

Abstract
Increasing matrix stiffness caused by the extracellular matrix (ECM) deposition surrounding cancer cells is accompanied by epithelial–mesenchymal transition (EMT). Here, we show that expression levels of EMT marker genes along with discoidin domain receptor 2 (DDR2) can increase upon matrix stiffening. DDR2 silencing by short hairpin RNA downregulated EMT markers. Promoter analysis and chromatin immunoprecipitation revealed that c-Myb and LEF1 may be responsible for DDR2 induction during cell culture on a stiff matrix. Mechanistically, c-Myb acetylation by p300, which is upregulated on the stiff matrix, seems to be necessary for the c-Myb-and-LEF1–mediated DDR2 expression. Finally, we found that the c-Myb–DDR2 axis is crucial for lung cancer cell line proliferation and expression of EMT marker genes in a stiff environment. Thus, our results suggest that DDR2 regulation by p300 expression and/or c-Myb acetylation upon matrix stiffening may be necessary for regulation of EMT and invasiveness of lung cancer cells.
Introduction
Fibrosis is related to fibroblast activation and mechanical remodelling of the extracellular matrix (ECM), resulting in tissue architecture destruction and increased matrix stiffness1, 2. ECM rigidity increased by fibrosis in tumour tissue has been found to be linked to pathological conditions including cancer cell proliferation and epithelial–mesenchymal transition (EMT)2, 3. In the case of pulmonary fibrosis in the lung tissue, for example, is major causes of lung cancer initiation4. A stiffened ECM in the tumour microenvironment confers elongated morphology onto the cells and promotes motility accompanied by expression of proliferation- and EMT-related factors such as Snail, Twist, and N-cadherin2, 5–7. Conversely, EMT involves promotion of tissue fibrosis, which reciprocally ensures a permissive microenvironment for cancer cell delamination from primary tumours and invasion through the ECM7. During invasion and metastasis, encounters between cancer cells and ECM components such as collagen are inevitable; in this regard, collagen receptors are important for cancer progression8, 9.
Discoidin domain receptors (DDRs), DDR1 and DDR2, are widely expressed collagen receptors8, 10, 11. Similar to other receptor tyrosine kinases, DDRs undergo receptor autophosphorylation after collagen binding, but this process is known to be unusually slow and involves prolonged stimulation11. DDRs consist of an extracellular region containing the discoidin domain, a transmembrane region connected to a cytoplasmic domain including an intracellular juxtamembrane region, and a catalytic domain. We have previously reported that a substantially longer intracellular juxtamembrane region of DDR2 plays a crucial role in DDR2 activation and lung cancer progression12. Because collagen is known to stimulate EMT1, 13, 14, DDR2 function in EMT during cancer progression cannot be ignored. Indeed, DDR2 contributes to metastasis of breast cancer cells that have undergone EMT via maintenance of SNAIL1 stability and activity9. DDR2 expression is also reported to mediate TGF-β–induced EMT in renal epithelial cells and lung cancers14. Nevertheless, the mechanisms controlling DDR2 expression during EMT are not clear.
In this study, we show that DDR2 expression accompanied with expression of EMT marker genes can increase during cell culture on a stiff matrix. Mechanistically, histone acetyltransferase (HAT) p300 upregulation on a stiff ECM increases the transcription factor c-Myb acetylation; this process appears to induce c-Myb binding along with LEF1 to the DDR2 promoter, resulting in DDR2 upregulation in a stiff environment. Finally, we show that c-Myb silencing may lead to DDR2 inhibition and invasion by lung cancer cells; these effects are reversed by ectopic expression of DDR2. Therefore, our results suggest that DDR2 upregulated by matrix stiffening can play a critical role in EMT marker expression and invasiveness.
Results
ECM stiffness affects cellular behaviour and gene expression levels
ECM stiffness is reported to affect cellular functions such as growth and differentiation in various cell types15–17. Recent evidence suggests that rigidity of the ECM surrounding cancer cells originating from lung or breast tissues mainly affects the expression of genes controlling proliferation and metastasis18; therefore, we studied the molecular mechanisms underlying the control of expression of these genes by ECM rigidity using non-small lung adenocarcinoma cell line, H1299. First, we cultured the cells on a collagen-coated soft (~0.5 kPa) or stiff (~40 kPa) polyacrylamide gel (PAG), which has the elasticity of normal or cancerous lung tissues19, respectively. Cells were found to be more spread and dispersed on the stiff matrix, indicating that these cells can sense and respond to the PAG stiffness (Fig. 1a). It has been reported that focal adhesion (FA) signalling is essential for cellular spreading on a substrate in response to sensing the matrix stiffness20; thus, we tested whether cell spreading on a stiff matrix is mediated by the canonical FA signalling (Fig. 1b,c). As previously reported for other cancer cell types5, 21, H1299 cells on the stiff matrix showed a fully developed vinculin-positive FA complex. Moreover, FA-related factors such as focal adhesion kinase (FAK) and extracellular signal-regulated kinase (ERK) were activated after matrix stiffening along with myosin light chain (MLC) phosphorylation. We showed that cell proliferation may be controlled by matrix stiffness (Fig. 1d), confirming that cellular growth is dependent on matrix rigidity as described in other reports18, 22.
Figure 1.

Stiffness of the ECM affects cellular behaviour and gene expression levels. (a) Phase contrast images of H1299 cells incubated on PAGs with the indicated stiffness or on glass. Scale bars, 50 μm. (b) Immunostaining of F-actin (red) and vinculin (green) in H1299 cells incubated under the indicated conditions (left); quantification of vinculin (n = 50) area for each condition (right). (c) Western blot analysis of pFAK, FAK, pERK, ERK, pMLC, and MLC in H1299 cells incubated under the indicated conditions. (d) Quantification of proliferation of H1299 cells incubated on a soft or stiff PAG with the indicated stiffness or on glass for the indicated periods. (e) Microarray gene expression analysis of H1299 cells incubated under the indicated conditions for 24 h. Heat map and gene ontology (GO) analysis of gene expression profiles of 557 upregulated genes with a ≥1.4× change in expression. (f) Heat map representation of microarray data showing the expression levels of several key EMT-related genes. (g) Quantitative RT-PCR (qRT-PCR) analysis of expression of EMT-related genes in H1299 cells incubated on a PAG with 0.5 or 40 kPa stiffness. qRT-PCR values were normalised to GAPDH. Data in (a), (b), (c), (d) and (g) represent the mean of three independent experiments ± SEM. * P < 0.05, ** P < 0.01.
To explore global gene expression patterns changed by matrix stiffness, we performed microarray analysis of samples obtained from H1299 cells cultured on a soft or stiff PAG. The increase in matrix stiffness upregulated various genes including proliferation-related genes as determined by gene ontology (GO) analysis (Fig. 1e); in particular, genes that are involved in EMT were enriched (associated with the GO term ‘cell proliferation’). In addition to increased expression of DDR2, we observed upregulation of TWIST1, TWIST2, and ACTA2 (gene of α smooth muscle actin) on the stiff matrix, whereas ZEB1, ZEB2, and CDH1 (gene of E-cadherin) among other EMT-related genes were slightly downregulated (Fig. 1f). Quantitative RT-PCR (qRT-PCR) results confirmed the microarray data, namely, that DDR2 and TWIST1 expression levels significantly and slightly increased, respectively, whereas CDH1 expression significantly decreased (Fig. 1g).
DDR2 is a key player in matrix stiffness–induced EMT
To ascertain whether DDR2 expression is regulated by matrix stiffness, we analysed DDR2 expression in H1299 cells incubated on PAGs of various stiffness or on glass. The results showed that DDR2 is specifically upregulated after matrix stiffening while expression of DDR1, another DDR isoform is independent on matrix stiffness in transcript and protein levels. (Fig. 2a and Supplementary Fig. 1a,b). Changing the composition of the ECM or serum in the culture medium did not affect the DDR2 expression (Supplementary Fig. 1c,d). These results imply that ECM stiffness is a major regulator of DDR2 expression but not DDR1.
Figure 2.
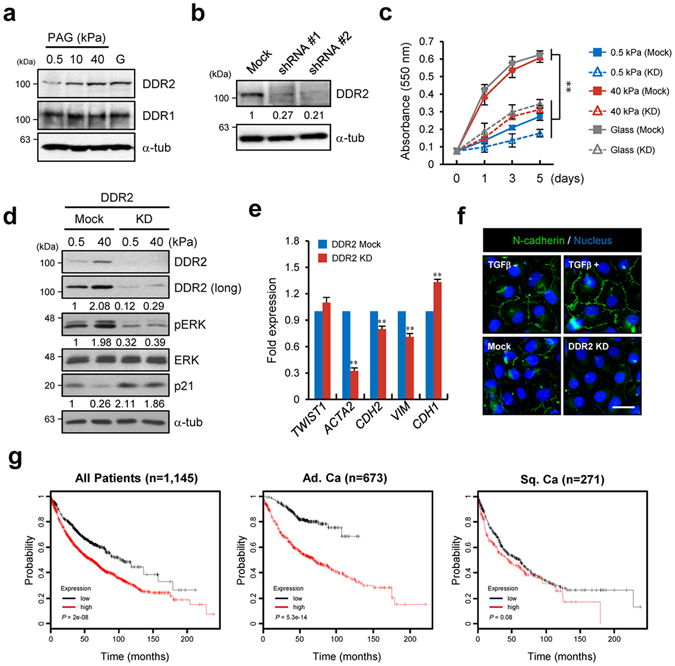
DDR2 is a key player in matrix stiffness–induced EMT. (a) Western blot analysis of DDR1 and DDR2 in H1299 cells incubated on a PAG with the indicated stiffness. (b) Western blot analysis of DDR2 in H1299 cells expressing mock or anti-DDR2 shRNA. (c) Quantification of proliferation of H1299 cells expressing mock or anti-DDR2 shRNA with indicated stiffness and duration. Data represent the mean of three independent experiments ± SEM. ** P < 0.01. (d) Western blot analysis of DDR2, pERK, ERK, and p21 in H1299 cells expressing mock or anti-DDR2 shRNA under the indicated conditions. (e) qRT-PCR analysis of expression of TWIST1, ACTA2, CDH2, VIM, and CDH1 in H1299 cells expressing mock or anti-DDR2 shRNA. Data represent the mean of three independent experiments ± SEM. ** P < 0.01. (f) Immunostaining of N-cadherin and the nucleus in H1299 cells expressing mock or anti-DDR2 shRNA. Mock shRNA–expressing H1299 cells in the presence of 10 ng ml−1 TGF-β served as positive controls. (g) Kaplan–Meier curves of overall survival among patients with lung cancer, including lung adenocarcinoma and squamous cell lung carcinoma.
Because DDR2 has been reported to regulate gene expression through activation of transcription factors9, 23, we determined whether DDR2 regulates EMT-related gene expression. After generation of DDR2 knockdown (KD) cell lines using short hairpin RNAs (shRNAs; Fig. 2b), we first tested the proliferative capacity on soft and stiff matrices because DDR2 is categorised as a proliferation-related gene. As expected, DDR2 KD cells lost the capacity for proliferation on the stiff matrix (Fig. 2c). Examination of pERK and p21 levels in DDR2 KD cells indicated that DDR2 expression on the stiff matrix may modulate signalling and expression of cell cycle–regulatory proteins (Fig. 2d). We found that DDR2 KD cells showed weak expression of EMT-related genes such as ACTA2, CDH2, VIM, and CDH1 (Fig. 2e). Immunocytochemical staining for N-cadherin revealed that N-cadherin expression was significantly decreased in DDR2 KD cells (Fig. 2f). Accordingly, these results suggested that DDR2 expression on a stiff matrix drives cancer cell proliferation and expression of a set of EMT-related genes. Furthermore, we evaluated the prognostic value of DDR2 using Kaplan–Meier Plotter24, an online meta-analysis tool for biomarker assessment (http://www.kmplot.com/analysis). According to this analysis, the expression level of DDR2 moderately correlates with overall survival of patients with lung cancer. Especially, it significantly correlates with a poor prognosis of adenocarcinoma, but DDR2 expression does not considerably correlate with squamous cell carcinoma (Fig. 2g). These results implied that DDR2 may act as an essential regulator in EMT related gene expression in stiff ECM environment.
c-Myb and LEF1 are responsible for DDR2 promoter activation
To understand the molecular mechanisms behind the matrix stiffness–dependent DDR2 expression, we analysed putative cis-acting elements in the DDR2 promoter. Sequence analysis of the DDR2 promoter predicted that 78 transcription factors possibly bind to the putative cis-acting elements in this promoter between positions −1659 and +27 (Supplementary Table 1). Among them, several cis-acting elements that have a high threshold value (>0.9) closely correlate with lung cancer progression (Supplementary Fig. 2). To determine which cis-acting elements are responsible for DDR2 expression, we constructed a DDR2 promoter (WT-Luc) containing the above-mentioned cis-acting elements with a high threshold value. WT-Luc activity measurement showed that the DDR2 promoter activity increased fourfold, just as under the conditions of a stiff matrix or glass (Fig. 3a). Expression of green fluorescent protein (GFP) under the DDR2 promoter control confirmed the results on the luciferase-mediated DDR2 promoter activity. DDR2 promoter-GFP construct expression was strong on a stiff matrix, while CMV-GFP vector expression was independent of matrix stiffness (Fig. 3b). We next sought to determine which cis-acting elements are associated with the DDR2 promoter activation; thus, reporter plasmids containing truncated fragments (Mut-1-Luc to Mut-6-Luc or Luc only) were generated. We showed that this promoter’s activity was reduced in cells transfected with Mut-4-Luc, and mostly downregulated in cells transfected with Mut-5-Luc (Fig. 3c). The Mut-4-Luc and Mut-5-Luc vectors lack 1435 and 1541 bp, respectively, at the 5′ end of the promoter. Therefore, our results suggest that the region between positions −505 and −118 containing the cis-acting element for the transcription factors FOXA1, c-Myb, ELF1, and LEF1 is crucial for DDR2 promoter activation on a stiff matrix (Fig. 3c).
Figure 3.
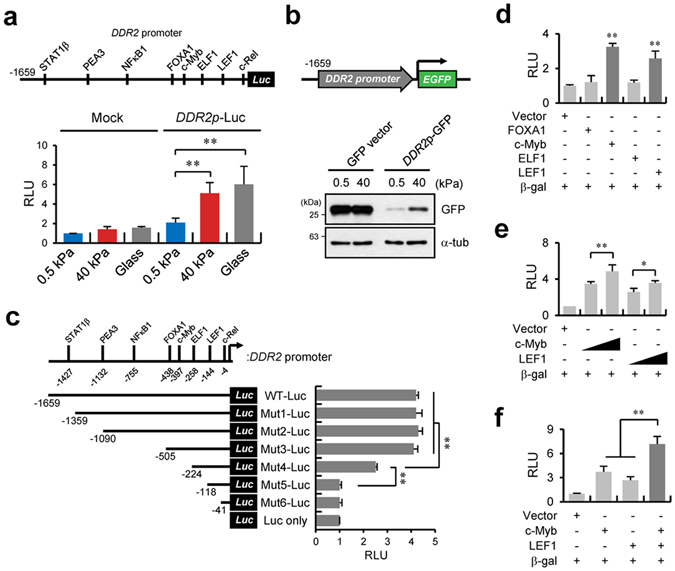
c-Myb and LEF1 are responsible for DDR2 promoter activity. (a) Schematic representation of the DDR2 promoter and possible relevant transcription factors (top). Relative expression of a mock or DDR2 wild-type (WT) promoter-driven luciferase reporter in H1299 cells incubated on a PAG with the indicated stiffness or on glass (bottom). (b) Western blot analysis of green fluorescent protein (GFP) under the control of the DDR2 WT promoter in H1299 cells incubated on a PAG with the indicated stiffness. (c) A schematic of the DDR2 WT promoter and six truncated constructs in reporter vectors (top). Relative expression of DDR2 WT or truncated promoter–driven luciferase reporters in H1299 cells incubated on a PAG with the indicated stiffness (bottom). (d) Relative expression of DDR2 WT promoter–driven luciferase reporters in FOXA1-, c-Myb-, ELF1-, or LEF1-overexpressing H1299 cells (bottom). (e,f) Relative expression of DDR2 WT promoter–driven luciferase reporters in c-Myb- or LEF1-overexpressing H1299 cells in a dose-dependent manner (e) and in co-overexpressing H1299 cells (f). Data in (a), (c), (d), (e) and (f) represent the mean of three independent experiments ± SEM. * P < 0.05,** P < 0.01.
Next, we attempted to determine which transcription factors (among the four) are responsible for DDR2 promoter activation under stiff-matrix conditions. A luciferase assay of the samples obtained from cells cotransfected with the DDR2 WT-Luc construct and the GFP-tagged construct encoding FOXA1, c-Myb, ELF1, or LEF1 indicated that c-Myb and LEF1 coexpression resulted in a four- and threefold increase in DDR2 WT-Luc activity, respectively (Fig. 3d). Dose-dependent overexpression of c-Myb and LEF1 confirmed that both transcription factors are specifically involved in the regulation of DDR2 promoter activity (Fig. 3e). Furthermore, DDR2 promoter activity was increased more than sevenfold during simultaneous expression of both transcription factors, as compared with cells transfected with each transcription factor individually (Fig. 3f). Collectively, these data suggest that c-Myb and LEF1 are responsible for the DDR2 promoter activation on a stiff matrix.
Increased cellular contractility on a stiff matrix is responsible for recruitment of c-Myb and LEF1 to the DDR2 promoter
To confirm that the recruitment of c-Myb and LEF1 to the DDR2 promoter depends on the matrix stiffness in the cell, we performed a chromatin immunoprecipitation (ChIP) assay for c-Myb and LEF1 under the soft- and stiff-matrix conditions. Figure 4a showed that the recruitment of c-Myb and LEF1 to the DDR2 promoter was significantly increased on a stiff matrix but not on soft matrix (Fig. 4a). The recruitment of c-Myb seemed to overlap with LEF1 recruitment and vice versa; this phenomenon is due to the interaction of these transcription factors25.
Figure 4.
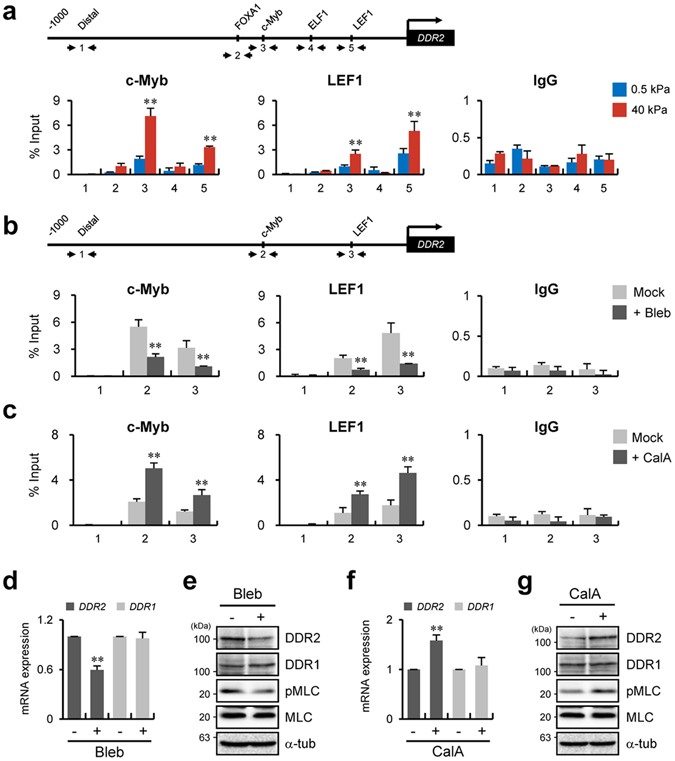
Increased cellular contractility on a stiff matrix is responsible for recruitment of c-Myb and LEF1 to the DDR2 promoter. (a) Schematic depiction of the DDR2 WT promoter with distal and putative binding sites for FOXA1, c-Myb, ELF1, and LEF1. Primer sets for chromatin immunoprecipitation (ChIP) analysis are indicated by the arrows in the scheme (top). ChIP analysis of the binding of c-Myb and LEF1 to the DDR2 WT promoter in H1299 cells under the indicated conditions. qRT-PCR analysis with primers specific to one of five regions in the DDR2 WT promoter as indicated in the scheme (bottom). (b,d,e) ChIP analysis of the binding of c-Myb and LEF1 to the DDR2 WT promoter in H1299 cells in the absence or presence of 10 μM blebbistatin on a PAG with 40-kPa stiffness (b). qRT-PCR analysis of DDR1 and DDR2 (d) and western blot analysis of DDR1, DDR2, MLC, and pMLC (e) under the same conditions as in panel b. (c,f,g) ChIP analysis of the binding of c-Myb and LEF1 to the DDR2 WT promoter in H1299 cells in the absence or presence of 2.5 nM calyculin A on a PAG with 0.5-kPa stiffness (c). qRT-PCR analysis of DDR1 and DDR2 (f) and western blot analysis of DDR1, DDR2, MLC, and pMLC (g) under the same conditions as in panel c. All data represent the mean of three independent experiments ± SEM. ** P < 0.01.
Because it was reported that a stiff substrate increases cellular contractility, whereas a soft substrate decreases the contractility20, it seemed possible that cellular contractility that is induced on a stiff matrix affects recruitment of c-Myb and LEF1 to the DDR2 promoter. To test this hypothesis, we cultured H1299 cells on a stiff matrix with a myosin inhibitor, blebbistatin, to decrease cellular contractility. Under these conditions, recruitment of c-Myb and LEF1 to the DDR2 promoter was significantly reduced (Fig. 4b). Conversely, enhancement of the cellular contractile activity by treatment with calyculin A (inhibitor of protein phosphatase 1) on a soft matrix increased the recruitment of the relevant transcription factors to the DDR2 promoter (Fig. 4c). In support of these results, expression of DDR2 was significantly decreased by blebbistatin treatment on the stiff matrix (Fig. 4d,e), whereas the downregulation of DDR2 on the soft matrix was considerably attenuated after treatment with calyculin A (Fig. 4f,g). Collectively, these findings suggest that DDR2 expression is regulated by recruitment of c-Myb and LEF1 to the promoter in a cooperative manner, and this recruitment is controlled by cellular contractility induced by matrix rigidity.
p300 regulates DDR2 promoter activity via c-Myb acetylation
Post-translational modifications (PTMs), such as methylation and acetylation, of a transcription factor are required for regulation of gene expression26. Particularly, it has been reported that acetylation of c-Myb is necessary for target gene expression27, 28; accordingly, we tested whether increased matrix stiffness induces acetylation of c-Myb and LEF1. Immunoprecipitation analysis of c-Myb and LEF1 from cell lysates prepared under soft- or stiff-matrix conditions indicated that the extent of c-Myb acetylation significantly increased on the stiff matrix, whereas LEF1 was not affected (Fig. 5a). It has been reported that p300 (encoded by EP300) and CBP (encoded by CREBBP) HATs are involved in c-Myb acetylation27, 28. Therefore, we tested which HAT is responsible for c-Myb acetylation after matrix stiffening. Because we found EP300 to be upregulated in the stiff-matrix condition in the microarray results, expression levels of EP300 and CREBBP at different levels of matrix stiffness were examined next. qRT-PCR results showed that EP300 expression increased in the stiff-matrix condition compared with the soft-matrix condition, while the expression of CREBBP was not changed at any stiffness (Fig. 5b). Next, we determined whether elevated expression of p300 on the stiff matrix also depends on the cellular contractility because cellular contractility increased the binding affinity of c-Myb for the DDR2 promoter (Fig. 4b,c). qRT-PCR results revealed that treatment with blebbistatin on the stiff matrix inhibited the EP300 expression, but this expression was markedly increased by treatment with calyculin A on the soft matrix. These findings indicated that increased expression of EP300 in the stiff-matrix condition may be due to an increase in cellular contractility (Fig. 5c). Cotransfection of p300 with c-Myb increased the DDR2 promoter activity significantly (10-fold), whereas coexpression of p300 with LEF1 did not affect the DDR2 promoter activity. These data support the idea that the acetylation of c-Myb by p300 is involved in DDR2 promoter activation (Supplementary Fig. 3).
Figure 5.
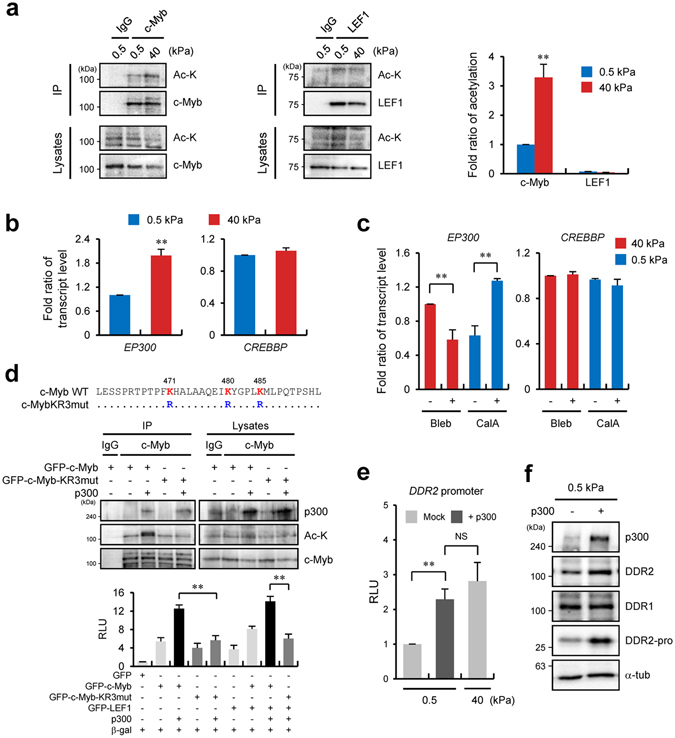
Histone acetyltransferase p300 regulates DDR2 promoter activity via c-Myb acetylation. (a) A coimmunoprecipitation assay of lysates from c-Myb- or LEF1-overexpressing HEK 293 T cells using the indicated antibodies under the indicated conditions (left) and quantification of acetylation of c-Myb and LEF1 (right). Labels above and to the right of blots refer to antibodies used for the immunoprecipitation and immunoblotting, respectively. (b) qRT-PCR analysis of the expression of EP300 and CREBBP in H1299 cells incubated on a 0.5- or 40-kPa PAG. (c) qRT-PCR analysis of expression of EP300 in H1299 cells incubated on a 40-kPa PAG with blebbistatin (10 μM) or a 0.5-kPa PAG with calyculin A (2.5 nM). (d) A partial protein sequence of c-Myb. Conserved lysine residues at positions 471, 480, and 485 are highlighted in red. Arginine residues (blue) denote mutated amino acid residues (top). Coimmunoprecipitation analysis of lysates from c-Myb-, c-MybKR3mutant-, or p300-overexpressing HEK 293T cells using the indicated antibodies. Labels above and to the right of the blots refer to antibodies used for the immunoprecipitation and immunoblotting, respectively (middle). Relative expression of DDR2 WT promoter–driven luciferase reporters in c-Myb-, c-MybKR3mut-, or p300-overexpressing H1299 cells as indicated (bottom). (e) Relative expression of DDR2 WT promoter–driven luciferase reporters in mock or p300-overexpressing H1299 cells incubated on a PAG with 0.5- or 40-kPa stiffness. (f) Western blot analysis of p300, DDR1, and DDR2 in p300-overexpressing H1299 cells incubated on a PAG with 0.5-kPa stiffness. DDR2-pro refers to GFP under the control of the DDR2 WT promoter. Data in (a), (b), (c) and (e) represent the mean of three independent experiments ± SEM. NS, non significant. ** P < 0.01.
Because it was reported that p300 can acetylate histone proteins, leading to chromatin remodelling and enhancement of transcription factor–DNA binding affinity29, we next tested whether acetylation of c-Myb by p300 directly participates in DDR2 expression. Because three lysine residues in c-Myb have been identified as acetylation sites of p30027, we generated a lysine-free c-Myb mutant (c-Myb-KR3mut) by substitution of lysines with arginines, and determined whether the c-Myb-KR3mut can activate the DDR2 promoter. The c-Myb-KR3mut protein neither induced the acetylation by p300 (Fig. 5d, top) nor increased the DDR2 promoter activity during coexpression with p300 (Fig. 5d, bottom), indicating that p300-mediated acetylation of c-Myb is crucial for DDR2 promoter activation. In addition, we found that coexpression of c-Myb-KR3mut with LEF1 did not yield the additive effect (of c-Myb and LEF1) on DDR2 promoter activity as compared with expression of wild-type c-Myb and LEF1 (strong additive effect on the promoter). These results imply that the cooperative effect of LEF1 and c-Myb on DDR2 promoter activity is dependent on c-Myb acetylation status.
The above results indicate that downregulation of DDR2 on the soft matrix is due to an insufficient extent of c-Myb acetylation, which is a consequence of low expression of p300 in the soft-matrix condition. Thus, overexpression of p300 may increase DDR2 expression on the soft matrix. As expected, cells ectopically expressing p300 showed increased DDR2 promoter activity even when these cells were cultured on the soft matrix (Fig. 5e). We next found that overexpression of p300 increased DDR2 expression and GFP synthesis driven by the modified luciferase vector shown in Fig. 3b (Fig. 5f). Collectively, these results suggested that higher expression of p300, c-Myb acetylation, and LEF1 are important for activation of DDR2 expression under stiff-matrix conditions.
c-Myb is associated with lung cancer progression because of activation of DDR2 expression
It has reported the importance of c-Myb on breast cancer progression30, 31, no report has yet been published regarding lung cancer. Since above results were obtained from the lung adenocarcinoma cell lines, we searched involvement of c-Myb in lung cancer progression using The Cancer Genome Atlas (TCGA) and Oncomine database. These analyses revealed that MYB expression (encoding the c-Myb) is increased in several cancers including breast, colon, and lung cancer as previously reported31–33, and MYB is overexpressed in lung cancer as compared with normal tissues (Fig. 6a,b).
Figure 6.

c-Myb is associated with lung cancer progression because of regulation of DDR2 expression. (a) Heat map representation of a TCGA pan-cancer RNA-Seq dataset from the Cancer Browser (Illumina HiSeq) showing the MYB, DDR2, and EP300 expression levels in various cancer tissues. BL, bladder; BR, breast; CX, cervix; CL, colon; EM, endometrium; GL, glioma; HN, head and neck; KN, kidney; LV, liver; MN, melanoma; OV, ovary; PC, pancreas; AG, adrenal gland; PR, prostate; RT, rectum; SC, sarcoma; TT, testis; TY, thyroid. (b) An Oncomine boxed plot showing MYB expression levels in human breast, colon, ovary, prostate, and lung normal and cancer tissues. N, normal; C, cancer. (c) Western blot analysis of DDR2, c-Myb, phospho (p)-ERK, and ERK in H1299 cells expressing mock or anti–c-Myb shRNA. KD: c-Myb knockdown. (d) qRT-PCR analysis of DDR1. DDR2, TWIST1, ACTA2, CDH2, VIM, CDH1, and MYB under the conditions shown in panel c. Data represent the mean of three independent experiments ± SEM. * P < 0.05, ** P < 0.01. (e) Western blot analysis of DDR2, c-Myb, pERK, and ERK in H1299 cells expressing mock or anti–c-Myb shRNA, or analysis of ectopically expressed DDR2 in H1299 cells expressing anti–c-Myb shRNA. (f,g) Quantification of proliferation (f) and invasion (g) shown by H1299 cells under the conditions shown in panel e. Data represent the mean of three independent experiments ± SEM. * P < 0.05,** P < 0.01. (h) qRT-PCR analysis of DDR2, TWIST1, ACTA2, CDH2, VIM, CDH1, and MYB under the conditions shown in panel e. Data represent the mean of three independent of three experiments ± SEM.; * P < 0.05, ** P < 0.01, # P < 0.01 as compared to c-Myb knockdown groups. (i) Immunostaining of N-cadherin and nuclei in H1299 cells under the conditions shown in panel e. The GFP signal indicates the cells ectopically expressing DDR2.
To study the importance of the c-Myb–DDR2 axis for cancer progression including proliferation and invasion, we generated a c-Myb KD H1299 cell line and analysed gene expression and signalling, which were defective in DDR2 KD cells, as shown in Fig. 2. The c-Myb KD caused significant DDR2 downregulation and ERK activation (Fig. 6c). Furthermore, the EMT-related gene expression pattern in c-Myb KD cells was found to be similar to that presented in Fig. 2e (Fig. 6d). To confirm that lower expression of these genes in c-Myb KD cells is due to the DDR2 downregulation, we ectopically expressed DDR2 in c-Myb KD H1299 cells and quantified the proliferation, invasion, and patterns of gene expression. Forced expression of DDR2 induced pERK expression without any effect on c-Myb expression (Fig. 6e). Moreover, the decrease in cellular proliferation and invasion shown by c-Myb KD cells was significantly reversed by DDR2 overexpression (Fig. 6f,g and Supplementary Fig. 4). Furthermore, we investigated cell invasion by c-Myb KD cells under 3D conditions using spheroids34. The invasiveness of H1299 cells in the 3D matrix was significantly decreased by addition of GM6001, a general MMP inhibitor, and DDR2 and c-Myb KD cell lines yielded similar results (Supplementary Fig. 5). These data suggested that lower expression of DDR2 and c-Myb inhibits cell invasion under not only conventional 2D conditions but also 3D conditions.
In addition, we found that downregulated EMT markers such as ACTA, CDH2, and VIM were considerably upregulated, whereas CDH1 was downregulated (Fig. 6h). Finally, immunostaining for N-cadherin indicated that the decrease in N-cadherin expression was reversed in DDR2-overexpressing cells (Fig. 6i). To confirm that these phenomena are not limited to H1299 cells, we generated a c-Myb KD in lung adenocarcinoma cell line A549, which is as metastatic as H1299 cells, but expresses wild-type p53. We found that expression of DDR2 also increased on the stiff matrix but expression of DDR1 was not changed. Also Increased DDR2 expression on 40 kPa PAGs was decreased by the c-Myb KD (Supplementary Fig. 6a,b). In addition, proliferation and invasiveness of A549 cells were significantly decreased by the c-Myb KD (Supplementary Fig. 6c,d). Therefore, the downregulation of c-Myb resulted in a decrease of DDR2 expression, leading to inhibition of cancer progression and downregulation of EMT marker genes.
It is known that integrin is a transmembrane receptor that mediates cellular responses to the rigidity of the ECM35. Because we found that cells are more spread on a stiff substrate, and canonical FA signalling is elevated depending on substrate stiffness, we tried to determine whether integrin is also involved in regulation of DDR2 expression and its induction depending on matrix stiffness. As a result of silencing of integrin β1, H1299 cells did not spread well on the stiff matrix (Supplementary Fig. 7a). Induction of ERK and MLC activity on the stiff matrix was also inhibited (Supplementary Fig. 7b). Besides, transcription levels of DDR2 and EP300 were markedly decreased by the KD of integrin β1 (Supplementary Fig. 7c,d), whereas transcription of the MYB gene was not changed significantly (Supplementary Fig. 7e). Besides regulation of DDR2 by c-Myb acetylation, these results may imply that integrin expression can be associated with the regulation of DDR2 expression by c-Myb and p300 depending on substrate stiffness.
Discussion
The increasing stiffness of cancer stroma is thought to provide a permissive environment for cancer development and EMT. Nonetheless, there are relatively few data on the mechanism behind the regulation of EMT progression by matrix stiffness. In this study, we demonstrated that DDR2 upregulation during cell culture on a stiff matrix may be crucial for expression of a set of EMT marker genes. DDR2 silencing significantly downregulated EMT marker genes, supporting the finding that DDR2 is a potent regulator of EMT marker gene expression in a stiff environment. Furthermore, we showed that p300 upregulation on a stiff matrix led to c-Myb acetylation, promoting c-Myb and LEF1 recruitment to the DDR2 promoter and leading to expression of DDR2 and EMT marker genes in the stiff stroma of a developing tumour (Fig. 7). DDR1 is reportedly involved in the TGF-β–induced EMT progression in lung adenocarcinoma and pancreatic cancer cells8, 14, but our microarray and qRT-PCR analyses showed that DDR1 expression is unchanged by matrix stiffness. Although no evidence of the relation between DDR2 expression and EMT has been reported, we showed that DDR2 – among EMT-related genes – is upregulated the most in the stiff-matrix condition (unlike DDR1). This finding implies that expression of EMT marker genes (associated with ECM stiffness) is related to DDR2. Thus, our results on mechanotranscriptional regulation of DDR2 expression by matrix stiffness seem to point to a new concept regarding how a mechanically permissive environment affects EMT and cancer progression.
Figure 7.

The proposed model of regulation of DDR2 expression by a combination of c-Myb, LEF1, and p300 in lung cancer. The overall model following from the results of this study suggestive of the mechanism behind the regulation of DDR2 expression via p300 and transcription factors c-Myb and LEF1 under the influence of ECM stiffness. On a soft matrix, physical binding between c-Myb or LEF1 and DDR2 promoter is relatively weak. p300 expression is also weak, leading to lowered acetylation of c-Myb. Consequently, DDR2 promoter is not very active under the soft-matrix conditions; therefore, cellular proliferation and invasiveness are low. On the other hand, direct binding of c-Myb, LEF1, and the DDR2 promoter is significantly increased on a stiff matrix. p300 expression is also induced, resulting in increased acetylation of c-Myb, which enhances DDR2 promoter activity. Thus, DDR2 expression is increased under the stiff-matrix conditions, thereby increasing cellular proliferation and invasiveness.
We showed that DDR2 expression in a stiff environment requires c-Myb activity. c-Myb is reportedly required for progression of some cancers including leukaemia and breast and colon cancers31–33, 36. c-Myb upregulation in these cancers highly correlates with cancer development and metastasis37. Specifically, the role of c-Myb in the proliferation and differentiation of breast and colorectal cancer cells is studied intensively30, 38, 39. In contrast, studies on c-Myb’s physiological importance and function in lung cancer have been relatively infrequent although the expression of c-Myb in lung cancer has been reported30, 37. We confirmed c-Myb expression in lung tumour tissue by exploring TCGA and the Oncomine database (Fig. 6a,b). Furthermore, a c-Myb KD dramatically inhibited proliferation and invasiveness of H1299 and A549 lung cancer cell lines, indicating that c-Myb is likely essential for lung cancer progression.
DNA-binding affinity and transactivation capacity of c-Myb are reportedly regulated by PTMs such as phosphorylation and acetylation27, 28, 40, 41. Acetylation of lysine residues in the negative regulatory domain (NRD) of c-Myb by p300 or CBP increases the DNA-binding affinity and transactivation capacity of c-Myb27, 28. Meanwhile, phosphorylation of serine residues 11 and 12 by casein kinase II decreases DNA-binding affinity42, whereas phosphorylation of serine residue 528 in the NRD lowers transactivation capacity of c-Myb43. Because our results showed that p300 expression but not CBP expression in H1299 cells is elevated on a stiff matrix, p300 may participate in the regulation of c-Myb activity via acetylation in the stiff-matrix condition. Our results revealed that c-Myb transactivation capacity is increased by coexpression with p300, whereas lysine mutants of c-Myb show decreased transactivation capacity induced by p300. These data indicate that c-Myb acetylation by p300 may be the mechanism underlying the increase in DDR2 expression in a mechanically stiff environment.
Aside from regulation by acetylation, c-Myb is reported to cooperate with other transcription factors to activate target promoters. For example, the recombination-activating gene (RAG) promoter in a B-cell line contains a binding site for LEF1, c-Myb, and Pax5; these three transcription factors bind to the promoter region cooperatively to activate the RAG promoter44. In a study on leukaemia cell survival, c-Myb was found to bind (together with LEF1) to promoter regions of anti-apoptotic genes Bcl2 and survivin45. Our results also indicated that c-Myb acts cooperatively with LEF1 to activate the DDR2 promoter. Deletion of the c-Myb–binding site partially decreased the DDR2 promoter activity, whereas simultaneous elimination of the LEF1-binding site completely suppressed the promoter activity. Additionally, promoter and ChIP analyses revealed that c-Myb and LEF1 are involved in the DDR2 promoter activation via their synergistic DNA binding (Figs 3f and 4a). In contrast, coexpression of a c-Myb lysine mutant with LEF1 did not increase DDR2 promoter activity (Fig. 5d), implying that the c-Myb acetylation by p300 prior to LEF1 binding is a prerequisite for DDR2 promoter activation.
A growing body of evidence indicates that the ECM rigidity influences gene expression via the regulation of cellular localisation of transcriptional coactivators. Expression of α smooth muscle actin (α-SMA) is regulated by nuclear translocation of myocardin-related transcription factor (MRTF), thereby leading to activation of its partner transcription factor, serum response factor (SRF)46, 47. Treatment with a myosin inhibitor, blebbistatin, or a Rho kinase inhibitor, Y27632, blocks nuclear translocation of MRTF; this change decreases SRF-dependent gene expression46, 47. Similarly, YAP and/or TAZ as coactivators for transcription factor TEAD also inhibit its nuclear localisation after a decrease in the cell contractility by treatment with blebbistatin or Y2763248–50. In addition to the coactivator, a transcription factor itself was also reported to relocate into the nucleus after matrix stiffness increases51–54. Thus, nuclear relocation of a transcription factor and/or its coactivator seems to be a common phenomenon for gene expression induced by matrix stiffness. For c-Myb and LEF1, however, we did not observe nuclear relocation under the influence of matrix stiffness (Supplementary Fig. 8). Instead, the prominent change affecting these transcription factors on a stiff matrix is the acetylation of c-Myb, and we showed that the acetyl-free c-Myb mutant failed to increase DDR2 promoter activity. Thus, these results suggest that the PTM of a transcription factor (in addition to regulation of nuclear localisation) can be important for the control of gene expression by matrix rigidity. Meanwhile, decreased actomyosin contractility reportedly leads to nuclear localisation of HDAC3, which catalyses deacetylation of histone H3 acetylated at lysine 9, thereby increasing chromatin compaction55. By contrast, HDAC3 cytoplasmic localisation causes chromatin loosening, thus triggering enhanced promoter activity of various genes, suggesting that epigenetic regulation at the genome level may be responsible for alteration of gene expression in response to matrix rigidity. Thus, identification of the molecular mechanism underlying PTMs of histone proteins and changes in chromatin structure may be worthwhile for further research into the effects of matrix mechanics on gene expression.
Methods
Cell culture and transfection
Non–small cell lung cancer H1299 and A549 cells were purchased from the Korean Cell Line Bank (Seoul, Korea). Human embryonic kidney 293T (HEK 293T) cells and lung normal epithelial HBE4 cells were acquired from the American Type Culture Collection (Manassas, VA, USA). HBE4 cells were cultured in the keratinocyte serum-free (KSF) medium (Gibco), H1299 and A549 cells were cultivated in RPMI 1640 (Invitrogen), and HEK 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% of foetal bovine serum (Hyclone). All cell cultures were maintained at 37 °C and 5% of CO2. Transient transfection procedures were performed using the TransFectin™ Lipid Reagent (Bio-Rad Laboratories Inc.) or a Neon® electroporation device (Invitrogen).
Plasmid construction
The pCMX-p300 plasmid was kindly provided by Professor Sangbeom Seo (Chung-Ang University, Korea). The construct of hemagglutinin-tagged LEF1 provided by Professor Kwonseop Kim (Chonnam National University, Korea)56 and plasmids encoding MYB, FOXA1, and ELF1 were prepared by a polymerase chain reaction (PCR) as reported previously12. The following primers were used: MYB forward 5′-CTAGCTAGCATGGCCCGAAGACCCCGG-3′, reverse 5′-CGGGGTACCATCATGACCAGCGTCCGGGC-3′; LEF1 forward 5′-CTAGCTAGCATGCCCCAACTCTCCGGA-3′, reverse 5′-CGGGGTACCAGGATGTAGGCAGCTGTCAT-3′;FOXA1 forward 5′-CTAGCTAGCATGTTAGGAACTGTGAAG-3′, reverse5′-CGGGGTACCGAGGAAGTGTTTAGGACGGG-3′; and ELF1 forward 5′-CTAGCTAGCATGGCTGCTGTTGTCCAA-3′, reverse 5′-CGGGGTACCCAAAAAGAGTTGGGTTCCAG-3′. The DNA fragment was digested with NheI and KpnI and introduced into the pEGFP-N1 expression vector (Clontech). All the clones were verified by DNA sequencing.
The lentiviral shRNA constructs against puromycin, human MYB (TRCN0000040060, with the hairpin sequence 5′-CCGGGCTCCTAATGTCAACCGAGAACTCGAGTTCTCGGTTGACATTAGGAGCTTTTTG-3′ targeting the coding sequence of MYB) and DDR2 #1 (TRCN0000121117, with the hairpin sequence 5′-CCGGCCCATGCCTATGCCACTCCATCTCGAGATGGAGTGGCATAGGCATGGGTTTTTG-3′ targeting the 3′ untranslated region of DDR2 mRNA) and DDR2 #2 (TRCN0000121121, with the hairpin sequence 5′-CCGGCCCTGGAGGTTCCATCATTTACTCGAGTAAATGATGGAACCTCCAGGGTTTTTG-3′ targeting the coding sequence of DDR2) were obtained from The RNAi Consortium (TRC) lentiviral shRNA library (Open Biosystems) and were cloned into the pLKO.1 vector (Addgene plasmid # 10878). Small interfering RNAs (siRNAs) targeting the coding sequence of integrin β1 (#1: 5′-CCCTCCAGATGACATAGAAA-3′) and the untranslated region of integrin β1 (#2: 5′-TAGGTAGCTTTAGGGCAATAT-3′) were synthesized by Genolution (Seoul, Korea).
The DDR2 promoter region (positions −1,659 to +27 relative to the transcription initiation site) was amplified from H1299 cell genomic DNA using the following primers: forward, 5′-CTAGCTAGCTCTAGTTGTCTCTGGCAA-3′; reverse, 5′-CCGCTCGAGGAGAGAAACCTCACATTC-3′. The amplicon was digested with NheI and XhoI and ligated into the pGL4.12 vector (Promega). Site-directed mutagenesis of the three acetylation sites of MYB, replacing AAACA or TTAAA of the AAACATGCACTTGCAGCTCAAGAAATTAAA site with CGACA or TTCGA, and AAG of the TACGGTCCCCTGAAG site with CGG, was carried out using the PfuUltra High-Fidelity DNA Polymerase (Agilent).
Generation of stable KD cell lines
These cell lines were generated using lentiviral plasmid vectors. Briefly, shRNA constructs were introduced by infection with lentiviruses. Concentrated viral supernatants were applied to target cells with 8 μg ml−1 polybrene. To create a stable cell line from H1299 cells, selection was performed in 1 μg ml−1 puromycin.
Immunofluorescence microscopy
Cell preparations were fixed for 15 min with 3.7% (w/v) paraformaldehyde in phosphate-buffered saline (PBS), then blocked with 2% (w/v) bovine serum albumin (BSA) in PBS for 1 h. The samples were permeabilised for 10 min with 0.5% (v/v) Triton X-100 in PBS. The cells were then incubated with a primary antibody for 1 h, followed by incubation with the appropriate secondary antibody for 1 h at room temperature. The samples were analysed using an Eclipse 80i fluorescence microscope (Nikon) or an LSM 700 confocal microscope (Carl Zeiss). Images were acquired by means of a digital camera (DS-Qi1Mc, Nikon) and NIS-Elements image analysis software (Nikon). The images were processed and pseudo-coloured using Photoshop 11.0 (Adobe Inc.).
PAG matrix preparation
The preparation of PAGs of various rigidity was performed as described elsewhere17, 57. We prepared PAGs ranging from 3% acrylamide/0.03% bis-acrylamide to 8% acrylamide/0.48% bis-acrylamide. PAGs were allowed to bind to 12- or 25-mm glass coverslips that had been pre-treated with 3-aminopropyltriethoxy-silane (Sigma-Aldrich). PAGs were then treated with 0.5 mg ml−1 sulfosuccinimidyl-6-[4′-azido-2′-nitrophenylamino]hexanoate (Thermo Fisher Scientific) and activated with ultraviolet light at 365 nm (2 × 10 min). The activated PAGs were coated with either 50 μg ml−1 neutralised collagen or a gelatine solution overnight at 4 °C. The gels were then thoroughly washed with PBS, equilibrated in DMEM or RPMI 1640, and used for luciferase assays, immunofluorescence microscopy, and RT-PCR assays.
RNA isolation and RT-PCR
Total RNA was extracted from H1299 cells using the RNAiso Plus Reagent (Takara). RNA samples were spectrophotometrically quantified at 260 nm, and only samples that had an OD260/OD280 ratio between 1.9 and 2.0 were used. First-strand cDNA was prepared from 1 μg of RNA using oligo(dT) and M-MLV reverse transcriptase (Thermo Fisher Scientific). Amplification was conducted using Pfu DNA polymerase (Takara). RT-PCR and qRT-PCR were conducted using the Ex-Taq DNA Polymerase (Takara) and SYBR Premix Ex-Taq II (Tli RNase H Plus, Takara), respectively. The thermal cycling protocol consisted of an initial denaturation step at 94 °C for 5 min, followed by 30 amplification cycles (94 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s) for RT-PCR or an initial denaturation step at 94 °C for 5 min, followed by 50 amplification cycles (94 °C 10 s, 57 °C 10 s, and 72 °C 30 s) for qRT-PCR. All the qRT-PCR experiments were conducted independently three times in triplicate.
Microarray analysis
This analysis was performed using H1299 cancer cells incubated on a soft (0.5 kPa) or stiff (40 kPa) PAG for 24 h by means of the Illumina HumanHT-12 v4 Expression BeadChip (Illumina), which includes a pool of unique bead types that correspond to 47,228 transcripts. Total RNA (0.55 μg) isolated from H1299 cells was reverse-transcribed and amplified, according to the protocols described in the Illumina Total Prep RNA Amplification Kit (Ambion). In vitro transcription was then carried out to generate cRNA (0.75 μg), which was hybridised onto each array and labelled with SA-Cy3 (FluoroLinkTM CyTM3). The array was then scanned using the Illumina Bead Array Reader Confocal Scanner. Array data export processing and analysis were carried out in the Illumina GenomeStudio v2009.2 software (Gene Expression Module v1.5.4). For qRT-PCR, 1 μg of total RNA was used to synthesise cDNA. cDNA synthesis was primed using an oligo(dT) primer (Promega), and the quantified cDNA was used for analysis of a set of EMT markers: mRNAs of DDR2, TWIST1, ACTA2, CDH2, VIM, CDH1, MYB, and GAPDH. The following primers were used: DDR2 forward 5′-CCACTATGCAGAGGCTGACA-3′, reverse 5′-CAGAGATGAACCTCCCCAAA-3′; TWIST1 forward 5′-GAGTCCGCAGTCTTACGAG-3′, reverse 5′-GAGGACCTGGTAGAGGAAGT-3′; ACTA2 forward 5′-TCCCTTGAGAAGAGTTACGA-3′, reverse 5′-CCCCTGATAGGACATTGTTA-3′; CDH2 forward 5′-ATGAAGAAGGTGGAGGAGAA-3′, reverse 5′-TTAATGAAGTCCCCAATGTC-3′; VIM forward 5′-ACATTGAGATTGCCACCTAC-3′, reverse 5′-GTTTCGTTGATAACCTGTCC-3′; CDH1 forward 5′-ATTCTGATTCTGCTGCTCTT-3′, reverse 5′-AACGTCGTTACGAGTCACTT-3′; MYB forward 5′-GCACCAGCATCAGAAGATGA-3′, reverse 5′-CATGACCAGCGTCCGGGC-3′; and GAPDH forward 5′-GAGTCAACGGATTTGGTCGT-3′, reverse 5′-TGTGGTCATGAGTCCTTCCA-3′.
The amplification reaction was conducted under the following conditions: 50 cycles of denaturation at 94 °C for 10 s, annealing at 57 °C for 10 s, and extension at 72 °C for 30 s. Dissociation curves were generated after each PCR run to ensure that a single product of the appropriate length was amplified. The mean threshold cycle (C T) and standard error were calculated from individual C T values obtained from three replicates per stage. The normalised mean C T was estimated as ΔC T by subtracting the mean C T of GAPDH from each value. ΔΔC T was calculated as the difference between the control ΔC T and the values obtained for each sample. The fold change in gene expression, relative to the no-treatment control, was calculated as 2−ΔΔCT.
Coimmunoprecipitation and western blotting
H1299 cells were transfected with one of the plasmids for 48 h. The resulting cell lysates were incubated with antibodies against c-Myb or LEF1, and next incubated with Protein A-agarose beads. Solubilised bead-bound materials were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked, washed, and incubated with primary and secondary antibodies as indicated. Signals were developed using the enhanced chemiluminescence reagent (GE Healthcare), and band density was measured using a Quantity One® system (Bio-Rad).
Luciferase assays
HEK 293T or H1299 cells were transfected with 0.5 μg of pGL3 carrying the firefly luciferase reporter gene (Promega) and 0.5 μg of pCMV-β-galactosidase (Clontech), along with our expression vector by means of the TransFectin™ Lipid Reagent. After 48 h, the transfected cells were lysed with reporter lysis buffer (Promega), and the lysates were analysed using a GloMax® Luminometer (Promega). Luciferase activity was normalised to β-galactosidase activity.
The ChIP assay
H1299 cells were incubated on a soft or rigid matrix for 24 h. The cells were cross-linked with 1% formaldehyde, which was added to the medium with incubation for 10 min at room temperature, followed by incubation with 125 mM glycine for 5 min at room temperature. The cells were then lysed in SDS lysis buffer, after which the samples were sonicated and subjected to immunoprecipitation using various antibodies. The immunoprecipitates were eluted and cross-links were reversed; after that, the DNA fragments were purified using the PCR Purification Kit (Axygen). The precipitated DNA was analysed by qRT-PCR using DDR2 promoter-specific primers. The following ChIP primers were used: MYB forward 5′-TGGTGGGAAGGGATTAAA-3′, reverse 5′-CTACCCCACACCTCATTTT-3′; LEF1 forward 5′-GCGGTGTCTTTAAGTCTGTC-3′, reverse 5′-TATTGCACATTTGGTTTCAA-3′; FOXA1 forward 5′-ATCCAGGACAGCAACAGAC-3′, reverse 5′- GCGGAGCTGACCTTTAG-3′; ELF1 forward 5′-AAAAATGAGGTGTGGGGTAG-3′, reverse 5′-TACAGCAAAACCCATTCATA-3′; and distal region forward 5′-CAAAGGTAAGCTCGGTCTTC-3′, reverse 5′-CCAGGCTCCAGCCTTAAC-3′.
3D spheroid invasion assay
This assay was carried out as previously described with minor modifications34. Briefly, 2 × 104 cells were seeded in suspension in agarose-coated plates. Then, the cells were coated with a type I collagen solution for 24 h. Next, a 1 mg ml−1 collagen mixture was poured into the wells and was incubated for polymerisation for 1 h. Spheroids were allowed to invade for 4 days, followed by fixation. Invasion was determined by measuring the circular area of the spheroid normalised to the original area of the spheroid. Fixed spheroids were stained for a proliferation marker with an anti-Ki-67 antibody (Cell signalling) and Alexa Fluor® 488 conjugated phalloidin (Invitrogen).
Quantification of cell proliferation with crystal violet
Cells (2 × 104) were seeded in a 35-mm dish and incubated for various periods. At the end of incubation, the cells were washed three times with PBS and fixed with 3.7% paraformaldehyde for 15 min, and then were stained with 0.1% crystal violet for 30 min, after which the cells were washed and allowed to air dry. After that, 2% SDS was added to each sample and incubated for 20 min with gentle shaking. The extracted dye was quantified at 550 nm using an ELISA (Epoch, BioTeK).
Statistical analysis
Differences between controls and treatment groups were evaluated by Student’s t test (between two groups) or by one-way analysis of variance (ANOVA; for three or more groups) in the GraphPad PRISM software. Data were expressed as mean ± standard error of the mean (SEM) of three independent experiments. Differences with P values less than 0.05 were considered statistically significant.
Electronic supplementary material
Acknowledgements
The hemagglutinin-tagged LEF1 plasmid was a kind gift from Professor Kwonseop Kim (Chonnam National University, Korea). This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2014R1A2A1A11050606 and NRF-2016R1A4A1008035).
Author Contributions
S.R. and D.K. designed the project. S.R., D.K., and E.Y. wrote the manuscript. D.K. and E.Y. conducted the experiments. S.R., D.K., and E.Y. analysed the results. J.J. and P.K. contributed to the experimental work. J.-W.K. provided advice on experimental design, and all the co-authors discussed the results and provided comments and feedback.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Daehwan Kim and Eunae You contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-07126-7
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine & growth factor reviews. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Wei SC, Yang J. Forcing through Tumor Metastasis: The Interplay between Tissue Rigidity and Epithelial-Mesenchymal Transition. Trends in cell biology. 2016;26:111–120. doi: 10.1016/j.tcb.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nature medicine. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar P, et al. Pulmonary fibrosis and lung cancer: risk and benefit analysis of pulmonary resection. J Thorac Cardiovasc Surg. 2003;125:1321–1327. doi: 10.1016/S0022-5223(03)00028-X. [DOI] [PubMed] [Google Scholar]
- 5.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Current opinion in cell biology. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer metastasis reviews. 2012;31:295–321. doi: 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang K, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15:677–687. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leitinger B. Transmembrane collagen receptors. Annu Rev Cell Dev Biol. 2011;27:265–290. doi: 10.1146/annurev-cellbio-092910-154013. [DOI] [PubMed] [Google Scholar]
- 11.Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Molecular cell. 1997;1:13–23. doi: 10.1016/S1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- 12.Kim D, Ko P, You E, Rhee S. The intracellular juxtamembrane domain of discoidin domain receptor 2 (DDR2) is essential for receptor activation and DDR2-mediated cancer progression. Int J Cancer. 2014;135:2547–2557. doi: 10.1002/ijc.28901. [DOI] [PubMed] [Google Scholar]
- 13.Menke A, et al. Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer research. 2001;61:3508–3517. [PubMed] [Google Scholar]
- 14.Walsh LA, Nawshad A, Medici D. Discoidin domain receptor 2 is a critical regulator of epithelial-mesenchymal transition. Matrix Biol. 2011;30:243–247. doi: 10.1016/j.matbio.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 16.Engler AJ, Sweeney HL, Discher DE, Schwarzbauer JE. Extracellular matrix elasticity directs stem cell differentiation. Journal of musculoskeletal & neuronal interactions. 2007;7:335. [PubMed] [Google Scholar]
- 17.Rhee S, Jiang H, Ho CH, Grinnell F. Microtubule function in fibroblast spreading is modulated according to the tension state of cell-matrix interactions. Proc Natl Acad Sci USA. 2007;104:5425–5430. doi: 10.1073/pnas.0608030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tilghman RW, et al. Matrix rigidity regulates cancer cell growth and cellular phenotype. PloS one. 2010;5:e12905. doi: 10.1371/journal.pone.0012905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang X, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. American journal of respiratory cell and molecular biology. 2012;47:340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science (New York. N.Y. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 21.Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO reports. 2014;15:1243–1253. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schrader J, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology (Baltimore. Md) 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, et al. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res. 2011;26:604–617. doi: 10.1002/jbmr.225. [DOI] [PubMed] [Google Scholar]
- 24.Gyorffy B, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 25.Hammerman PS, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer discovery. 2011;1:78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Molecular cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomita A, et al. c-Myb acetylation at the carboxyl-terminal conserved domain by transcriptional co-activator p300. Oncogene. 2000;19:444–451. doi: 10.1038/sj.onc.1203329. [DOI] [PubMed] [Google Scholar]
- 28.Sano Y, Ishii S. Increased affinity of c-Myb for CREB-binding protein (CBP) after CBP-induced acetylation. The Journal of biological chemistry. 2001;276:3674–3682. doi: 10.1074/jbc.M006896200. [DOI] [PubMed] [Google Scholar]
- 29.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/S0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 30.Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer. 2008;8:523–534. doi: 10.1038/nrc2439. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, et al. c-Myb Enhances Breast Cancer Invasion and Metastasis through the Wnt/beta-Catenin/Axin2 Pathway. Cancer research. 2016;76:3364–3375. doi: 10.1158/0008-5472.CAN-15-2302. [DOI] [PubMed] [Google Scholar]
- 32.Drabsch Y, et al. Mechanism of and requirement for estrogen-regulated MYB expression in estrogen-receptor-positive breast cancer cells. Proc Natl Acad Sci USA. 2007;104:13762–13767. doi: 10.1073/pnas.0700104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malaterre J, et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci USA. 2007;104:3829–3834. doi: 10.1073/pnas.0610055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim D, et al. mDia1 regulates breast cancer invasion by controlling membrane type 1-matrix metalloproteinase localization. Oncotarget. 2016;7:17829–17843. doi: 10.18632/oncotarget.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross TD, et al. Integrins in mechanotransduction. Current opinion in cell biology. 2013;25:613–618. doi: 10.1016/j.ceb.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sandberg ML, et al. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Developmental cell. 2005;8:153–166. doi: 10.1016/j.devcel.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 37.Ramsay RG, Barton AL, Gonda TJ. Targeting c-Myb expression in human disease. Expert opinion on therapeutic targets. 2003;7:235–248. doi: 10.1517/14728222.7.2.235. [DOI] [PubMed] [Google Scholar]
- 38.Drabsch Y, Robert RG, Gonda TJ. MYB suppresses differentiation and apoptosis of human breast cancer cells. Breast Cancer Res. 2010;12:R55. doi: 10.1186/bcr2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramsay RG, et al. Colon epithelial cell differentiation is inhibited by constitutive c-myb expression or mutant APC plus activated RAS. DNA and cell biology. 2005;24:21–29. doi: 10.1089/dna.2005.24.21. [DOI] [PubMed] [Google Scholar]
- 40.Bies J, Feikova S, Bottaro DP, Wolff L. Hyperphosphorylation and increased proteolytic breakdown of c-Myb induced by the inhibition of Ser/Thr protein phosphatases. Oncogene. 2000;19:2846–2854. doi: 10.1038/sj.onc.1203613. [DOI] [PubMed] [Google Scholar]
- 41.Zhou Y, Ness SA. Myb proteins: angels and demons in normal and transformed cells. Frontiers in bioscience (Landmark edition) 2011;16:1109–1131. doi: 10.2741/3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luscher B, Christenson E, Litchfield DW, Krebs EG, Eisenman RN. Myb DNA binding inhibited by phosphorylation at a site deleted during oncogenic activation. Nature. 1990;344:517–522. doi: 10.1038/344517a0. [DOI] [PubMed] [Google Scholar]
- 43.Bies J, Wolff L. Oncogenic activation of c-Myb by carboxyl-terminal truncation leads to decreased proteolysis by the ubiquitin-26S proteasome pathway. Oncogene. 1997;14:203–212. doi: 10.1038/sj.onc.1200828. [DOI] [PubMed] [Google Scholar]
- 44.Jin ZX, et al. Lymphoid enhancer-binding factor-1 binds and activates the recombination-activating gene-2 promoter together with c-Myb and Pax-5 in immature B cells. J Immunol. 2002;169:3783–3792. doi: 10.4049/jimmunol.169.7.3783. [DOI] [PubMed] [Google Scholar]
- 45.Zhou F, Zhang L, van Laar T, van Dam H, Ten Dijke P. GSK3beta inactivation induces apoptosis of leukemia cells by repressing the function of c-Myb. Molecular biology of the cell. 2011;22:3533–3540. doi: 10.1091/mbc.E11-06-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan L, et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Molecular biology of the cell. 2007;18:1083–1097. doi: 10.1091/mbc.E06-07-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/S0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 48.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Developmental cell. 2008;14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 50.Zhang L, et al. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Developmental cell. 2008;14:377–387. doi: 10.1016/j.devcel.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei SC, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015;17:678–688. doi: 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen WC, Lin HH, Tang MJ. Matrix-Stiffness-Regulated Inverse Expression of Kruppel-Like Factor 5 and Kruppel-Like Factor 4 in the Pathogenesis of Renal Fibrosis. The American journal of pathology. 2015;185:2468–2481. doi: 10.1016/j.ajpath.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 53.Ishihara S, et al. Substrate stiffness regulates temporary NF-kappaB activation via actomyosin contractions. Experimental cell research. 2013;319:2916–2927. doi: 10.1016/j.yexcr.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 54.Mammoto A, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009;457:1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jain N, Iyer KV, Kumar A, Shivashankar GV. Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc Natl Acad Sci USA. 2013;110:11349–11354. doi: 10.1073/pnas.1300801110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ki H, et al. Overexpressed LEF-1 proteins display different nuclear localization patterns of beta-catenin in normal versus tumor cells. Cell biology international. 2006;30:253–261. doi: 10.1016/j.cellbi.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 57.Tse, J. R. & Engler, A. J. Preparation of hydrogel substrates with tunable mechanical properties. Current protocols in cell biology/editorial board. Juan S. Bonifacino… [et al.] Chapter 10, Unit 10 16 (2010). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.