

Abstract
Molecular characterization of PAH deficiency has been proven essential in establishing treatment options. We examine the diagnostic accuracy of two genetic assays to predict BH4 responsiveness: to determine whether the AV sum test or mutation-status assessment test can obviate the need for BH4 loading in Chinese patients. The overall predicted response in 346 patients was 31.65% by the AV sum test and 25.43% by the other assay; both percentages were lower than 51.06% derived from loading results in 94 patients. Responders were compound heterozygotes with definite BH4 responsive mutations, while non-responders had null/null ones; some consistently with specific mutations and genotypes. The sensitivity and specificity of the assays were 81.1% and 92.5% for the AV sum, and 82.9%, 97.3% for the other. An AV sum cutoff >2 has a positive predictive value (PPV) of 90.9%, while the presence of at least one BH4 responsive mutation has a PPV of 97.1%. The two approaches showed good concordance. Our data confirmed that the mutation-status assessment has a higher diagnostic accuracy in predicting response for Chinese patients than the AV sum test. BH4-responsiveness may be predicted or excluded from patients’ molecular characteristics to some extent, thus some patients may avoid the initial loading.
Introduction
Phenylalanine hydroxylase (PAH; NP_000268, EC 1.14.16.1) deficiency (MIM# 261600) is an autosomal recessive disorder that results from intolerance to the dietary intake of phenylalanine (Phe)1. The mainstay of treatment is a Phe-restricted diet2, but its life-long application with stable levels between 120 and 360 umol/L3, 4 is a challenge that results in high rates of non-compliance and breakdown, which are known to increase with age5. Studies6, 7 have been conducted to examine the relationship between cerebral complications of the disease and the increase or the fluctuations in Phe concentrations, and it is demonstrated that liberalization of Phe control at any age, especially during the first 10 to 12 years of life8, 9, has led to deficits of cognitive and executive functions10, 11, impairment of working memory12 and other psychological and neurologic abnormalities13.
In 2007, the United States Food and Drug Administration (FDA) approved sapropterin dihydrochloride (Kuvan®, formerly known as Phenoptin), a synthetic form of tetrahydrobiopterin (BH4), the first oral pharmacotherapy for PAH deficiency. Two randomized controlled trials14, 15 showed that in some patients the drug significantly lowered blood Phe concentration with no serious adverse side-effects and a long term retrospective study16 indicated that sapropterin improved their Phe tolerance. Additional research17, 18 revealed that sapropterin therapy could increase the stability of blood Phe levels. This group of patients was then classified as ‘tetrahydrobiopterin (BH4)-responsive hyperphenylalaninemia (HPA)/PKU19, a new subtype of PAH deficiency.
Therefore, it is of crucial importance to detect this subtype of patients correctly and to determine the individual sensitivity to BH4; a 30% reduction or more in blood Phe concentration 24 hours after administration of sapropterin is the most commonly used biochemical definition of a positive response20. Since the corresponding work of the test is onerous, finding a rational testing modality is imperative for further application. As PAH gene (NG_008690.1) mutations have been characterized, the association of these mutations with disease phenotypes has been uncovered. It was acknowledged that although different biochemical phenotypes were observed in identical genotypes, their BH4 responsiveness was always the same except in a very small percentage of patients21–23, and BH4-responsiveness was a common trait among subjects with the milder forms of PAH deficiency22, 24, 25. This observation significantly increased research activities on the feasibility of using genetic analysis to assess the response26–30 and a consensus was reached that such research might help to guide clinical decisions and promote personalized medicine in managing PAH deficiency for specific populations.
The prevalence of PAH deficiency in Chinese population is about 1 in 11,572 live births31. Previously, our team carried out genetic analyses for patients from diverse regions of China32, 33, investigated the mutational spectrum, and established the genotype-phenotype correlations. These studies comprise the initial steps in the development of an optimal molecular diagnostic algorithm. In the current study, we used two assays to predict the prevalence of BH4 response in a cohort of 346 Chinese PAH deficient patients. The assigned values (AV) sum approach34 is a genotype severity tool in which the predicted phenotype for a patient was expressed numerically as the sum of two mutations’ AVs. The results for the predicted phenotypes for all our patients were derived from Guldberg et al.34 (see the Appendix) and our previous paper32. The other approach is based on the speculation that the presence of some specific mutations on at least one PAH gene copy, which is repeatedly found to be associated with BH4-responsiveness, would be sufficient for positive BH4 response. The performances of both tests in predicting the BH4 response were respectively validated by the clinical BH4 loading results in 94 of them. The accuracy of both analyses in differentiating between BH4 responsive and non-BH4 responsive patients was assessed; whether the AV sum test or mutation-status assessment test can obviate the need for BH4 loading was discerned.
Results
The 346 PAH deficient patients (Table S1) were classified into three phenotypic categories, 172 (172/346, 49.71%) as classic PKU (cPKU), 156 (156/346, 45.09%) as mild PKU (mPKU), and 18 (18/346, 5.20%) as mild hyperphenylalaninemia (MHP), indicating half of our population as carrying the milder form. All patients displayed 186 different genotypes based on 109 mutations, 53 of which were classified as null32, 33. Among the 346 individuals, 35 (35/346, 10.12%) were homozygous [23 for p.Arg243Gln (c.728G > A), 5 for p.Ex6-96A > G (c.611A > G), 3 for p.Arg413Pro (c.1238G > C), 1 for p.Tyr166* (c.498C > A), p.Arg241Cys (c.721C > T), p.Gly257Val (c.770G > T) and p.Leu385Pro (c.1154T > C), respectively], and the remaining patients were compound heterozygotes (311/346, 89.88%). Homozygosity was found in 9.12% of our population and 8 mutations accounted for over two-thirds of the 692 alleles, with p.Arg243Gln (c.728G > A) at a 24.06% frequency. Thus, the investigation of common mutations or allelic combinations in accordance to BH4 responsiveness was of great interest to Chinese patients in general. Based on the AV sum information32, 34, 88 out of the 278 individuals (refer to Method: Determining the PAH Genotype Severity Using the AV Sum and Classification of BH4 Response) have an AV sum >2 (88/278, 31.65%), thus being potentially responsive. While for the assessment of the mutation BH4-responsiveness-status, 88 out of the 346 patients (88/346, 25.43%) were potentially responsive.
BH4 Loading Test
The outcome of the BH4 loading test in 94 patients was summarized in Table 1. Among these patients, 48 (48/94, 51.06%) were shown to be BH4-responders: 5 (5/48, 10.42%) were cPKU, 37 (37/48, 77.08%) were mPKU, and 6 (6/48, 12.50%) were MHP. The non-responders were 46 (46/94, 48.94%): 29 were (29/46, 63.04%) cPKU, 17 (17/46, 39.96%) were mPKU, and none (0/46, 0%) was MHP. It was found that 9 (9/46, 19.57%) of the non-responders had a Phe reduction rate less than 20% at 8 hours after the loading and between 20% and 30% at 24 hours. Fiege B et al.35 defined this situation as “slow responsiveness”. Interestingly, in our study, these late responsive patients have cPKU (6) and mPKU (3).
Table 1.
BH4 loading test in 94 Chinese PAH Deficient patients.
| Patient ID | Pre-Phea | Phenotypeb | Protein variation | AV sumc | Groupd | Phe reduction 24 h (%) | BH4 R statuse | under BH4, Phe became ≤360 umol/Lf |
|---|---|---|---|---|---|---|---|---|
| M914 | 249 | MHP | p.[Arg241Cys]; [Gly247Arg] | ? | II | 65.63% | R+ | yes |
| M1080 | 660 | mPKU | p.[IVS4-1G > A]; [Arg241Cys] | 5 | II | 87.14% | R+ | yes |
| M1085 | 469.8 | mPKU | p.[Arg241Cys]; [Val388Met] | 6 | II | 85.16% | R+ | yes |
| M1127 | 258 | MHP | p.[Arg241Cys]; [Arg241Cys] | 8 | II | 87.33% | R+ | yes |
| M1229 | 607.2 | mPKU | p.[Arg241Cys]; [IVS7+2 T > A] | 5 | II | 77.30% | R+ | yes |
| M1294 | 627.6 | mPKU | p.[Arg241Cys]; [IVS7+1 G > A] | 5 | II | 92.22% | R+ | yes |
| M1300 | 600.6 | mPKU | p.[Ex6-96 A > G]; [Arg241Cys] | 5 | II | 84.58% | R+ | yes |
| M1314 | 588 | mPKU | p.[Ex6-96 A > G]; [Arg241Cys] | 5 | II | 65.18% | R+ | yes |
| M1343 | 562.2 | mPKU | p.[Arg241Cys]; [; [Leu255Ser] | 5 | II | 83.85% | R+ | yes |
| M1362 | 451.8 | mPKU | p.[Phe161Ser]; [Arg241Cys] | 6 | II | 85.13% | R+ | yes |
| M1374 | 660 | mPKU | p.[Arg241Cys]; [Arg400Lys] | ? | II | 73.41% | R+ | yes |
| M1451 | 489 | mPKU | p.[Arg111*]; [Arg241Cys] | 5 | II | 87.23% | R+ | yes |
| M1501 | 1320 | cPKU | p.[Arg243Gln]; [IVS12+4 A > G] | 2 | I | 60.24% | R+ | yes |
| M1604 | 959.4 | mPKU | p.[Arg241Cys]; [Arg243Gln] | 5 | II | 58.48% | R+ | yes |
| M1675 | 510 | mPKU | p.[Arg111*]; [Arg241Cys] | 5 | II | 69.52% | R+ | yes |
| M1686 | 420 | mPKU | p.[Arg241Cys]; [Arg243Gln] | 5 | II | 92.04% | R+ | yes |
| M1709 | 384 | mPKU | p.[Ex6-96 A > G]; [Gln419Arg] | 5 | III | 88.70% | R+ | yes |
| M1731 | 408 | mPKU | p.[Ser70del]; [Arg241Cys] | 5 | II | 60.09% | R+ | Yes |
| M1772 | 228 | MHP | p.[Arg155His]; [Val399Val] | 5 | III | 79.15% | R+ | yes |
| M1837 | 924 | mPKU | p.[Ex6-96 A > G]; [Arg241Cys] | 5 | II | 68.90% | R+ | yes |
| M1853 | 822 | mPKU | p.[Ser70del]; [Arg158Gln] | 2 | I | 72.51% | R+ | yes |
| M1950 | 342 | MHP | p.[Ex6-96 A > G]; [Arg241Cys] | 5 | II | 75.20% | R+ | yes |
| M810 | 849 | mPKU | p.[Arg241Cys]; [Arg413Pro] | 5 | II | 82.91% | R+ | yes |
| M835 | 833.4 | mPKU | p.[Arg241Cys]; [IVS7+2 T > A] | 5 | II | 54.26% | R+ | yes |
| M840 | 780 | mPKU | p.[Ser70del]; [Val388Met] | 3 | II | 46.79% | R+ | yes |
| M855 | 780 | mPKU | p.[Arg243Gln]; [Ile324Asn] | ? | III | 47.62% | R+ | yes |
| M862 | 360 | MHP | p.[Ex6-96 A > G]; [Arg241Cys] | 5 | II | 79.67% | R+ | yes |
| M865 | 900 | mPKU | p.[IVS4-1G > A]; [721 C > T] | 5 | II | 99.75% | R+ | yes |
| M870 | 330 | MHP | p.[Ser70del]; [Arg261Gln] | 3 | II | 72.57% | R+ | yes |
| M878 | 960 | mPKU | p.[Ser70del]; [Arg243Gln] | 2 | I | 52.21% | R+ | yes |
| M882 | 804 | mPKU | p.[Glu183Gly]; [Arg241Cys] | ? | II | 53.33% | R+ | yes |
| M896 | 660 | cPKU | p.[Arg241Cys]; [Glu286Lys] | ? | II | 80.34% | R+ | yes |
| M904 | 534 | mPKU | p.[Phe121Leu]; [Arg241Cys] | ? | II | 89.62% | R+ | yes |
| M915 | 1680 | cPKU | p.[IVS4-1G > A]; [Tyr356*] | 2 | I | 56.96% | R+ | yes |
| M922 | 627 | mPKU | p.[Arg243Gln]; [Met276Lys] | ? | III | 64.73% | R+ | yes |
| M952 | 576 | mPKU | p.[Arg241Cys]; [Glu286Lys] | ? | II | 85.74% | R+ | yes |
| M987 | 552 | mPKU | p.[Arg241Cys]; [Gly247Arg] | ? | II | 53.67% | R+ | yes |
| M1105 | 616.8 | mPKU | p.[Arg241Cys]; [Arg243Gln] | 5 | II | 32.25% | R+ | no |
| M1437 | 1020 | mPKU | p.[Pro275Ala]; [Tyr356*] | ? | III | 36.72% | R+ | no |
| M1480 | 720 | mPKU | p.[Arg243Gln]; [Arg261Gln] | 3 | II | 42.26% | R+ | no |
| M1483 | 1260 | cPKU | p.[Arg243Gln]; [Val399Val] | 2 | I | 30.12% | R+ | no |
| M1785 | 1980 | cKU | p.[Ex6-96 A > G]; [Val388Met] | 3 | II | 34.24% | R+ | no |
| M1829 | 880.8 | mPKU | p.[Phe161Ser]; [Val399Val] | 3 | III | 31.03% | R+ | no |
| M793 | 1740 | cPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 39.22% | R+ | no |
| M857 | 938.4 | mPKU | p.[Arg243Gln]; [Val388Met] | 3 | II | 39.55% | R+ | no |
| M1334 | 1080 | mPKU | p.[Arg243Gln]; [Gly247Val] | 2 | I | 34.17% | R+ | no |
| M1340 | 1080 | mPKU | p.[Arg243Gln]; [Ala434Asp] | 3 | III | 37.42% | R+ | no |
| M996 | 894 | mPKU | p.[Ala156Pro]; [Arg408Gln] | ? | II | 58.49% | R+ | no |
| M1268 | 1830 | cPKU | p.[Arg243Gln]; [Cys357*] | 2 | I | 12.79% | R− | no |
| M1269 | 2057.4 | cPKU | p.[Arg111*]; [Arg413Pro] | 2 | I | 9.24% | R− | no |
| M1304 | 1500 | cPKU | p.[Tyr166*]; [Ex6-96 A > G] | 2 | I | 25.01% | R− | no |
| M1438 | 1320 | cPKU | p.[IVS4-1G > A]; [Arg413Pro] | 2 | I | 0.42% | R− | no |
| M1463 | 510 | mPKU | p.[Arg111*]; [Val399Val] | 2 | I | 17.00% | R− | no |
| M1516 | 1020 | mPKU | p.[Phe161Ser]; [Arg243Gln] | 3 | I | 21.82% | R− | no |
| M1518 | 1296 | cPKU | p.[Arg111*]; [Met276Arg] | ? | III | 0.00% | R− | no |
| M1565 | 1680 | cPKU | p.[Arg243Gln]; [Arg243Gln] | 2 | I | 15.83% | R− | no |
| M1575 | 2400 | cPKU | p.[Arg111*]; [Arg243Gln] | 2 | I | 26.46% | R− | no |
| M1590 | 1373.4 | cPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 5.93% | R− | no |
| M1623 | 1108.2 | mPKU | p.[Arg243Gln]; [Arg243Gln] | 2 | I | 11.19% | R− | no |
| M1630 | 1920 | cPKU | p.[Arg243Gln]; [Tyr356*] | 2 | I | −0.48% | R− | no |
| M1649 | 900 | mPKU | p.[Arg261*]; [Tyr356*] | 2 | I | −20.00% | R− | no |
| M1651 | 1500 | cPKU | p.[Arg243Gln]; [Ala434Asp] | 3 | III | 12.74% | R− | no |
| M1660 | 816 | mPKU | p.[Arg243Gln]; [Arg243Gln] | 2 | I | 8.77% | R− | no |
| M1674 | 500.4 | mPKU | p.[Tyr356*]; [Ala434Asp] | 3 | III | 8.86% | R− | no |
| M1710 | 931.2 | mPKU | p.[Ex6-96 A > G]; [Arg413Pro] | 2 | I | 3.71% | R− | no |
| M1714 | 1506 | cPKU | p.[Arg111*]; [Tyr356*] | 2 | I | 1.73% | R− | no |
| M1733 | 1071 | mPKU | p.[Arg243Gln]; [Val399Val] | 2 | I | 8.59% | R− | no |
| M1744 | 1440 | cPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 16.52% | R− | no |
| M1747 | 1504.8 | cPKU | p.[Gln232*]; [Arg243Gln] | 2 | I | 10.29% | R− | no |
| M1753 | 694.2 | mPKU | p.[Ex6-96 A > G]; [Tyr356*] | 2 | I | 11.67% | R− | no |
| M1755 | 1115.4 | mPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 9.59% | R− | no |
| M1770 | 1651.8 | cPKU | p.[IVS4-1G > A]; [Ile406Thr] | ? | III | 5.81% | R− | no |
| M1775 | 1335 | cPKU | p.[Arg243Gln]; [Arg243Gln] | 2 | I | 29.54% | R− | no |
| M1782 | 1218 | cPKU | p.[IVS4-1G > A]; [Arg243Gln] | 2 | I | 20.44% | R− | no |
| M1799 | 1141.2 | mPKU | p.[Arg111*]; [Tyr356*] | 2 | I | 0.77% | R− | no |
| M1820 | 666 | mPKU | p.[Arg243Gln]; [Arg252Gln] | 2 | III | 13.24% | R− | no |
| M1846 | 1800 | cPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 13.54% | R− | no |
| M1891 | 1260 | cPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 0.00% | R− | no |
| M1916 | 1320 | cPKU | p.[His64*]; [Ala156Pro] | ? | III | 12.07% | R− | no |
| M1952 | 624 | mPKU | p.[Arg111*]; [Arg243Gln] | 2 | I | 0.00% | R− | no |
| M744 | 2100 | cPKU | p.[Arg243Gln]; [Arg243Gln] | 2 | I | 2.00% | R− | no |
| M899 | 2040 | cPKU | p.[Ex6-96 A > G]; [IVS6-1G > A] | 2 | I | 22.58% | R− | no |
| M908 | 660 | mPKU | p.[Glu280Lys]; [Ala434Asp] | 3 | III | 27.46% | R− | no |
| M911 | 1734 | cPKU | p.[Arg158Trp]; [Val399Val] | ? | III | 10.71% | R− | no |
| M920 | 1006.2 | mPKU | p.[Gly257Val]; [Arg408Gln] | ? | II | 15.86% | R− | no |
| M936 | 1920 | cPKU | p.[IVS4-1G > A]; [Arg413Pro] | 2 | I | 24.32% | R− | no |
| M960 | 1260 | cPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 0.00% | R− | no |
| M1316 | 876 | mPKU | p.[Ex6-96 A > G]; [Arg243Gln] | 2 | I | 29.40% | R− | no |
| M1339 | 1920 | cPKU | p.[Arg413Pro]; [Arg413Pro] | 2 | I | 11.89% | R− | no |
| M1342 | 726.6 | mPKU | p.[Gly247Val]; [Cys357Tyr] | ? | III | 2.22% | R− | no |
| M1349 | 1218 | cPKU | p.[Arg111*]; [Val399Val] | 2 | I | 19.51% | R− | no |
| M1401 | 564 | mPKU | p.[Ex6-96 A > G]; [Ex6-96 A > G] | 2 | I | −44.77% | R− | no |
| M1412 | 1260 | cPKU | p.[Ex6-96 A > G]; [Arg261*] | 2 | I | 9.34% | R− | no |
| M1420 | 1260 | cPKU | p.[Arg176*]; [IVS6-1G > A] | 2 | I | 15.33% | R− | no |
aThe highest plasma Phe concentration (umol/L)before treatment, and all of which applied in this study were the maximum pretreatment values.
bPhenotypes were stratified as mild hyperphenylalaninemia (MHP), mild phenylketonuria (mPKU), and classical PKU (cPKU) based on the pretreatment plasma Phe values.
cAn arbitrary value (AV) if available was assigned to each mutation: AV = 1 for classical PKU mutation; AV = 2 for moderate PKU mutation; AV = 4 for mild PKU mutation, and AV = 8 for MHP mutation. Phenotypes resulting from a combination of the two mutant alleles were expressed as the sum of the two mutations’ AVs.
dThe groups for the patients based on the assessment of the BH4-responsiveness-status of the detected mutations. I for “Non BH4 responsive”; II for “BH4 responsive”; III for “Undefined BH4 responsive”.
eBased on BH4 loading test; R: response; R+: responder; R−: non-responder.
According to the phenotypic scheme, response to BH4 was detected in 15.15% (5/33) of cPKU, in 67.27% (37/55) of mPKU and in all the 6 MHP patients. The differences in the percentages of responders among the three phenotypic groups were statistically significant (χ2 = 28.56, df = 2, p < 0.001). As shown in Table 1, the reduction of Phe from the baseline among BH4 responders ranged between 30.12% and 99.75%, revealing a considerable degree of inter-subject variability. Most patients (37/48, 77.08%) had normal or nearly normal (≤360 umol/L) Phe levels 24 hours following the BH4 challenge whereas 11 responders had not. The Phe for 3 out of 5 cPKU responders and 28 out of 37 mPKU responders reached the normal range while all 6 MHP patients showed pronounced Phe reductions, indicating that the rate of Phe reduction was largely dependent on the phenotypic class of the patients. For the group of 37 responders (Fig. 1a), blood Phe level was 642.32 ± 294.23 umol/L during pre-treatment and 688.65 ± 216.23 umol/L at time 0 hour of BH4 loading. The latter number was reduced to 170.62 ± 83.80 umol/L at 24 hours after loading. Data analysis of this group suggested a significant correlation between pre-treatment Phe and the percentage of Phe reduction after challenge (r = 0.378, P < 0.05) as opposed to before-BH4 (time at 0 hour) Phe and the extent of response which did not seem to have a statistically significant correlation (r = 0.08, P > 0.05) (The statistical correlation between pre-treatment Phe and before-BH4 (time at 0 hour) was excluded (r = −0.209, P > 0.05)). The group of the other 11 responders (Fig. 1b) had a pre-treatment Phe concentration of 1110.00 ± 413.54 umol/L and 1238.82 ± 710.33 umol/L at 0 hour of BH4 loading. The latter number was reduced to 733.07 ± 454.64 umol/L at the end of the test. Significant correlations existed between pre-treatment Phe and post-loading Phe level (time at 24 hour) (r = 0.777, P < 0.01), and between pre-BH4 Phe and Phe level at 24 hours (r = 0.981, P < 0.001), which was inconsistent with the view derived from the study of Leuzzi, V. et al.36.
Figure 1.
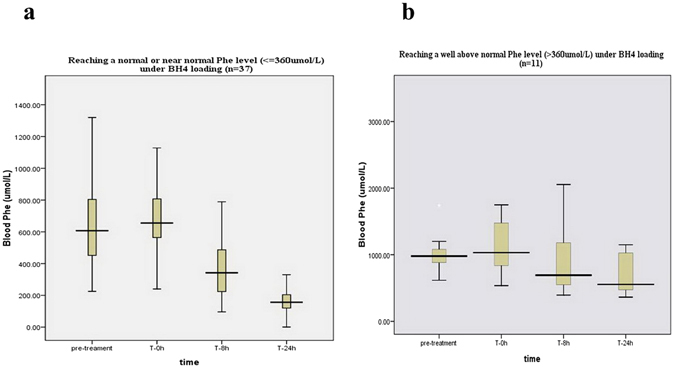
The length of the boxes indicates the interquartile space (P25–P75); the horizontal line into the box represents the median (P50), and the error bars indicate the adjacent values, that is, the maximum and minimum values of the distribution, which may not be considered abnormal.
Classification of Mutations and Genotypes for BH4 Response
Genetic analysis revealed 45 mutations and identified 66 genotypes in these 94 patients (Table 2). Among 43 patients with 2 pre-defined null mutations, 7 patients were identified as responders: p.[Arg243Gln]; [Gly247Val], p.[Arg243Gln]; [IVS12 + 4A > G], p.[Arg243Gln]; [Val399Val], p.[Ser70del]; [Arg158Gln], p.[Ex6-96A > G]; [Arg243Gln], p.[Ser70del]; [Arg243Gln], p.[IVS4-1G > A]; [Tyr356*]. For 9 mutations involved, the intrinsic BH4 responsiveness might be ambiguous due to category overlap. Among the 35 patients presenting at least 1 mutation that had been reported to associate with BH4 responsiveness, only 1 patient, i.e. p.[Gly257Val]; [Arg408Gln], did not respond to the challenge. Among 16 patients with the presence of mutations without definite BH4 responsiveness, all of them were functionally hemizygous which could help us to assess the intrinsic BH4 responsiveness of the missense mutation present on the other chromosome. From the derivation of our data, we classified p.Arg241Cys (c.721C > T), p.Val388Met (c.1162G > A), and p.Arg261Gln (c.782G > A) as BH4 responsive mutations, p.Arg111* (c.331C > T), p.[IVS6-1G > A] (c.707-1G > A) and p.Arg413Pro (c.1238G > C) as constantly non-BH4 responsive variants. Both classifications were unambiguous because the response was consistent in two or more functionally hemizygous patients. Since p.Arg408Gln (c. 1223G > A) was found in two compound-heterozygous patients, and the following mutations including p.Arg155His (c.464G > A), p.Arg158Gln (c.473G > A), p.Pro275Ala (c.823C > G), p.Met276Lys (c.827T > A), p.Ile324Asn (c.971T > A) and p.Gln419Arg (c.1256A > G) were found in only one functionally hemizygous patient, the assignment of these variants to any specific BH4 responsive category may therefore be uncertain.
Table 2.
Genotype versus BH4 response in 94 Chinese PAH Deficient patients.
| Genotypea | No | responder | non-responder | Inconsistencies (No.) |
|---|---|---|---|---|
| null/null | ||||
| p.[Arg243Gln]; [IVS12 + 4 A > G] | 1 | 1cPKU | 0 | 1 |
| p.[Ex6-96 A > G]; [Arg243Gln] | 8 | 1cPKU | 5cPKU/2mPKU | 1 |
| p.[IVS4-1G > A]; [Tyr356*] | 1 | 1cPKU | 0 | 1 |
| p.[Arg243Gln]; [Gly247Val] | 1 | 1mPKU | 0 | 1 |
| p.[Arg243Gln]; [Val399Val] | 2 | 1mPKU | 1mPKU | 1 |
| p.[Phe161Ser]; [Arg243Gln] | 1 | 1mPKU | 0 | 1 |
| p.[Ser70del]; [Arg243Gln] | 1 | 1mPKU | 0 | 1 |
| p.[Arg243Gln]; [Cys357*] | 1 | 0 | 1cPKU | 0 |
| p.[Arg111*]; [Arg413Pro] | 1 | 0 | 1cPKU | 0 |
| p.[Tyr166*]; [Ex6-96 A > G] | 1 | 0 | 1cPKU | 0 |
| p.[Arg413Pro]; [Arg413Pro] | 1 | 0 | 1cPKU | 0 |
| p.[Ex6-96 A > G]; [Arg261*] | 1 | 0 | 1cPKU | 0 |
| p.[Arg176*]; [IVS6-1G > A] | 1 | 0 | 1cPKU | 0 |
| p.[IVS4-1G > A]; [Arg413Pro] | 2 | 0 | 2cPKU | 0 |
| p.[Arg243Gln]; [Arg243Gln] | 5 | 0 | 3cPKU/2mPKU | 0 |
| p.[Arg111*]; [Arg243Gln] | 2 | 0 | 1cPKU/1mPKU | 0 |
| p.[Arg243Gln]; [Tyr356*] | 1 | 0 | 1cPKU | 0 |
| p.[Arg111*]; [Tyr356*] | 2 | 0 | 1cPKU/1mPKU | 0 |
| p.[Gln232*]; [Arg243Gln] | 1 | 0 | 1cPKU | 0 |
| p.[IVS4-1G > A]; [Arg243Gln] | 1 | 0 | 1cPKU | 0 |
| p.[Ex6-96 A > G]; [IVS6-1G > A] | 1 | 0 | 1cPKU | 0 |
| p.[Arg158Trp]; [Val399Val] | 1 | 0 | 1cPKU | 0 |
| p.[Arg111*]; [Val399Val] | 2 | 0 | 1cPKU/1mPKU | 0 |
| p.[Arg261*]; [Tyr356*] | 1 | 0 | 1mPKU | 0 |
| p.[Ex6-96 A > G]; [Arg413Pro] | 1 | 0 | 1mPKU | 0 |
| p.[Ex6-96 A > G]; [Tyr356*] | 1 | 0 | 1mPKU | 0 |
| p.[Ex6-96 A > G]; [Ex6-96 A > G] | 1 | 0 | 1mPKU | 0 |
| Total | 43 | 7 | 36 | 7 |
| at least one mutation with definite BH4 responsiveness | ||||
| p.[Ex6-96 A > G]; [Val388Met] | 1 | 1cPKU | 0 | 0 |
| p.[IVS4-1G > A]; [Arg241Cys] | 2 | 2mPKU | 0 | 0 |
| p.[Arg241Cys]; [Arg243Gln] | 3 | 3mPKU | 0 | 0 |
| p.[Arg241Cys]; [IVS7 + 2 T > A] | 2 | 2mPKU | 0 | 0 |
| p.[Arg241Cys]; [IVS7 + 1 G > A] | 1 | 1mPKU | 0 | 0 |
| p.[Ex6-96 A > G]; [Arg241Cys] | 5 | 3mPKU/2MHP | 0 | 0 |
| p.[Arg241Cys]; [Arg400Lys] | 1 | 1mPKU | 0 | 0 |
| p.[Arg243Gln]; [Arg261Gln] | 1 | 1mPKU | 0 | 0 |
| p.[Ser70del]; [Val388Met] | 1 | 1mPKU | 0 | 0 |
| p.[Arg243Gln]; [Val388Met] | 1 | 1mPKU | 0 | 0 |
| p.[Glu183Gly]; [Arg241Cys] | 1 | 1mPKU | 0 | 0 |
| p.[Arg241Cys]; [Glu286Lys] | 2 | 2mPKU | 0 | 0 |
| p.[Ala156Pro]; [Arg408Gln] | 1 | 1mPKU | 0 | 0 |
| p.[Arg241Cys]; [Val388Met] | 1 | 1mPKU | 0 | 0 |
| p.[Arg241Cys]; [Arg241Cys] | 1 | 1MHP | 0 | 0 |
| p.[Arg241Cys]; [Leu255Ser] | 1 | 1mPKU | 0 | 0 |
| p.[Phe161Ser]; [Arg241Cys] | 1 | 1mPKU | 0 | 0 |
| p.[Arg111*]; [Arg241Cys] | 2 | 2mPKU | 0 | 0 |
| p.[Ser70del]; [Arg241Cys] | 1 | 1mPKU | 0 | 0 |
| p.[Arg241Cys]; [Arg413Pro] | 1 | 1mPKU | 0 | 0 |
| p.[Ser70del]; [Arg261Gln] | 1 | 1MHP | 0 | 0 |
| p.[Phe121Leu]; [Arg241Cys] | 1 | 1mPKU | 0 | 0 |
| p.[Arg241Cys]; [Gly247Arg] | 2 | 1mPKU/1MHP | 0 | 0 |
| p.[Gly257Val]; [Arg408Gln] | 1 | 0 | 1mPKU | 1 |
| Total | 35 | 34 | 1 | 1 |
| mutations without definite BH4 responsiveness | ||||
| p.[Arg243Gln]; [Ala434Asp] | 2 | 1mPKU | 1cPKU | / |
| p.[Ser70del]; [Arg158Gln] | 1 | 1mPKU | 0 | / |
| p.[Ex6-96 A > G]; [Gln419Arg] | 1 | 1mPKU | 0 | / |
| p.[Arg155His]; [Val399Val] | 1 | 1MHP | 0 | / |
| p.[IVS4-1G > A]; [Ile406Thr] | 1 | 0 | 1cPKU | / |
| p.[His64*]; [Ala156Pro] | 1 | 0 | 1cPKU | / |
| p.[Gly247Val]; [Cys357Tyr] | 1 | 0 | 1mPKU | / |
| p.[Phe161Ser]; [Arg243Gln] | 1 | 0 | 1mPKU | / |
| p.[Arg111*]; [Met276Arg] | 1 | 0 | 1mPKU | / |
| p.[Arg243Gln]; [Arg252Gln] | 1 | 0 | 1mPKU | / |
| p.[Glu280Lys]; [Ala434Asp] | 1 | 0 | 1mPKU | / |
| p.[Tyr356*]; [Ala434Asp] | 1 | 0 | 1mPKU | / |
| p.[Pro275Ala]; [Tyr356*] | 1 | 1mPKU | 0 | / |
| p.[Arg243Gln]; [Ile324Asn] | 1 | 1mPKU | 0 | / |
| p.[Arg243Gln]; [Met276Lys] | 1 | 1mPKU | 0 | / |
| Total | 16 | 7 | 9 | |
aThe genotypes are described at protein level.
Table 2 presents the 66 different genotypes identified in these patients: 34 different genotypes belonging to the responder group, 29 belonging to the non-responder group, and 3 belonging to both groups. Genotypes occurring in two or more patients showed consistency in BH4 loading results: p.[IVS4-1G > A]; [Arg241Cys], p.[Arg241Cys]; [Arg243Gln], p.[Arg241Cys]; [IVS7 + T > A], p.[Ex6-96A > G]; [Arg241Cys], p.[Arg241Cys]; [Glu286Lys], p.[Arg111*]; [Arg241Cys] and p.[Arg241Cys]; [Gly247Arg] in responders; p.[Arg243Gln]; [Arg243Gln], p.[IVS4-1G > A]; [Arg413Pro], p.[Arg111*]; [Arg243Gln], p.[Arg111*]; [Tyr356*] and p.[Arg111*]; [Val399Val] in non-responders; p. [Ex6-96A > G]; [Arg243Gln], p.[Arg243Gln]; [Ala434Asp] and p.[Arg243Gln]; [Val399Val] belonged to both groups.
The Diagnostic Performance of PAH Genotype AV Sum to Predict BH4 Response
The PAH genotypes, AV sums, and clinical BH4 responsive status of 77 patients (refer to Method: Determining the PAH Genotype Severity Using the AV Sum and Classification of BH4 Response) were shown in Table 1. The majority of responders (81.08%, 30/37) had an AV sum >2, indicating that at least one mutation is moderate or mild in severity. The remaining seven responders carried two severe mutations (AV = 1) and were given an AV sum of 2. BH4 responders represented a specific heterogeneous group, with the AV sum ranging from 2 to 8, 8 being the highest AV sum present in a homozygote for p.Arg241Cys. In contrast, the AV sum for the majority of non-responders (92.5%, 37/40) was 2, indicating a severe PAH genotype. The other three non-responders who carried a moderate mutation in combination with a severe one had an AV sum of 3.
Then, we analyzed ROC curve (Fig. 2) to verify the efficiency of this approach and to determine the cutoff value of the AV sum that differentiated PAH deficient patients with and without BH4 response. The AUC value was 0.891 (95% confidence interval (95% CI) = 0.811–0.972)). The optimal cutoff level of the AV sum, determined by the curve to calculate sensitivity and specificity, was set at 2.5. Since AV sums ranged from 2 to 8, with a whole number indicating the patient phenotype, we used AV sum >2 as the cutoff. The sensitivity and specificity we found at this level was 81.1% and 92.5%, which likely resulted in a positive predictive value (PPV) as opposed to a negative predictive value (NPV) of 90.9% and 84.1% (Table 3).
Figure 2.
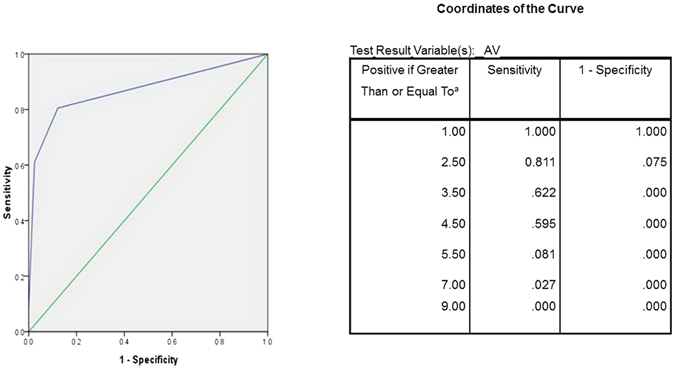
The test result variable(s): AV has at least one tie between the positive actual state group and the negative actual state group. *The smallest cutoff value is the minimum observed test value minus 1, and the largest cutoff value is the maximum observed test value plus 1. All the other cutoff values are the averages of two consecutive ordered observed test values.
Table 3.
Diagnostic performance of two predicted tests, estimated using the BH4 loading as the gold standard.
| Sensitivity (%) | Specificity (%) | PPV (%) | NPV (%) | Accuracy (%) | |
|---|---|---|---|---|---|
| AV sum (cut-off: >2) | 81.1% | 92.5% | 90.9% | 84.1% | 87.01% |
| BH4-responsiveness-status of the mutations | 82.9% | 97.3% | 97.1% | 83.7% | 89.74% |
PPV: positive predictive value; NPV: negative predictive value; OP: odd product; A: true positive (TP); B: false negative (FN); C: false positive (FP); D: true negative (TN).
Sensitivity = True positive rate (TPR) = A/(A + B) = TP/(TP + FN).
Specificity = True negative rate (TNR) = D/(C + D) = TN/(FP + TN).
PPV = precision = A/(A + C) = TP/(TP + FP).
NPV = TN/(TN + FN).
Accuracy = (A + D)/(A + B + C + D).
The Diagnostic Performance of Assessment of the Mutation BH4-Responsiveness-Status to Predict BH4 Response
Based on the BH4-responsiveness-status of every mutation, in this cohort of 94 patients, 43 patients carried known null mutations on both alleles, excluding BH4 responsiveness in 45.74% (43/94); 35 patients carried at least one reported mutation with consistent BH4 responsiveness, indicating a high probability of BH4 responsiveness in 37.23% (35/94); and the remaining 16 patients without defined BH4 responsive mutations (Table 2). We note that the observed phenotype of the patient did not always match his predicted phenotype. As shown in Table 2, the observed BH4 response/non-response results matched that of the expected in 70 out of 78 patients (89.74%, 70/78), and the discrepancies were most apparent in patients with expected non-BH4 responsiveness (16.28%, 7/43).
Table 3 shows the diagnostic accuracy of this approach in predicting PAH deficient patients’ clinical BH4 response status. The sensitivity, specificity, PPV and NPV related to the presence of a BH4 responsive variant were 82.9%, 97.3%, 97.1% and 83.7% respectively.
Finally, we used Kappa test to examine the consistency between two approaches in classification of the clinical BH4 response status. Kappa test revealed that the results of the AV sum approach showed good consistency with those of the mutation BH4-responsiveness-status assay (Kappa test, kappa = 0.71, P < 0.001).
Discussion
Sapropterin has been available as a non-dietary therapy option for patients with BH4-responsive PKU since 2007 in the United States, 2008 in the European Union, and 2010 in Canada. It is prescribed to patients 4 years of age and onwards4. The implementation of pharmacological intervention at a younger age has been recently indicated to be feasible and beneficial37. It is also noted that sapropterin could be considered as a treatment option in pregnant women with PKU who cannot achieve the recommended ranges of blood Phe level with dietary therapy alone38. So a careful selection of patients who are eligible for the therapy using sound procedures (with low false-negatives and false-positives) is imperative. The goal of this study is to investigate the characteristics of BH4-responsiveness in Chinese patients and to determine the predictive value of genetic analysis for BH4-responsiveness in order to identify the appropriate candidates for this medication.
Our BH4 loading results demonstrated that BH4 responsiveness was mostly associated with milder phenotypes (43/48, 89.58%) except 5 (5/48, 10.42%) BH4 responders being cPKU, and the rate of Phe reduction was largely dependent on phenotypic classes with 28 mPKU responders, 6 MHP responders and 3 cPKU responders reaching normal or nearly normal (≤360 umol/L) Phe levels 24 hours post load. These findings supported the statement that mild phenotypes respond better to BH4 challenge22. Therefore, we speculated that half our patients (50.29%, 174/346) might be potential candidates for future pharmacologic intervention with concomitant relief or withdrawal of the burdensome diet therapy. In our group, we noted that there were individuals (8/18, 44.44%) who were previously untreated and in whom a positive response to BH4 was identified. The results of our study, coupled with the findings proposed by Moseley KD39, Koch R40 and Grosse SD41, led to the consideration that sapropterin should be an appropriate treatment option for some Chinese patients, and that it might be useful as an adjuvant in maintaining blood Phe levels, thus hindering fluctuations. Inhibiting the fluctuations in blood Phe levels has been recognized as neuro-protective42, particularly during infancy, early childhood, and pregnancy. Moreover, sapropterin may allow BH4-responsive patients, who were later diagnosed, to make progress in their development.
Our previous work32 has proven that genetic diagnosis was useful for evaluating the biochemical phenotypes. In this study, we estimated the accuracy of two commonly used genetic diagnostic tests for their statistical prediction of BH4 responsiveness. The sensitivity and specificity relative to the BH4 responsiveness status of the detected mutations in predicting clinical response were 82.9% and 97.3%, respectively, a little higher than those of the AV sum tool, 81.1% and 92.5%. Our results showed that the AV sum approach appeared to provide a lesser degree of sensitivity for identifying patients, which was not consistent with the view proposed by Quirk et al.43, who concluded that the AV sum tool was viable for identifying definitive responders with a predictive sensitivity of 89.5%, as derived from their clinical results of a cohort of 58 patients.
Furthermore, we evaluated the effects of the two tests in 346 Chinese patients. Our data identified 31.65% of our cohort as potentially BH4-responsive using the AV sum test and 25.43% by the other. But the overall frequency of BH4-responsiveness in 94 patients who took the loading test was 51.06%, almost two times higher than the predicted data. The following reasons might contribute to this discrepancy. In our study, only p.Arg241Cys (c.721C > T), p.Val388Met (c.1162G > A) and p.Arg261Gln (c.782G > A) were confirmed to be constantly responsive; p.Arg243Gln (c.728G > A) and p.Arg413Pro (c.1238G > C) which were defined as responsive by Zurfluh et al.22, were defined as null mutations. Apart from the different criteria for mutation classification due to population-based findings, a unique genotypic feature of high heterogeneity was partly related to it. The mutation spectrum observed in China33 showed a characteristic of the combination of a small number of common mutations and a very high number of rare ones. Based on our observation, for the most common 8 mutations, p.Arg241Cys (c.721C > T) was consistently associated with BH4 responsiveness; p.Arg243Gln (c.728G > A) was associated with non-BH4 responsiveness except for 5 patients; p.Ex6-96A > G (c.611A > G), p.[IVS4-1G > A] (c.442-1G > A), and p.Val399Val (c.1197A > T) as splicing mutations, were equally distributed in both categories; while p.Arg413Pro (c.1238G > C), p.Arg111* (c331C > T) and p.[IVS6-1G > A] (c. 707-1G > A) were consistent with their non-BH4 responsiveness in every patient, which might be the reason that the lower “concordance rate” (83.72%) was observed in patients within the expected non-BH4 responsive group. In our study, the patients with the rare mutations were excluded from the AV sum analysis or were assigned to the “Undefined BH4 responsive” group since most rare mutations had not been analyzed in vitro and were of unknown BH4 responsiveness.
In terms of optimizing the use and allocation of public health resources, the BH4 response detection procedure based on genotyping is an imperative step for our population since there is an estimated not-too-low potential incidence of BH4-responsiveness in China, yet the drug used for treatment is of high cost with a limited supply. We will present detailed reasons below: first, a genotype-predicted prevalence of BH4-responsiveness is almost two times lower than the result obtained by loading tests, this may lead to lower false positive results, thus eliminating the need for challenging every patient; second, the information of established BH4 responsiveness or non-responsiveness in single allele and mutational combinations derived from our research allows Chinese patients with those genotypes to avoid prior 24 h BH4 loading tests; third, since up to more than 10% of classic patients responded to loading test, 32.73% of mild ones did not respond, and 19.57% of the non-responders were “slow responsive” based on our data, these patients may need a longer period of time for testing to show observable BH4 long-term efficiency; the response in these patients is a difficult target to evaluate properly and the current BH4 loading test should be optimized accordingly to determine the patients who are feasible subjects for complete diet liberalization or who can increase their intake of natural protein with sapropterin as an adjuvant option.
In this way, the inconsistency of our work and the genetic analysis importance in Chinese patients stressed the need for a population-specialized approach to the evaluation of BH4 responsiveness. As a next step, we aimed to verify the clinical relevance of our personalized procedure (Fig. 3) and to analyze the effects of different sapropterin concentrations, dosages, and durations on the outcome of BH4 loading tests in individuals with different genotypes in a prospective study, which might serve as a way to establish individualized treatment for Chinese PAH deficiency patients.
Figure 3.
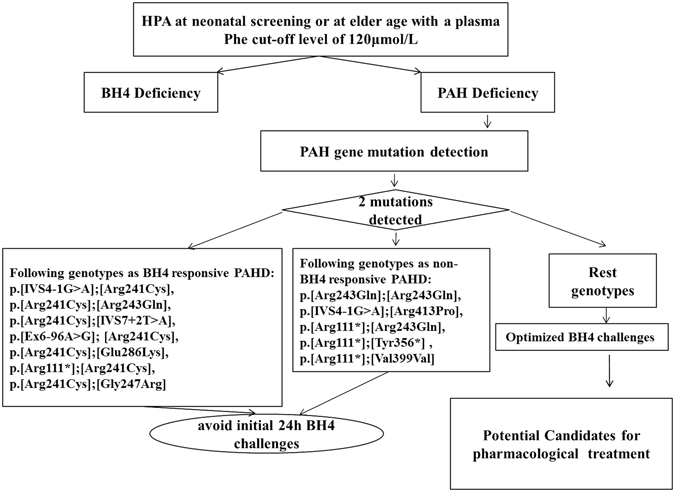
HPA: hyperphenylalaninemia; BH4 deficiency: genetic defects in the path way of BH4 synthesis or recycling leading to secondary PAH deficiency and elevated blood Phe levels; PAH deficiency: the inborn error of Phe metabolism caused by deficient activity of phenylalanine hydroxylate; Initial 24 h BH4 challenges: a BH4-loading test with a dose of 20 mg/kg body weight and duration of 24 h; Optimized BH4 challenges: more personalized testing procedures with different dosage and duration to address individual BH4 responsiveness in PAH deficient patients.
Methods
Population Study
Our study included 346 unrelated Chinese patients (171 males and 175 females, with ages ranging from 1 month to 13 years) from the Department of Pediatric Endocrinology and Genetic Metabolism of Xin Hua Hospital affiliated with Shanghai Jiao Tong University School of Medicine between January, 2006 and December, 2012 (Table S1). These patients were found to have two presumably causative mutations in the PAH gene. In all patients, PAH deficiency had been detected by either a national screening (56.94%, 197/346) or by the presence of neurological deterioration observed at an older age (43.06%, 149/346) with a plasma Phe cutoff level of 120 μmol/L. Patients with defect in the synthesis and recycling of BH4 were excluded by analysis of urinary pterins and dihydropteridine reductase activity in erythrocytes. Of these patients, 94 whose informed consents were obtained took part in BH4 loading test (40 males and 54 females, ages ranging from 1 month to 5.6 years; 18 patients presented neurological deterioration at an older age while the others were detected with a national screening). Table S2 showed that there were no significant differences between the demographic characteristics of patients who took the BH4 loading tests and those who did not.
The metabolic phenotype for each patient was stratified according to the individual’s plasma Phe concentration before treatment. All of the Phe levels were maximum values (Phe range of 132–2937 umol/L) during pretreatment. Phe was measured using a fluorometric method until 2003 followed by tandem-mass spectrometry. Patients were classified as cPKU (Phe more than 1200 μmol/L), mPKU (Phe, 360 to 1200 μmol/L), and MHP (Phe less than 360 μmol/L) according to the Chinese HPA consensus updated in 20143.
The Ethics Committee of Xinhua Hospital affiliated with Shanghai Jiao Tong University School of Medicine approved this study and informed consents were subsequently obtained from the parents of the patients enrolled. The methods were performed in accordance with the relevant approved guidelines.
BH4 Loading Test
The BH4 loading tests that were used as the gold standard diagnostic method for BH4 response in this study were performed between 2005 and 2009 following the routine protocol20 combined with our local regulation44. During the entire testing period, patients had no dietary restrictions. Blood Phe was measured by dried blood spot analysis at times 0-2-4-6-8 and 24 hours after oral administration of 20 mg/kg KUVAN™ (sapropterin dihydrochloride); Biomarin Pharmaceutical Inc. Novato, CA) in 68 patients whose initial plasma Phe results were ≥600 umol/L. For 26 patients whose Phe levels were <600 umol/L, L-Phe (100 mg/kg; Shanghai Pujiang Institute of Applied Biochemistry, China) was given initially, followed by sapropterin (20 mg/kg). Patients’ blood samples were taken at times 0-1-2 and 3 hours after Phe loading and at 0-2-4-6-8 and 24 hours after BH4 loading. Responsiveness to BH4 was calculated as a percentage of blood Phe reduction 24 hours after sapropterin administration. A reduction of at least 30% indicated a positive response. No side effects were observed during the test. Plasma Phe concentrations were determined by tandem-mass spectrometry (API 4000 LC/MS/MS System, Shimadzu, Tokyo, Japan).
Mutation Analysis
Genomic DNA was isolated from peripheral blood samples, 13 exons and related intronic boundaries of the PAH gene were amplified. All PCR products were scanned for mutations by direct sequence analysis, most of which were previously done32, 33. All experiments were conducted according to the standard protocol45, 46, and the details were described in our previous paper33. Mutations were referred to by their description at the DNA level and protein level (http://www.hgvs.org/mutnomen). Since the mutations present in 68 individuals lacked expression data, they were excluded from the analyses that depended on in vitro expression information. For mutation classification in relation to BH4-responsiveness, we used the criteria developed by Zurfluh et al.22, supplemented by the data from BIOPKU database and other published papers21, 23.
Assigning Groups Based on the Assessment of the BH4-Responsiveness-Status of the Detected Mutations
A mutation/allele was regarded as BH4-responsive if it appeared either as homozygous or compound heterozygous form associated with a known null mutation in BH4 responders22. BH4-responsiveness was expected in patients with a BH4-responsive mutation on at least one PAH gene copy. We assigned each patient to one of three groups according to the following criteria47:
“Non-BH4 responsive” group: if both alleles of the patient were null mutations;
“BH4 responsive” group: if the patient carried a consistent BH4 responsive mutation on at least one allele;
“Undefined BH4 responsive” group: a patient with the presence of at least one mutation encoding a protein with known residual activity but having inconsistent or pending information on BH4 response.
Of 346 patients, 88 patients were assigned to the “BH4 responsive” group, 189 to the “Non-BH4 responsive” group, and the remaining 69 to the “Undefined BH4 responsive” group. We thus attempted to predict the BH4 responsiveness of 346 patients from the respective groups that were assigned based on the information of the single allele.
Determining the PAH Genotype Severity Using the AV Sum and Classification of BH4 Response
We used an arbitrary assigned value (AV) approach stated by Guldberg et al.34 to assess the PAH genotype severity of our patients, and the resulting predicted phenotypes from a combination of the two mutant alleles were represented as the sum of the two mutations’ AVs in 278 patients (data were shown in our previous paper)32. The ideal cutoff level of AV sum used for predicting the response was determined by the receiver operating characteristic (ROC) curve analysis. Of 278 patients, 88 with an AV sum >2 were classified as “AV sum responders” and 190 with an AV sum = 2 were classified as “AV sum non-responders”.
We investigated the prevalence of BH4 response in those 278 patients who had an AV sum data.
Assessing the Diagnostic Value of Both Classification Approaches in Predicting BH4 Response
The ability of the AV sum to differentiate the patient’s clinical BH4 response status was validated in 77 BH4-challenged patients having AV estimates for both mutations. Those patients were simultaneously dichotomized into “BH4 responder” and “non-BH4 responder” groups based on their clinical response classification derived from the BH4 loading test and their AV sum. In order to quantify the ability of AV sum to classify BH4 response, sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were calculated. The ROC curve is a binary tool with five degrees of rating: Excellent (0.9 to 1), good (0.8 to 0.9), fair (0.7 to 0.8), poor (0.6 to 0.7), and not discriminating (0.5 to 0.6)48.
The ability of the detected mutations’ BH4 responsiveness-status to discriminate the patient’s clinical BH4 responses status was confirmed in 94 patients loaded with BH4. The patients were designated to one of the three groups (mentioned in Methods: assigning groups based on the assessment of the BH4-responsiveness-status of the detected mutations). The clinical distribution was based on the BH4 loading test. Sensitivity, specificity, PPV, and NPV were calculated to evaluate performance relevant to clinical management.
We used Kappa test to compare the consistency of the two methods (Kappa ≤ 0.20, poor; 0.2 < Kappa ≤ 0.40, fair; 0.4 < Kappa ≤ 0.60, moderate; 0.6 < Kappa ≤ 0.80, good; 0.8 < Kappa, excellent agreement)49.
Statistical Analysis
All statistical analyses were performed using SPSS 17.0 (SPSS Inc., Chicago, Illinois). Initially, all data were analyzed using the Kolmogorov-Smirnov test to assess whether the data were normally distributed. Quantitative data were reported as mean ± standard deviation; normally distributed parameters were compared using the ANOV and non-normally distributed parameters were compared using the Mann-Whitney U test. Qualitative data were compared using the chi-square test; a p-value < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
This work was supported by grants from Shanghai City Health Bureau project (20124104) and The National Natural Science Foundation of China (81200654).We greatly acknowledge Xiaoming Tu (Department of Biostatistics, School of Basic Medical Sciences, Nanjing medical University), in data management and processing, Kendall Muzzarelli (Department of Biochemistry and Molecular Biology, School of Medicine, Wayne State University) for reviewing our manuscript. We also thank Ya-fen Zhang, Zhi-wen Gong and Jian-De Zhou (Xinhua Hospital affiliated to shanghai Jiao Tong University School of Medicine) in the laboratory work.
Author Contributions
Xuefan Gu and Tianwen Zhu conceived and designed the experiments. Jun Ye, Lianshu Han, Wenjuan Qiu, Huiwen Zhang, and Lili Liang, prepared the samples and performed the loading tests. Tianwen Zhu analyzed the data and wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-06462-y
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mitchell JJ, Trakadis YJ, Scriver CR. Phenylalanine hydroxylase deficiency. Genetics in medicine: official journal of the American College of Medical Genetics. 2011;13:697–707. doi: 10.1097/GIM.0b013e3182141b48. [DOI] [PubMed] [Google Scholar]
- 2.Bickel H, Gerrard J, Hickmans EM. Influence of phenylalanine intake on phenylketonuria. Lancet. 1953;265:812–813. doi: 10.1016/S0140-6736(53)90473-5. [DOI] [PubMed] [Google Scholar]
- 3.Ye, J. G. & X. F. A consensus of the diagnosis and treatment of hyperphenylalaninemia. Chin J Pediatr52, 5 (2014). [PubMed]
- 4.Vockley J, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genetics in medicine: official journal of the American College of Medical Genetics. 2014;16:188–200. doi: 10.1038/gim.2013.157. [DOI] [PubMed] [Google Scholar]
- 5.Trefz FK, et al. Management of adult patients with phenylketonuria: survey results from 24 countries. European journal of pediatrics. 2015;174:119–127. doi: 10.1007/s00431-014-2458-4. [DOI] [PubMed] [Google Scholar]
- 6.Hood A, Grange DK, Christ SE, Steiner R, White DA. Variability in phenylalanine control predicts IQ and executive abilities in children with phenylketonuria. Molecular genetics and metabolism. 2014;111:445–451. doi: 10.1016/j.ymgme.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hood A, et al. Prolonged exposure to high and variable phenylalanine levels over the lifetime predicts brain white matter integrity in children with phenylketonuria. Molecular genetics and metabolism. 2015;114:19–24. doi: 10.1016/j.ymgme.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Recommendations on the dietary management of phenylketonuria. Report of Medical Research Council Working Party on Phenylketonuria. Archives of disease in childhood68, 426 (1993). [DOI] [PMC free article] [PubMed]
- 9.Vockley J, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genetics in medicine: official journal of the American College of Medical Genetics. 2014;16:188. doi: 10.1038/gim.2013.157. [DOI] [PubMed] [Google Scholar]
- 10.Waisbren SE, He J, McCarter R. Assessing Psychological Functioning in Metabolic Disorders: Validation of the Adaptive Behavior Assessment System, Second Edition (ABAS-II), and the Behavior Rating Inventory of Executive Function (BRIEF) for Identification of Individuals at Risk. JIMD reports. 2015;21:35–43. doi: 10.1007/8904_2014_373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regnault A, et al. Development and psychometric validation of measures to assess the impact of phenylketonuria and its dietary treatment on patients’ and parents’ quality of life: the phenylketonuria - quality of life (PKU-QOL) questionnaires. Orphanet journal of rare diseases. 2015;10:59. doi: 10.1186/s13023-015-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White DA, Nortz MJ, Mandernach T, Huntington K, Steiner RD. Age-related working memory impairments in children with prefrontal dysfunction associated with phenylketonuria. Journal of the International Neuropsychological Society: JINS. 2002;8:1–11. doi: 10.1017/S1355617702811018. [DOI] [PubMed] [Google Scholar]
- 13.Pietz J, et al. Psychiatric disorders in adult patients with early-treated phenylketonuria. Pediatrics. 1997;99:345–350. doi: 10.1542/peds.99.3.345. [DOI] [PubMed] [Google Scholar]
- 14.Levy HL, et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet. 2007;370:504–510. doi: 10.1016/S0140-6736(07)61234-3. [DOI] [PubMed] [Google Scholar]
- 15.Trefz FK, et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. The Journal of pediatrics. 2009;154:700–707. doi: 10.1016/j.jpeds.2008.11.040. [DOI] [PubMed] [Google Scholar]
- 16.Keil S, et al. Long-term follow-up and outcome of phenylketonuria patients on sapropterin: a retrospective study. Pediatrics. 2013;131:e1881–1888. doi: 10.1542/peds.2012-3291. [DOI] [PubMed] [Google Scholar]
- 17.Burton BK, Bausell H, Katz R, Laduca H, Sullivan C. Sapropterin therapy increases stability of blood phenylalanine levels in patients with BH4-responsive phenylketonuria (PKU) Molecular genetics and metabolism. 2010;101:110–114. doi: 10.1016/j.ymgme.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 18.Humphrey M, Nation J, Francis I, Boneh A. Effect of tetrahydrobiopterin on Phe/Tyr ratios and variation in Phe levels in tetrahydrobiopterin responsive PKU patients. Molecular genetics and metabolism. 2011;104:89–92. doi: 10.1016/j.ymgme.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Muntau AC, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. The New England journal of medicine. 2002;347:2122–2132. doi: 10.1056/NEJMoa021654. [DOI] [PubMed] [Google Scholar]
- 20.Michals-Matalon K, Bhatia G, Guttler F, Tyring SK, Matalon R. Response of phenylketonuria to tetrahydrobiopterin. The Journal of nutrition. 2007;137:1564S–1567S. doi: 10.1093/jn/137.6.1564S. [DOI] [PubMed] [Google Scholar]
- 21.Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Molecular genetics and metabolism. 2011;104(Suppl):S2–9. doi: 10.1016/j.ymgme.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 22.Zurfluh MR, et al. Molecular genetics of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Human mutation. 2008;29:167–175. doi: 10.1002/humu.20637. [DOI] [PubMed] [Google Scholar]
- 23.Heintz C, Cotton RG, Blau N. Tetrahydrobiopterin, its mode of action on phenylalanine hydroxylase, and importance of genotypes for pharmacological therapy of phenylketonuria. Human mutation. 2013;34:927–936. doi: 10.1002/humu.22320. [DOI] [PubMed] [Google Scholar]
- 24.Bernegger C, Blau N. High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: a study of 1,919 patients observed from 1988 to 2002. Molecular genetics and metabolism. 2002;77:304–313. doi: 10.1016/S1096-7192(02)00171-3. [DOI] [PubMed] [Google Scholar]
- 25.Lindner M, et al. Tetrahydrobiopterin sensitivity in German patients with mild phenylalanine hydroxylase deficiency. Human mutation. 2003;21:400. doi: 10.1002/humu.9117. [DOI] [PubMed] [Google Scholar]
- 26.Ho G, et al. The Molecular Bases of Phenylketonuria (PKU) in New South Wales, Australia: Mutation Profile and Correlation with Tetrahydrobiopterin (BH4) Responsiveness. JIMD reports. 2014;14:55–65. doi: 10.1007/8904_2013_284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polak E, et al. Phenylalanine hydroxylase deficiency in the Slovak population: genotype-phenotype correlations and genotype-based predictions of BH4-responsiveness. Gene. 2013;526:347–355. doi: 10.1016/j.gene.2013.05.057. [DOI] [PubMed] [Google Scholar]
- 28.Karacic I, et al. Genotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian patients with phenylalanine hydroxylase (PAH) deficiency. Molecular genetics and metabolism. 2009;97:165–171. doi: 10.1016/j.ymgme.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Sarkissian CN, et al. Chaperone-like therapy with tetrahydrobiopterin in clinical trials for phenylketonuria: is genotype a predictor of response? JIMD reports. 2012;5:59–70. doi: 10.1007/8904_2011_96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeannesson-Thivisol E, et al. Genotype-phenotype associations in French patients with phenylketonuria and importance of genotype for full assessment of tetrahydrobiopterin responsiveness. Orphanet journal of rare diseases. 2015;10:158. doi: 10.1186/s13023-015-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhan JY, Qin YF, Zhao ZY. Neonatal screening for congenital hypothyroidism and phenylketonuria in China. World journal of pediatrics: WJP. 2009;5:136–139. doi: 10.1007/s12519-009-0027-0. [DOI] [PubMed] [Google Scholar]
- 32.Zhu T, et al. Variations in genotype-phenotype correlations in phenylalanine hydroxylase deficiency in Chinese Han population. Gene. 2013;529:80–87. doi: 10.1016/j.gene.2013.07.079. [DOI] [PubMed] [Google Scholar]
- 33.Zhu T, et al. Mutational spectrum of phenylketonuria in the Chinese Han population: a novel insight into the geographic distribution of the common mutations. Pediatric research. 2010;67:280–285. doi: 10.1203/PDR.0b013e3181c9fb85. [DOI] [PubMed] [Google Scholar]
- 34.Guldberg P, et al. A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. American journal of human genetics. 1998;63:71–79. doi: 10.1086/301920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiege B, et al. Extended tetrahydrobiopterin loading test in the diagnosis of cofactor-responsive phenylketonuria: a pilot study. Molecular genetics and metabolism. 2005;86(Suppl 1):S91–95. doi: 10.1016/j.ymgme.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Leuzzi V, et al. The spectrum of phenylalanine variations under tetrahydrobiopterin load in subjects affected by phenylalanine hydroxylase deficiency. Journal of inherited metabolic disease. 2006;29:38–46. doi: 10.1007/s10545-006-0096-3. [DOI] [PubMed] [Google Scholar]
- 37.Burton BK, et al. Tetrahydrobiopterin therapy for phenylketonuria in infants and young children. The Journal of pediatrics. 2011;158:410–415. doi: 10.1016/j.jpeds.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 38.Grange DK, et al. Sapropterin dihydrochloride use in pregnant women with phenylketonuria: an interim report of the PKU MOMS sub-registry. Molecular genetics and metabolism. 2014;112:9–16. doi: 10.1016/j.ymgme.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 39.Moseley KD, Ottina MJ, Azen CG, Yano S. Pilot study to evaluate the effects of tetrahydrobiopterin on adult individuals with phenylketonuria with measurable maladaptive behaviors. CNS spectrums. 2015;20:157–163. doi: 10.1017/S1092852914000455. [DOI] [PubMed] [Google Scholar]
- 40.Koch R, Guttler F, Blau N. Mental illness in mild PKU responds to biopterin. Molecular genetics and metabolism. 2002;75:284–286. doi: 10.1006/mgme.2001.3289. [DOI] [PubMed] [Google Scholar]
- 41.Grosse SD. Late-treated phenylketonuria and partial reversibility of intellectual impairment. Child development. 2010;81:200–211. doi: 10.1111/j.1467-8624.2009.01389.x. [DOI] [PubMed] [Google Scholar]
- 42.Cleary M, et al. Fluctuations in phenylalanine concentrations in phenylketonuria: a review of possible relationships with outcomes. Molecular genetics and metabolism. 2013;110:418–423. doi: 10.1016/j.ymgme.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Quirk ME, Dobrowolski SF, Nelson BE, Coffee B, Singh RH. Utility of phenylalanine hydroxylase genotype for tetrahydrobiopterin responsiveness classification in patients with phenylketonuria. Molecular genetics and metabolism. 2012;107:31–36. doi: 10.1016/j.ymgme.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye J, et al. Clinical and gene detection of tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Chinese J of Practical Pediatrics. 2005;20:3. [Google Scholar]
- 45.John SW, Weitzner G, Rozen R, Scriver CR. A rapid procedure for extracting genomic DNA from leukocytes. Nucleic acids research. 1991;19:408. doi: 10.1093/nar/19.2.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nollau P, Wagener C. Methods for detection of point mutations: performance and quality assessment. IFCC Scientific Division, Committee on Molecular Biology Techniques. Clinical chemistry. 1997;43:1114–1128. [PubMed] [Google Scholar]
- 47.Bik-Multanowski M, et al. Molecular genetics of PKU in Poland and potential impact of mutations on BH4 responsiveness. Acta biochimica Polonica. 2013;60:613–616. [PubMed] [Google Scholar]
- 48.Akobeng AK. Understanding diagnostic tests 3: Receiver operating characteristic curves. Acta Paediatr. 2007;96:644–647. doi: 10.1111/j.1651-2227.2006.00178.x. [DOI] [PubMed] [Google Scholar]
- 49.Chmura Kraemer H, Periyakoil VS, Noda A. Kappa coefficients in medical research. Statistics in medicine. 2002;21:2109–2129. doi: 10.1002/sim.1180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.