppress the cell growth, differentiation, proliferation, and apoptosis in various diseases. Such lncRNAs include: HOTAIRM1 26, DANCR 27, GAS5 28, LINC-PINT 29, HEIH 30, RMRP 31, which have found to be overexpressed in primary monocytes of the XLA patients compared to the healthy subjects. By comparing to healthy subjects, our analysis also detected significant decreased in expression of lncRNA TUG1 in the XLA patients. Downregulation of TUG1 has been reported to inhibit osteosarcoma cell proliferation and promote apoptosis32. Similar observation regarding the dysregulated expression of these lncRNAs seen in the primary monocytes of XLA patients, suggesting their possible role in regulating monocyte cell cycle and apoptosis in XLA patients. The analysis of DE novel lincRNAs also revealed that some DE novel lincRNAs were co-located with DE protein-coding genes related to immune system. In particular, the novel DE lncRNA TCONS_00030433, which was significantly downregulated in the XLA patients, interacted with VAV3, of which its expression was also detected to be downregulated in the XLA patients. VAV3 is known to be involved in “Fc gamma R-mediated phagocytosis” pathway46. These results would suggest the possible role of TCONS_00030433 in the regulation of “Fc gamma R-mediated phagocytosis” pathway in primary monocytes of XLA patients.
In summary, our analysis based on deep RNA-Seq datasets revealed that the expression profiles of protein-coding genes were significantly altered in primary monocytes of XLA patients compared to the healthy subjects. Regardless type of mutations, loss of function mutation in BTK genes eventually induced dysfunction of BTK protein, block the development of B-cell and caused the disease. The functional analysis of differentially expressed (DE) protein-coding genes showed that the impaired immune function and increased susceptibility to apoptosis in monocytes of XLA patients were due to BTK deficiency. This showed that BTK is not only involved in the development and function of B cells, but it may play an important role in establishing the immunity function of monocytes. Our study also revealed the differential expression patterns of lncRNAs in primary monocytes of XLA patients compared to healthy subjects and predicted their potential role in regulation of monocyte cell cycle and apoptosis in XLA patients. However, the specific biological functions and molecular mechanisms of these lncRNAs in monocytes of XLA patients required additional evaluation. Moreover, further large scale studies utilising integrated omics approaches would advance our knowledge and understanding on the impaired immune function of monocytes in XLA patients.
Methods
Sample collection and purification
Ethics approval to conduct this study was obtained from Medical Research and Ethics Committee (MREC), Malaysia with reference number NMRR-13-972-16921. Three unrelated male patients with XLA disease and 3 unrelated healthy male subjects were selected in this study. The written informed consent from all subjects for the use of their blood samples were obtained. The blood samples were handled according to the guidelines of the Helsinki Declaration. The entire experiment was conducted in accordance with the guidelines of Medical Research and Ethics Committee (MREC), Malaysia. The healthy subjects were non-smokers, did not have any medical illness, did not prescribed with any long-term medication and did not receive any vaccination at least 6 months before the study. The patients were diagnosed to have XLA disease based on the criteria of the World Health Organization Scientific Group for PIDs: low levels of circulating B cells (measured by levels of CD19+ B cells in blood samples), reduced or absent of immunoglobulins in serum and a typical clinical history with recurrent bacterial infection or a positive family history47. All selected patients had deficient monocyte BTK expression, as evaluated by flow cytometry. BTK gene mutations in all patients were confirmed by Sanger sequencing. All the patients were in a stable clinical situation without fever and not hospitalized when the blood was taken. They were under monthly IVIG therapy. The blood samples from patients were collected before the administration of IVIG. The 10 mL of peripheral blood from each healthy subjects and XLA patients were collected in EDTA tubes. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-based density gradient centrifugation. The classical monocytes (CD14++CD16−) were isolated from PBMCs by negative selection method using monocyte isolation kit II (Miltenyi Biotec, Germany). The BD FACSCanto II Flow Cytometer (BD Biosciences, USA) was used to perform the purity check of the isolated monocytes.
RNA extraction, library preparation and sequencing
Total RNA was extracted from monocytes using RNeasy mini-kit followed by DNase treatment (QIAGEN, Germany). The quality and quantity of RNA was measured using NanoDrop 2000 (Thermo Fisher Scientific Inc, USA) and Qubit 2.0 RNA Broad Range Assay (Invitrogen, USA). The RNA purity was checked using Agilent Bioanalyzer RNA Nano chip Bioanalyzer (Agilent Technologies, USA). All samples had RNA Integrity Number (RIN) higher than 9. Messenger RNA isolation and cDNA synthesis were performed using TruSeq RNA Sample Preparation Kit (Illumina, USA) and Super Script II Reverse Transcriptase (Invitrogen, USA). The synthesized cDNA was quantified using Qubit 2.0 DNA Broad Range Assay (Invitrogen, USA). A minimum of 40ng cDNA was fragmented using Covaris S220 (Covaris INC, USA) to a targeted size of 200–300 bp. The fragmented cDNA was then end-repaired, ligated to Illumina TruSeq adaptors, and PCR-enriched using TruSeq RNA sample preparation kit (Illumina, USA). The final sequencing libraries were quantified using KAPA kit (KAPA Biosystem, USA) on Agilent Stragene Mx-3005p quantitative PCR (Agilent, USA) and sizes were confirmed using Agilent Bioanalyzer High Sensitivity DNA Chip (Agilent, USA). The RNA libraries were sequenced on Illumina HiSeq 2000 Platform (Illumina, USA) to generate 2 × 100 bp paired-end sequencing reads. The quality of sequences in fastq format was evaluated using FASTQC48. Low quality bases and adaptors were trimmed from the sequences using Trimmomatic49.
Alignment and transcript assembly
Quality trimmed sequences from all samples were separately mapped to the human reference genome sequence (Ensemble GRCh38.79) using HISAT (version 0.1.4)50 with GENCODE junctions as a guided reference annotation and the transcript assembly was performed with StringTie (version 1.3.3)51 using a GENCODE reference annotation (version 22). The transcripts abundance was estimated as fragments per kilobases of exon per million fragments mapped (FPKM)52. The assembled transcripts of XLA patients and healthy subject were separately merged by using Cuffmerge (a part of cufflinks, version 2.2.1)52. The merged assembled files were then compared with a GENCODE reference annotation file (version 22) to obtain the number of expressed protein-coding genes and lncRNAs in the XLA patients and healthy subjects.
Identification of novel long intergenic non-coding RNAs (lincRNAs)
Using an established pipeline for identification of lncRNA24, we analyzed the expression of putative novel long intergenic non-coding RNAs (lincRNAs) in primary monocytes. Briefly, transcripts with coverage of 3 x and above were filtered from the patients and healthy subjects’ transcripts assembled files. Then the filtered transcript files of each dataset were separately merged to form a non-redundant set of transcripts using Cuffmerge. The expression level of each transcript was quantified using Cuffquant (a part of Cufflinks, version 2.2.1)52 and normalized for total number of reads using Cuffnorm (a part of Cufflinks, verstion 2.2.1)52. The transcripts were mapped to the known gene annotation from GENCODE (version 22) and categorized as protein-coding, lncRNAs, novel and others (such as tRNAs, microRNAs, pseudogenes, etc). All known transcripts were filtered out while novel transcripts were retained. Only novel transcripts with more than 200 nucleotides which are intergenic to any GENCODE transcript were considered for further analysis. The coding potential of transcripts was then evaluated using Coding Potential Assessment Tool (CPAT)53. Coding probability cut-off of 0.375 was used to mark any putative protein coding sequences and the sequences were excluded from our analysis. The nucleotide sequences of putative novel lincRNAs were searched for any matching sequences against the non-redundant (NR) database from NCBI using BLASTN and no significant homology was found.
Differential gene expression analysis
Differential gene expression analysis was performed by employing Cuffdiff (a part of Cufflinks, version 2.2.1)52. The DE genes were identified with q-value ≤ 0.01 and log2 fold-change ≥ 1 or ≤ −1. Hierarchical clustering of the expression profiles of detected DE protein-coding genes and DE lncRNAs were done with the heatmap function in the R “NMF” package54 using Pearson Correlation.
GO and KEGG pathway analysis of DE protein-coding genes
To investigate the biological importance of the identified DE protein-coding genes, DAVID (Database for Annotation, Visualization and Integrated Discovery) functional annotation tool55 was used to perform Gene Ontology (GO) analysis56. DAVID uses modified Fisher’s exact test (known as EASE)57 to measure enrichment against a background gene list and adjusting the resulting p-values (adj.P-value) using a Benjamini-Hochberg method58. The GO analysis was restricted to the category of biological processes as it is the most prominent for evaluation of genes function. The DAVID category-GOTERM_BP_FAT level was selected for displaying the results. The minimum number of genes for enrichment in each category was set at 2 and the significance cutoff was adj.P-value < 0.01. Subsequently, the pathway analysis of identified protein-coding genes was conducted by applying the Kyoto Encyclopedia of Genes and Genomes (KEGG) database59 in DAVID tool using the same parameters.
Co-location analysis DE lncRNAs with protein-coding genes
lncRNAs are presumed to regulate the expression of their neighboring genes (co-located genes)60. To predict the function of DE lncRNAs through their co-located genes, the genomic coordinates of DE lncRNAs were imported to Genomic Regions Enrichment of Annotations Tool (GREAT)61. The GO and KEGG pathway analysis of DE lncRNAs co-located genes was performed using the DAVID functional annotation tool.
The identified co-located genes were then matched with DE protein-coding genes to obtain a lncRNAs co-located genes which were also differentially expressed between XLA patients and healthy subjects62.
Gene interaction network construction
In order to predict the DE protein-coding genes interaction, the protein-protein interaction network was constructed using Cytoscape plug-in GeneMANIA63 for genes which were significantly enriched (adj.P-value < 0.01) in upregulated and downregulated KEGG pathways. In addition, gene interaction networks between two subgroups of genes (DE lncRNA and their co-located DE protein-coding genes) were also constructed using Cytoscape64. The possible protein-protein interactions between DE protein-coding genes in each subgroup was predicted using the Cytoscape plug-in GeneMANIA. The network edges automatically added for lncRNAs that were close in genomic space to the DE protein-coding genes.
Validation of RNA-Seq results by qRT-PCR
The expression levels of selected DE protein-coding and DE lncRNAs were validated using Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) analysis. First-strand cDNA was synthesized from 300 ng of RNA from each sample by using the High Capacity RNA to cDNA Kits (Applied Biosystems, USA). All the primers and probes for Taqman® Real-time PCR (Life Technologies, USA) were designed by Applied Biosystems (Supplementary Table S7). Expression of target genes was assessed using the QuantStudio™ 12 K Flex Real-Time PCR System. The conditions of the PCR cycle conditions were: 50 °C for 2 minutes, 95 °C for 20 seconds, followed by 40 cycles of 95 °C for 3 seconds, and 40 cycles of 60 °C for 30 seconds. Each gene was analyzed in triplicates for each sample. PPIA (Peptidylprolyl Isomerase A) gene was used as an endogenous control. Fold-changes in gene expression between samples were calculated using 2−ΔΔCt method65. The Student’s t-test was used to evaluate the expression differences of selected protein-coding genes and lncRNAs between XLA patients and healthy subjects. P-value < 0.05 was considered as statistically significance.
Availability of data and materials
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO series accessions number GSE80095 and GSE89980.
Electronic supplementary material
Acknowledgements
We would like to thank the Director General of Health Malaysia for his approval to publish this paper. Special thanks to Chang Lee Wei, Research Officer at CRYSTAL, University of Malaya for helping out with submission process. This research work was supported by the Ministry of Higher Education Malaysia - University of Malaya High Impact Research (HIR) Grant: UM.S/P/HIR/MOHE/30, Ministry of Higher Education Malaysia Fundamental Research Grant Scheme (FRGS): FP050-2016, University of Malaya Research Grant (UMRG): RP004C-13AFR and University of Malaya PPP Postgraduate Grant: PG086-2013B.
Author Contributions
H.M., A.M.R. and C.C.T. collected the samples, performed the laboratory experiments. H.M. and W.M.L. performed the computational analyses. H.M. drafted the manuscript. A.M.R., S.B.M. and A.F.M. designed and supervised the study and critically reviewed the manuscript. All authors have final approval on the manuscript to be published.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Hoda Mirsafian and Adiratna Mat Ripen contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06342-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Vihinen M, et al. Mutations of the human BTK gene coding for bruton tyrosine kinase in X-linked agammaglobulinemia. Hum Mutat. 1999;13:280–285. doi: 10.1002/(SICI)1098-1004(1999)13:4<280::AID-HUMU3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 2.Vetrie D, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nat. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 3.Ochs HD, Smith CI. X-linked agammaglobulinemia. A clinical and molecular analysis. Medicine. 1996;75:287–99. doi: 10.1097/00005792-199611000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Honda F, et al. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol. 2012;13:369–378. doi: 10.1038/ni.2234. [DOI] [PubMed] [Google Scholar]
- 5.Bao Y, et al. Tyrosine Kinase Btk Is Required for NK Cell Activation. J. Biol Chem. 2012;287:23769–23778. doi: 10.1074/jbc.M112.372425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koprulu AD, Ellmeier W. The role of Tec family kinases in mononuclear phagocytes. Crit Rev Immunol. 2009;29:317–333. doi: 10.1615/CritRevImmunol.v29.i4.30. [DOI] [PubMed] [Google Scholar]
- 7.Mukhopadhyay S, et al. Macrophage effector functions controlled by Bruton’s tyrosine kinase are more crucial than the cytokine balance of T cell responses for microfilarial clearance. J. Immunol. 2002;168:2914–2921. doi: 10.4049/jimmunol.168.6.2914. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt NW, Thieu VT, Mann BA, Ahyi A-NN, Kaplan MH. Bruton’s tyrosine kinase is required for TLR-induced IL-10 production. J. Immunol. 2006;177:7203–7210. doi: 10.4049/jimmunol.177.10.7203. [DOI] [PubMed] [Google Scholar]
- 9.Lee KG, et al. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci. 2012;109:5791–5796. doi: 10.1073/pnas.1119238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doyle SL, Jefferies CA, Feighery C, O’Neill LAJ. Signaling by Toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. J Biol Chem. 2007;282:36953–36960. doi: 10.1074/jbc.M707682200. [DOI] [PubMed] [Google Scholar]
- 11.Horwood NJ, et al. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol. 2006;176:3635–3641. doi: 10.4049/jimmunol.176.6.3635. [DOI] [PubMed] [Google Scholar]
- 12.Jefferies CA, et al. Bruton’s Tyrosine Kinase Is a Toll/Interleukin-1 Receptor Domain-binding Protein That Participates in Nuclear Factor B Activation by Toll-like Receptor 4. J Biol Chem. 2003;278:26258–26264. doi: 10.1074/jbc.M301484200. [DOI] [PubMed] [Google Scholar]
- 13.Amoras AL, Kanegane H, Miyawaki T, Vilela MM. Defective Fc-, CR1- and CR3-mediated monocyte phagocytosis and chemotaxis in common variable immunodeficiency and X-linked agammaglobulinemia patients. J Investig Allergol Clin Immunol. 2003;13:181–188. [PubMed] [Google Scholar]
- 14.Geng H, Tan XD. Functional diversity of long non-coding RNAs in immune regulation. Genes & Diseases. 2016;3:72–81. doi: 10.1016/j.gendis.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guttman M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nat. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jebali S, et al. Landscape of transcription in human cells. Nat. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tano K, et al. MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 2010;584:4575–4580. doi: 10.1016/j.febslet.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Fu XL, et al. Analysis of long non-coding RNA expression profiles in pancreatic ductal adenocarcinoma. Sci Rep. 2016;6:33535. doi: 10.1038/srep33535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson R, et al. The Human Accelerated Region 1 noncoding RNA is repressed by REST in Huntington’s disease. Physiol Genomics. 2010;41:269–274. doi: 10.1152/physiolgenomics.00019.2010. [DOI] [PubMed] [Google Scholar]
- 20.Hrdlickova B, et al. Expression profiles of long non-coding RNAs located in autoimmune disease-associated regions reveal immune cell-type specificity. Genome Med. 2014;6:88. doi: 10.1186/s13073-014-0088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, et al. Differentially expressed lncRNAs and mRNAs identified by microarray analysis in GBS patients vs healthy controls. Sci Rep. 2016;6:21819. doi: 10.1038/srep21819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez JA, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell. 2013;152:743–754. doi: 10.1016/j.cell.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirsafian H, Ripen AM, Manaharan T, Mohamad SB, Merican AF. Toward a Reference Gene Catalog of Human Primary Monocytes. OMICS. 2016;20:627–634. doi: 10.1089/omi.2016.0124. [DOI] [PubMed] [Google Scholar]
- 24.Mirsafian H, et al. Long non-coding RNA expression in primary human monocytes. Genomics. 2016;108:37–45. doi: 10.1016/j.ygeno.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Chear CT, et al. A novel Bruton’s tyrosine kinase gene (BTK) invariant splice site mutation in a Malaysian family with X-linked agammaglobulinemia. Asian Pac J Allergy Immunol. 2013;31:320. doi: 10.12932/AP0304.31.4.2013. [DOI] [PubMed] [Google Scholar]
- 26.Wan L, et al. HOTAIRM1 as a potential biomarker for diagnosis of colorectal cancer functions the role in the tumour suppressor. J Cell Mol Med. 2016;20:2036–2044. doi: 10.1111/jcmm.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong X, Gu P, Xu S, Lin X. Long non-coding RNA-DANCR in human circulating monocytes: a potential biomarker associated with postmenopausal osteoporosis. Biosci Biotechnol Biochem. 2015;79:732–737. doi: 10.1080/09168451.2014.998617. [DOI] [PubMed] [Google Scholar]
- 28.Tu ZQ, Li RJ, Mei JZ, Li XH. Down-regulation of long non-coding RNA GAS5 is associated with the prognosis of hepatocellular carcinoma. Int J Clin Exp Pathol. 2014;7:4303–4309. [PMC free article] [PubMed] [Google Scholar]
- 29.Marín-Béjar O, et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013;14:R104. doi: 10.1186/gb-2013-14-9-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang F, et al. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology. 2011;54:1679–1689. doi: 10.1002/hep.24563. [DOI] [PubMed] [Google Scholar]
- 31.Elling R, Chan J, Fitzgerald KA. Emerging role of long noncoding RNAs as regulators of innate immune cell development and inflammatory gene expression. Eur J Immunol. 2016;46:504–512. doi: 10.1002/eji.201444558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young TL, Matsuda T, Cepko CL. The Noncoding RNA Taurine Upregulated Gene 1 Is Required for Differentiation of the Murine Retina. Curr Biol. 2005;15:501–512. doi: 10.1016/j.cub.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 33.Mohamed AJ, et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228:58–73. doi: 10.1111/j.1600-065X.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 34.Xia-Fang C, et al. Clinical characteristics and genetic profiles of 174 patients with X-linked agammaglobulinemia. Medicine (Baltimore). 2016;95:e4544. doi: 10.1097/MD.0000000000004544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Gorter DJJ, et al. Bruton’s Tyrosine Kinase and Phospholipase Cγ2 Mediate Chemokine-Controlled B Cell Migration and Homing. Immunity. 2007;26:93–104. doi: 10.1016/j.immuni.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 36.Alugupalli KR, Akira S, Lien E, Leong JM. MyD88- and Bruton’s tyrosine kinase-mediated signals are essential for T cell-independent pathogen-specific IgM responses. J Immunol. 2007;178:3740–3749. doi: 10.4049/jimmunol.178.6.3740. [DOI] [PubMed] [Google Scholar]
- 37.Ezell SA, et al. Differential regulation of mTOR signaling determines sensitivity to AKT inhibition in diffuse large B cell lymphoma. Oncotarget. 2016;7:9163–9174. doi: 10.18632/oncotarget.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soliman GA. The role of mechanistic target of rapamycin (mTOR) complexes signaling in the immune responses. Nutrients. 2013;5:2231–2257. doi: 10.3390/nu5062231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. 2015;15:599–614. doi: 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keating R, McGargill MA. mTOR Regulation of Lymphoid Cells in Immunity to Pathogens. Front Immunol. 2016;7:180. doi: 10.3389/fimmu.2016.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griguer CE, Oliva CR, Gillespie GY. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J Neurooncol. 2005;74:123–133. doi: 10.1007/s11060-004-6404-6. [DOI] [PubMed] [Google Scholar]
- 43.Zhou L, et al. First evidence of overlaps between HIV-Associated Dementia (HAD) and non-viral neurodegenerative diseases: proteomic analysis of the frontal cortex from HIV+ patients with and without dementia. Mol Neurodegener. 2010;5:27. doi: 10.1186/1750-1326-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5:147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 45.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hall AB, et al. Requirements for Vav Guanine Nucleotide Exchange Factors and Rho GTPases in FcγR- and Complement-Mediated Phagocytosis. Immunity. 2006;24:305–316. doi: 10.1016/j.immuni.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Anker, M. Addressing sex and gender in epidemic-prone infectious diseases. Genève (Suisse): World Health Organization (2007).
- 48.Andrews, S. FastQC: a quality control tool for high throughput sequence data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
- 49.Bolger AM, Lohse M, Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74–e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2005;11:367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dennis G, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 56.Ashburner M, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hosack DA, Dennis G, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4(10):R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. R Stat Soc Series B Stat Methodol. 289–300 (1995).
- 59.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ørom UA, et al. Long Noncoding RNAs with Enhancer-like Function in Human Cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao W, et al. Integrated Analysis of Long Noncoding RNA and Coding RNA Expression in Esophageal Squamous Cell Carcinoma. Int J Genomics. 2013;2013:480534. doi: 10.1155/2013/480534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Montojo J, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO series accessions number GSE80095 and GSE89980.