

ABSTRACT
Sox2 is known to be important for neuron formation, but the precise mechanism through which it activates a neurogenic program and how this differs from its well-established function in self-renewal of stem cells remain elusive. In this study, we identified a highly conserved cyclin-dependent kinase (Cdk) phosphorylation site on serine 39 (S39) in Sox2. In neural stem cells (NSCs), phosphorylation of S39 enhances the ability of Sox2 to negatively regulate neuronal differentiation, while loss of phosphorylation is necessary for chromatin retention of a truncated form of Sox2 generated during neurogenesis. We further demonstrated that nonphosphorylated cleaved Sox2 specifically induces the expression of proneural genes and promotes neurogenic commitment in vivo. Our present study sheds light on how the level of Cdk kinase activity directly regulates Sox2 to tip the balance between self-renewal and differentiation in NSCs.
KEYWORDS: neural stem cells, cell cycle regulation, self-renewal, differentiation, Sox2, Cdks, cyclin-dependent kinases
INTRODUCTION
Sox2 is a member of the SRY-related family of transcription factors, which are characterized by a conserved HMG box DNA binding domain (1). Targeted disruption of Sox2 resulted in early embryonic lethality shortly after implantation due to impaired development of the epiblast and extraembryonic ectoderm (2, 3), while ablation of Sox2 in embryonic stem cells (ESCs) caused inappropriate differentiation into trophoectoderm-like cells (4, 5). More importantly, forced expression of Sox2 together with Oct4, Klf4, and c-Myc is capable of reprogramming differentiated somatic cells into induced pluripotent stem (iPS) cells (6–8), highlighting the importance of Sox2 in the establishment and maintenance of an undifferentiated state. Upon commitment to more restricted cell lineages, Sox2 expression is retained in the developing central nervous system (CNS) (9), where it serves as one of the earliest pan-neural marker (10–12), supports neural stem cell (NSC) self-renewal throughout development and into adulthood (13, 14), and shares overlapping functions with two other members of the structurally related group B1 proteins: Sox1 and Sox3 (15–17). Constitutive expression of Sox2 inhibited neuronal differentiation and promoted precursor cell characteristics, while decreasing of Sox2 signaling induced cell cycle withdrawal and triggered premature neurogenesis (13).
Despite its well-documented role in the maintenance of an undifferentiated state in stem cells, Sox2 is paradoxically also required for the formation and maturation of differentiated neurons (N). This was based on several independent observations of cases in which targeted disruption of Sox2 in brain and eyes of mice led to impaired generation of specific neuronal subtypes (18–21). For example, conditional loss of Sox2 with nestin-Cre in the developing mouse brain did not affect the self-renewal capacity of NSCs due to compensation by Sox3 but attenuated its neurogenic potential (19). Hypomorphic/null compound mutants expressing 25 to 30% of wild-type levels of Sox2 in the brain resulted in proliferative defects in precursor cells of the hippocampus and periventricular zone, and even more prominent neurological defects, including neurodegeneration and intraneuronal aggregates, in neurons of the thalamic and striatal parenchyma (21). Analysis of NSCs isolated from these Sox2-hypomorphic mice suggested that stem cell proliferation was relatively unaffected but neuronal differentiation was severely compromised by the Sox2 deficiency (20). Together with a recent study which demonstrated that the forced expression of Sox2 alone was sufficient for the conversion of reactive glial cells into induced neurons following traumatic injury to the cerebral cortex (22), these findings allow one to conclude that Sox2 is indeed required for the specification of neuronal cell fate in addition to the maintenance of neural stem cell identity.
The posttranslational modifications of Sox2 are garnering attention as key mechanisms to modulate its activity. For example, SUMO (small ubiquitin-like modifier) modification of mouse Sox2 at lysine 247 (K247) was suggested to negatively regulate its transcriptional activity by impairing its DNA binding ability (23). An analysis of human ESC phosphoproteome revealed that SUMOylation of Sox2 is dependent on prior phosphorylation at one or more of three consecutive serine residues—S249, S250, and S251—located immediately downstream of the SUMOylation site (24). Therefore, this serine triplet forms the phosphorylation-dependent SUMOylation motif similar to that defined for other Sox family members (25–27). Acetylation by p300/CBP (CREB [cyclic AMP-responsive element-binding protein]-binding protein) at lysine 75 (K75), which lies within the leucine-rich nuclear export signal, was shown to increase the interaction of Sox2 with Crm1, a component of the nuclear export machinery. Binding to Crm1 resulted in the nuclear exclusion of Sox2, followed by its ubiquitination and proteasomal degradation (28). While the modifications described thus far suppress Sox2 activity, recent studies have identified others that potentiate Sox2 activity. For example, phosphorylation by Akt at a threonine residue (T118 in mouse Sox2) situated right after the HMG box was reported to counteract methylation by Set7 at the subsequent lysine residue (K119), thereby preventing the generation of a methyl motif that is recognized by the E3 ubiquitin-protein ligase WWP2 (WW domain-containing protein 2) (29, 30). Indeed, a variety of posttranslational modifications of Sox2 act concertedly to fine-tune its level or activity. However, these have all been characterized for pluripotent ESCs, and it is currently not known whether similar modifications also play an important role in the regulation of Sox2 in other somatic stem cells. Furthermore, the residues within Sox2 that have been assigned as targets of posttranslational modifications are conserved among related Sox family members, suggesting that they probably represent a common mechanism through which the activities of all Sox proteins are controlled. Considering that Sox2 has unique roles in the developing CNS that cannot be compensated by Sox1 and Sox3 (15), additional modifications may exist which regulate the ability of Sox2 to function in diverse processes.
Cyclin-dependent kinases (Cdks) belong to a family of serine/threonine kinases whose best-characterized functions are in controlling cell cycle progression (31). During the development of the CNS, there is a gradual decline in the overall rate of cell cycle progression as NSCs switch from divisions that generate additional stem cells (expansion) to divisions that generate committed progenitors or postmitotic cells (differentiation) (32, 33). Several studies (including our own) have shown that a change in Cdk/cyclin activity itself is sufficient to alter the specification of neural cell fate (34–37; for a review, see reference 38). Therefore, given the importance of Cdks in controlling the switch from proliferation to differentiation during neurodevelopment and the role of Sox2 as a transcription factor required in both NSCs and neurons, we hypothesize that Cdks may posttranslationally modify Sox2 as a mechanism to regulate the transition from dividing NSCs to nondividing neurons in accordance with the cell cycle status. To confirm this, we have identified Sox2 as a direct substrate of Cdks. Phosphorylation occurs at S39 and is required for the optimal suppression of neuronal differentiation in proliferating NSCs. We found that upon the induction of neurogenesis, Sox2 is cleaved by serine proteases. A cleavage product containing the HMG domain is selectively retained on chromatin once the phosphate group at S39 is lost together with the drop in Cdk kinase activity as cells differentiate and withdraw from the cell cycle. Importantly, we demonstrate that this truncated form is physiologically active and specifically drives the expression of a subset of target genes originally bound by its full-length counterpart: the neurogenic genes. We provide mechanistic insights on how the loss of phosphorylation at Sox2-S39 is key to its ability to drive neuron formation.
RESULTS
Cdks phosphorylate Sox2 at serine 39.
To investigate whether Cdks can modify Sox2 posttranslationally, we first searched for Cdk phosphorylation sites in Sox2. Using an in silico functional site prediction program (39), we identified a candidate at Sox2 serine 39 (S39) (Fig. 1A, purple box), which precedes the HMG box DNA binding domain (Fig. 1A, green box). The amino acid residues serine (S), proline (P), aspartic acid (D), and arginine (R) are a perfect match with the consensus sequence for Cdk recognition, (S/T)PX(R/K), where S/T is the serine/threonine phosphorylation site, X is any amino acid, and R/K is a basic residue arginine/lysine (40, 41). The presence of a putative cyclin-binding motif after the HMG box (Fig. 1A, blue box), coupled with the high surface accessibility (42) and extensive sequence conservation across different Sox2 species (Fig. 1A), further enhances the likelihood of S39 phosphorylation by Cdk/cyclin complexes. This phosphorylation site (S39) is specific to Sox2 and cannot be found in other Sox family members.
FIG 1.

Identification of a Cdk phosphorylation site on Sox2 serine 39. (A) Alignment of Sox2 protein sequences from different species. Only the N-terminal region containing the putative Cdk phosphorylation site on serine 39 (purple), the HMG box (green), and the predicted cyclin-binding motif (blue) are shown. Protein sequences are from the following (with NCBI Protein database accession numbers in parentheses): Mus musculus (NP_035573), Rattus norvegicus (NP_001102651), Homo sapiens (NP_003097), Gallus gallus (NP_990519), Equus caballus (NP_001137271), Bos taurus (NP_001098933), Macaca mulatta (NP_001136412), Sus scrofa (NP_001116669), and Xenopus tropicalis (NP_998869). Red indicates nonconserved residues. (B) In vitro kinase assay where active Cdk2/cyclin A, Cdk1/cyclin A, or Cdk1/cyclin B complexes were incubated with recombinant purified GST-Sox2 or GST-Sox2-S39A. No substrate was added into the lanes marked by a minus sign. All lanes contain [γ-32P]ATP. GST, glutathione S-transferase. (C) Western blot analysis of recombinant purified GST-Sox2 or GST-Sox2-S39A incubated without (−) or with (+) Cdk2/cyclin A complexes and probed with antibodies against total Sox2 (top blot) or our homemade phospho-Sox2P-S39 (bottom blot). (D) Western blot analysis of extracts prepared from ROSA26-CreERT2 expressing NSCs with the indicated genotypes using antibodies against Sox2, phospho-Sox2P-S39, Cdk1, and Hsp90. A 20-ng/ml concentration of 4OHT was added to induce Cre-mediated excision of floxed (fl) sequences and therefore loss of Cdk1 expression. Hsp90 served as a loading control. Lanes 1 and 4 as well as 5 and 8 show replicates of the same samples. (E) Quantification of the intensities of the bands for total Sox2 and phospho-S39-Sox2. The ratio of total Sox2 over phospho-S39-Sox2 is displayed and indicates that Sox2 is completely phosphorylated under normal conditions.
To test whether Sox2-S39 is indeed a target for Cdk phosphorylation, in vitro kinase assays were performed using an array of recombinant purified Cdk/cyclin complexes and Sox2 as the substrate in the presence of [γ-32P]ATP. High levels of radiolabeled phosphate were detected when Sox2 was incubated with Cdk2/cyclin A, Cdk1/cyclin A, or Cdk1/cyclin B (Fig. 1B, lanes 1, 4, and 7). Mutation of S39 in Sox2 to alanine (Sox2-S39A) completely abolished the incorporation of radioactive phosphate (Fig. 1B, lanes 2, 5, and 8), suggesting that Cdk-mediated phosphorylation of Sox2 occurs on S39. These results are consistent with those of a study by Ouyang et al., who reported Sox2-S39 phosphorylation by Cdk2-containing complexes in human ESCs (43). Although they did not use Cdk1 complexes in their in vitro kinase assays, they did note that mitotic ESCs with strong Cdk1 activity contained the highest level of Sox2-S39 phosphorylation (43).
To detect the presence of phosphorylated Sox2 in vivo, we raised phospho-specific antibodies against Sox2P-S39. Antibody specificity was determined using recombinant purified Sox2 or Sox2-S39A in the presence or absence of Cdk2/cyclin A. In the absence of phosphorylation, no signals were detected from either protein, suggesting that the phospho-specific antibody does not recognize unmodified Sox2 (Fig. 1C, lanes 1 and 2). Upon Cdk2/cyclin A phosphorylation, a signal was picked up for Sox2 (Fig. 1C, lane 3) that was not present when S39 was mutated to alanine (Fig. 1C, lane 4), implying that the phospho-specific antibody binds only to a phosphate group that was introduced at position 39. Following antibody validation, we proceeded to analyze NSCs that lacked different combinations of Cdks in order to determine which Cdks phosphorylate Sox2 in cells (Fig. 1D). To inactivate Cdk1, a floxed allele was used (44) in combination with a ROSA26 locus expressing Cre-recombinase fused to an estrogen receptor T2 moiety (ROSA26-CreERT2). Upon the addition of 4-hydroxy-tamoxifen (4OHT), CreERT2 translocates into the nucleus to mediate recombination of loxP sites (45) and induces the deletion of Cdk1. As previously shown (35), combined loss of Cdk2 and Cdk4 led to a reduced protein expression of Sox2 and therefore Sox2P-S39 compared to Cdk1flox/flox NSCs, which are functionally equivalent to the wild type in the absence of 4OHT (Fig. 1D, compare lanes 1 and 4 to lanes 2 and 3). Interestingly, in the absence of 4OHT the levels of Sox2P-S39 were comparable between Cdk1flox/flox Cdk2−/− Cdk4+/− (essentially Cdk2KO) and Cdk1flox/flox Cdk2+/− Cdk4+/− (control) NSCs (Fig. 1D, lanes 2 and 3), suggesting that Cdk1 can compensate for the loss of Cdk2 and/or that Cdk2 may not be a potent kinase for Sox2-S39. Seventy-two hours after treatment with 4OHT; the depletion of Cdk1 caused a substantial decrease in Sox2 phosphorylation, while the total Sox2 protein levels decreased less than 2-fold (Fig. 1D, lanes 5 to 8). The signal for Sox2P-S39 was slightly stronger in Cdk1−/− than in Cdk1−/− Cdk2−/− NSCs (Fig. 1D, lanes 6 and 7), indicating that Cdk2 may play a role in the phosphorylation of Sox2. Finally, the additional removal of Cdk4 in Cdk1−/− Cdk2−/− NSCs did not further decrease the phospho-signal (Fig. 1D, lanes 5, 7, and 8), indicating that Sox2 is most likely not a major substrate of Cdk4-associated complexes in NSCs. In addition, in vitro kinase reactions were unsuccessful in phosphorylating Sox2 with Cdk4 or Cdk6/cyclin D complexes (data not shown). Quantification of the data in Fig. 1D indicated that Sox2 is fully phosphorylated (lanes 1 to 4), but in the absence of Cdk1 and Cdk2, the ratio of total Sox2 to S39-phosphorylated Sox2 drops below 0.4 (Fig. 1E). Our data thus provide evidence for the existence of Cdk-directed phosphorylation at Sox2-S39 in NSCs.
Phosphorylation of Sox2 inhibits neurogenesis.
To gain insights into the biological function of Sox2-S39 phosphorylation, we determined the consequences associated with the expression of Sox2 or its mutants (S39A or S39D) in undifferentiated NSCs and upon induction of differentiation. Previous studies have indicated that elevating the levels of Sox2 in embryonal carcinoma cells and ESCs unexpectedly inhibited the expression of Sox2:Oct3/4 target genes and triggered differentiation (46, 47), suggesting that the levels of Sox2 in stem cells are dynamically regulated and precisely controlled within a narrow range such that both increased and decreased levels of Sox2 affect differentiation (48, 49). In our retroviral system and culture conditions that actively sustain self-renewal, infected NSCs typically express approximately double the amount of Sox2 or its mutants (Fig. 2A) and can be propagated as neurospheres in the presence of puromycin as a selection marker. Quantitative PCR (qPCR) analysis of known Sox2 target genes (50) revealed a significant increase in Nmyc transcripts following the overexpression of Sox2 (Fig. 2B). However, the degrees of upregulation were comparable between Sox2 and its mutants, suggesting that the phosphorylation status of Sox2 did not influence the expression of Nmyc. The remaining Sox2 target genes analyzed, including Rbpj, Gli2, Gli3, Jag1, and Tulp3 (Fig. 2G to K), did not show substantial differential regulation by forced Sox2 expression, implying that the levels of exogenous Sox2 in the present study were not sufficient to induce a change in the underlying transcription of these genes in the stem cell state. Overexpression of Sox2 or its mutants also did not affect the expression of a proneural gene such as the NeuroG2 gene (Fig. 2C).
FIG 2.

Phosphorylated Sox2 inhibits neuronal differentiation. (A to C) qPCR analysis of the mRNA expression levels of Sox2 (A), Nmyc (B), and NeuroG2 (C) in NSCs overexpressing full-length Sox2 or its mutants (S39D or S39A) under self-renewing conditions, where proliferation is dominant. Each bar represents the average fold change compared to vector control (n = 2 to 4) after normalization to β-actin, Hsp90, and cyclophilin A expression. *, P < 0.05. (D to F) qPCR analysis of the mRNA expression levels of Gfap (D), NeuroD1 (E), and NeuroG2 (F) in NSCs overexpressing full-length Sox2 or its mutants (S39D or S39A) and differentiated with 10% FBS for 6 days. Each bar represents the average fold change compared to vector control (n = 2 or 3) after normalization to β-actin, Hsp90, and cyclophilin A expression. *, P < 0.05. (G to K) qPCR analysis of the expression levels of Rbpj (G), Gli2 (H), Gli3 (I), Jag1 (J), and Tulp3 (K) in NSCs overexpressing full-length Sox2 or its mutants (S39D or S39A). Each bar represents the average fold change compared to vector control (n = 3) after normalization to β-actin, Hsp90, and cyclophilin A expression. (L) In vivo mouse embryo electroporation of full-length Sox2, full-length Sox2-S39A, and control (RFP) as shown in Fig. 7G to J. AP, apical progenitor; BP, basal progenitor; N, neuron.
Given that qPCR analysis of undifferentiated NSCs did not yield major differences between Sox2 and its mutants, we decided to investigate how infected NSCs might react to the presence of a signal to differentiate. A previous study reported that in NSCs, proliferation was not affected by Sox2 overexpression but that differentiation into astrocytes and neurons was inhibited (51). This observation can probably be explained by the existence of a surveillance mechanism in NSCs that keeps total Sox2 levels in check (as described above; see also references 48 and 49). Hence, any biological effects of Sox2 overexpression are manifested only when the levels of endogenous Sox2 start to decline at the initiation of differentiation. Differentiation was induced by the addition of 10% fetal bovine serum (FBS)–Dulbecco modified Eagle medium (DMEM), a condition that promotes the generation of astrocytes and neurons (52). Six days postinduction, cells were collected and the expression of lineage-specific markers was determined using qPCR. Consistent with previous reports (51, 53), overexpression of Sox2 inhibited differentiation into astrocytes and neurons, as demonstrated by the decreased expression of the astrocyte marker Gfap (Fig. 2D) and the neuronal markers NeuroD1 and NeuroG2 (Fig. 2E and F) compared to vector control. Sox2-S39D, mimicking phosphorylated Sox2 at S39, behaved in a similar fashion as wild-type Sox2 and also suppressed the formation of astrocytes and neurons. Remarkably, upon Sox2-S39A overexpression, astrocytes were likewise inhibited but neurons were generated at a rate comparable to that of the vector control, as evidenced by the lack of suppression of NeuroG2 expression (Fig. 2F). Therefore, our results indicate that the phosphorylation of Sox2 at S39 is important for its role in the prevention of neuronal differentiation but does not affect the formation of astrocytes.
Phosphorylation of Sox2 at S39 does not affect its protein stability and DNA binding efficiency.
To understand how phosphorylation of Sox2 could alter its function, we studied several processes, including protein degradation, nuclear localization, and in vitro DNA binding. We first characterized the turnover of Sox2 in NSCs to investigate the effect of phosphorylation on Sox2 stability. MG132, a proteasome inhibitor that prevents the degradation of ubiquitin-conjugated proteins, induced the accumulation of Sox2 (Fig. 3A), while cycloheximide, an inhibitor of de novo protein synthesis, caused a gradual reduction in Sox2 (Fig. 3B). When NSCs expressing myc-tagged Sox2 or its mutants (S39D or S39A) were treated with MG132, all 3 exogenous proteins accumulated in a similar fashion (Fig. 3C), indicating that phosphorylation had no major influence on the rate of Sox2 degradation. While we were able to show that the ubiquitin-proteasome machinery is involved in the degradation of Sox2 in NSCs, at present there is no evidence to suggest that this process is affected by the phosphorylation status of Sox2 at S39. Considering recent reports demonstrating multiple Cdk-targeted residues on Sox2 (43), we cannot exclude the possibility that more than one phosphorylation site acts concertedly to regulate the stability of Sox2 in NSCs or that Sox2 stability is regulated differentially in ESCs and NSCs. Evidence for multiple phosphorylation sites in Sox2 using ESCs has recently been published (54).
FIG 3.

Protein stability and DNA binding capacity of Sox2 and its mutants. (A and B) Western blot analysis of extracts prepared from NSCs treated with 10 μM MG132 (A) or 10 μg/ml of cycloheximide (CHX) (B) for the indicated length of time and probed with antibodies against Sox2. Hsp90 served as a loading control. (C) Western blot analysis of extracts prepared from NSCs infected with empty vectors (−) or vectors expressing Sox2-myc (WT), Sox2-S39D-myc, or Sox2-S39A-myc and treated with 10 μM MG132 for the indicated length of time. Cyclin A2 (cycA2) and p27 served as positive controls for MG132 treatment. Hsp90 served as a loading control. (D) Recombinant purified full-length GST-Sox2 or GST-Sox2-S39A was phosphorylated by Cdk1/cyclin A2 complexes in vitro and used for EMSA at the indicated concentrations with double-stranded 5′-, Cy5-labeled DNA as a probe. The probe contained canonical motifs for Sox (CATTGTC) and Oct (ATGCAAAT) in tandem with no spacer and G's and C's as boundary nucleotides. F, free DNA; B, protein-DNA complex. (E) EMSA as described for panel D using truncated Sox2 (Sox233–141) or its mutants (S39D or S39A) at the indicated concentrations.
Given that phosphorylation at S39 alone did not regulate degradation of Sox2, an alternative explanation for the observed decline in Sox2 whenever phosphorylation is inhibited could be a reduction in Sox2 expression. Since Sox2 is known to bind to its own enhancer to stimulate its expression in an autoregulatory loop that is essential for the stabilization of cell state (55–57), phosphorylation could affect its transactivation activity to control its abundance. We first tested the nuclear accessibility of Sox2 and its mutants using immunofluorescence. As expected, Sox2 was found in the nucleus in NSCs and Sox2P-S39 produced a similar staining pattern (data not shown), suggesting that phosphorylated Sox2 is also retained within the nucleus. To determine if nonphosphorylated Sox2 is mislocalized, myc-tagged Sox2-S39A was expressed in NSCs and detected with anti-myc-tag antibodies. Sox2-S39A was clearly limited to the nucleus, where the phosphomimetic Sox2-S39D was also targeted. Therefore, our results suggest that the nuclear localization of Sox2 is not affected by phosphorylation at S39.
Since there was no effect of phosphorylation on the recruitment of Sox2 to the nucleus, we next studied the DNA binding affinities of Sox2 and its mutants by electrophoretic mobility shift assay (EMSA). The DNA probe was a synthetic sequence specifically chosen to contain idealized binding motifs for Sox (CATTGTC) and Oct (ATGCAAAT) in tandem with no spacer and flanked by random G's and C's as boundary nucleotides to minimize binding anomalies due to cryptic elements at the periphery (58, 59). Recombinant purified Sox2 subjected to prior phosphorylation by Cdk1/cyclin A2 bound this element at concentrations above 50 nM, as demonstrated by the appearance of a slower-migrating band consisting of protein-DNA complexes (B) in addition to the faster-running free probe (F) (Fig. 3D). When phosphorylation was inhibited with the S39A mutant, an identical gel shift pattern was observed, indicating that the absence of a phosphate moiety at S39 did not alter the DNA binding capacity of Sox2 in this context. A similar experiment was conducted using Sox2 and its mutants (S39D and S39A) expressed as minimal forms containing the HMG box and a few additional residues flanking both ends (Fig. 3E). Again, these proteins showed no differences in DNA binding efficiencies, suggesting that the association of Sox2 with its minimal canonical DNA motif is not controlled by phosphorylation at S39 alone in vitro.
Sox2 is cleaved by serine proteases at the onset of neuronal differentiation.
Since functional differences between Sox2 and its mutants were observed only upon differentiation, we reasoned that subsequent analyses of the role of Sox2-S39 phosphorylation should be centered on the process of neurogenesis. This is also an event where Cdk kinase activity changes drastically as stem cells exit the cell cycle and nonphosphorylated forms of Sox2 are expected to exist physiologically. A previous study suggested that in progenitors committed to neurogenic divisions, Sox2 is cleaved by serine proteases as a way to eliminate Sox2 and relieve the inhibition on neurogenesis (53). Using NSCs expressing an N-terminally hemagglutinin (HA)-tagged Sox2 (HA-Sox2), a specially formulated medium was added to direct optimal differentiation toward the neuronal lineage (35). During the early hours (12 h) following neuronal differentiation, three specific cleavage products of Sox2 were visible (Fig. 4A, second blot, lane 8), and their formation preceded the upregulation of NeuroD (Fig. 4A, third blot, lanes 7 to 12), a proneural basic helix-loop-helix (bHLH) transcription factor required for the acquisition of the neuronal fate (60). As time after neuron induction increased, full-length Sox2 protein levels progressively declined (Fig. 4A, top blot, lanes 7 to 12). Since this was not matched by a corresponding increase in cleaved forms of Sox2, Sox2 is also degraded upon differentiation, most likely through ubiquitin-mediated degradation (54). Of more importance to our study, we were able to show that the cleavage of Sox2 seen at the 12-h time point was mediated by serine proteases, since treatment with an irreversible serine protease inhibitor, AEBSF [4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride], prevented Sox2 cleavage in a dose-dependent manner (Fig. 4A, second blot, lanes 1 to 4) and rescued the decrease of full-length Sox2 protein levels after 12 h of neuronal differentiation (Fig. 4A, top blot, lanes 1 to 4). At the highest concentration used (1 mM), AEBSF abolished the expression of NeuroD (Fig. 4A, third blot, lane 1), suggesting that serine protease activity is needed for neurogenesis to ensue, although unspecific side effects of this inhibitor cannot be excluded.
FIG 4.

Sox2 is cleaved by serine proteases during neuronal differentiation. (A) Western blot analysis of extracts prepared from NSCs infected with empty vectors (−) or vectors expressing N-terminally HA-tagged Sox2 (HA-Sox2). Cells were differentiated into neurons for 12 h in the presence of AEBSF at the indicated concentrations (lanes 1 to 4), or differentiated into neurons and collected at the indicated times after neuron induction (lanes 8 to 12). Two different exposures after probing with anti-HA tag antibodies are shown. Hsp90 served as a loading control. (B) Western blot analysis of lysates prepared from various organs of embryos or pups at the indicated stages of development. The brain was extracted whole or subdivided into the cortex and mid- and hindbrain (M/H). Two different exposures after probing with antibodies raised against the N-terminal region of Sox2 (Abcam; ab59776) are shown. Hsp90 served as a loading control. (C) Immunoprecipitation using HA beads followed by Western blotting of extracts prepared from NSCs infected with empty vector (−) or vectors expressing HA-Sox2 (WT), HA-Sox2-S39D, or HA-Sox2-S39A. Cells were undifferentiated or differentiated into neurons for 12 h. Two antibodies were used, one raised against the HA tag (left) and the other raised against the N-terminal region of Sox2 (Abcam; ab59776) (right), with identical results. Short and long exposures for each blot are shown.
To determine whether the cleavage of Sox2 during neurogenesis occurs in vivo, we selected an antibody that was raised against the N-terminal region of Sox2 in order to recognize cleavage fragments that retain the HMG domain. Unfortunately, we did not have access to antibodies against the C terminus of Sox2 and therefore could not track the C-terminal fragment. Cleaved forms of Sox2 were found in the brain, where Sox2 expression is abundant but to a much lesser extent in the liver and kidneys (Fig. 4B). More importantly, cleaved forms of Sox2 increased with the rise in neuronal output as the embryo matured and accumulated to higher levels in the midbrain and hindbrain, regions with greater neuron density, than in the cortex. Our data suggest that Sox2 cleavage is possibly a physiological event that coincides with the generation of neurons during neurodevelopment.
We next investigated whether Sox2 cleavage is affected by phosphorylation at S39. Using NSCs expressing HA-Sox2 or its mutants (S39D or S39A), we immunoprecipitated the exogenous proteins before differentiation and after 12 h of neuron induction. Regardless of the residue at position 39, cleavage of various Sox2 forms occurred with equal efficiencies, suggesting that Cdk-mediated phosphorylation may not control Sox2 cleavage (Fig. 4C). The two antibodies used (one against the HA tag and one against the N-terminal region of Sox2) gave identical results, reinforcing our claim that specific cleavage products of Sox2 are formed in progenitors that are committed to the neuronal lineage. Although cleavage was not dependent on phosphorylation at S39, all Sox2 fragments were noticeably nonphosphorylated, as the phospho-specific antibody gave a signal that corresponded only to full-length Sox2 in undifferentiated cells (data not shown). This may suggest that in actively proliferating NSCs, Sox2 is continuously phosphorylated at S39 by Cdks but in differentiating cells, cleaved forms of Sox2 are generated that become dephosphorylated due to the concurrent decline in Cdk kinase activity and possibly due to the activity of phosphatases.
Nonphosphorylated cleaved Sox2 binds neurogenic genes.
Since Sox2 is a transcription factor that binds to DNA, we tested whether the cleavage products of Sox2 generated during neurogenesis are still able to associate with DNA. Three different C-terminal truncations of Sox2 were produced to mimic as closely as possible the expected molecular sizes of the endogenous cleavage fragments (Fig. 5A, right side). Each of these truncations bearing a C-terminal myc tag was introduced into NSCs as a serine or a nonphosphorylatable alanine at position 39. In NSCs with high Cdk kinase activity, S39 is expected to be phosphorylated continuously, and indeed, with the expression of fragments of smaller size, the molecular weight difference due to an additional phosphate group was distinguishable as a slower-migrating band (Fig. 5A, lanes 1 and 2). Following cell lysis under conditions that allowed the separation of soluble proteins (Fig. 5A, lanes 3 and 4) from those that were chromatin bound (Fig. 5A, lanes 5 and 6), the two larger fragments of Sox2 (Sox21–235 and Sox21–159) were found to be strongly associated with DNA regardless of their phosphorylation status (Fig. 5A, top and middle rows). However, Sox21–113, which had lost the entire region after the HMG box, was dissociated from chromatin unless phosphorylation at S39 was inhibited (Fig. 5A, bottom row, lanes 5 and 6). Interestingly, the Sox21–113 that was retained on chromatin did not run as a shifted band unlike those in the whole-cell lysate (WCL) and soluble fraction, again indicating that it was the dephosphorylated form that was capable of binding to DNA. Chromatin extraction was repeated with the S39D phosphomimetic mutant to emphasize that the presence of a phosphate group similar to that in the phosphorylated wild type (WT) did impair the localization of Sox21–113 onto chromatin (Fig. 5B, top blot, lanes 10 to 12). Although Sox21–113-S39A was able to bind DNA, efficiency was low compared to that of full-length wild-type Sox2 and most of the protein appeared in the soluble fraction (Fig. 5B, top blot, lanes 6 to 8). In addition, we also noticed that this DNA binding ability is restricted to NSCs, since the expression of Sox21–113-S39A in NIH 3T3 fibroblasts did not yield any signals in the chromatin fraction (Fig. 5C, top blot, lanes 22 to 24) even though the two larger fragments were enriched in a manner that was independent of phosphorylation at S39 (Fig. 5C, top blot, lanes 18 to 21). This suggests that in NSCs, an exclusive interaction partner that is responsible for targeting Sox21–113-S39A to chromatin is expressed. As described above, the cleaved forms of Sox2 are mainly hypophosphorylated in differentiating NSCs due to the declining levels of Cdk kinase activity. Therefore, the retention of a nonphosphorylated cleavage product (Sox21–113-S39A) on chromatin may be a natural occurrence during the formation of neurons and its purpose warrants further investigation.
FIG 5.
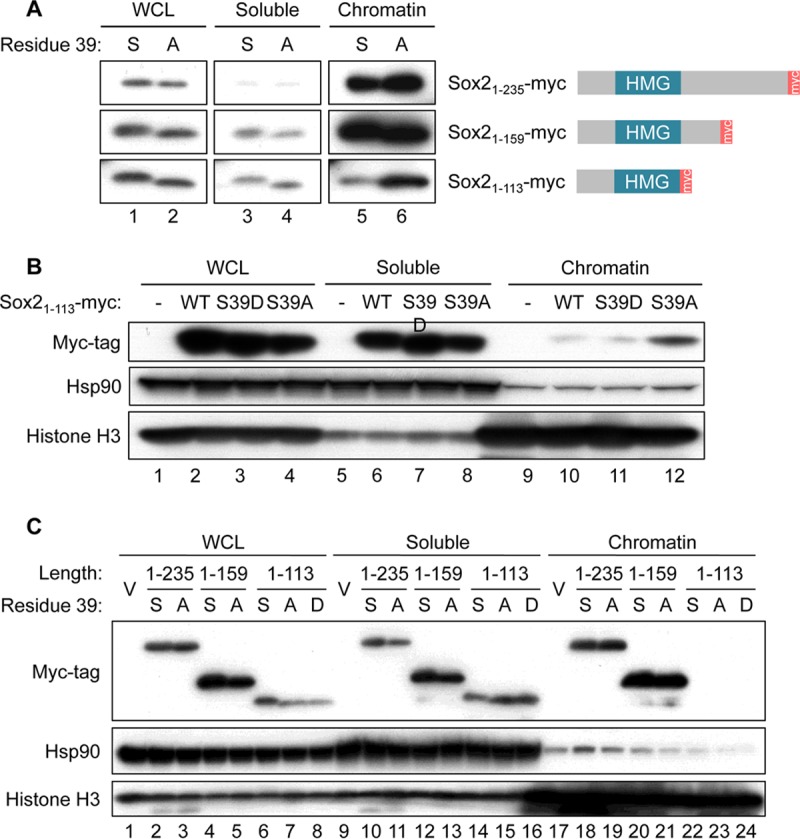
Lack of phosphorylation on S39 allows Sox21–113 to remain bound on chromatin. (A) Chromatin extraction and Western blot analysis of extracts prepared from NSCs expressing three different C-terminal truncations of Sox2: Sox21–235, Sox21–159, and Sox21–113, all containing a C-terminal Myc tag and detected using antibodies against the Myc tag. Each Sox2 truncated form had a serine (S) or an alanine (A) residue at position 39. Chromatin extraction was performed with CSK buffer to obtain a soluble fraction and a chromatin-bound fraction. WCL, whole-cell lysate. (B) Chromatin extraction as in panel A and Western blot analysis of extracts prepared from NSCs infected with empty vector (−) or vectors expressing Sox21–113-myc (WT), Sox21–113-S39D-myc, or Sox21–113-S39A-myc. Hsp90 and histone H3 served as loading controls for the soluble and chromatin-bound fractions, respectively. (C) Chromatin extraction and Western blot analysis of extracts prepared from NIH 3T3 cells infected with empty vectors (V) or vectors expressing C-terminally Myc-tagged Sox21–235, Sox21–159, or Sox21–113. Each Sox2 truncated form had a serine, an alanine, or an aspartic acid (D) residue at position 39. Hsp90 and histone H3 served as loading controls for the soluble and chromatin-bound fractions, respectively.
After establishing that Sox21–113-S39A is still bound to chromatin, we performed chromatin immunoprecipitations followed by massively parallel DNA sequencing (ChIPseq) to interrogate the genome-wide binding profiles of exogenously introduced Sox2, Sox2-S39A, and Sox21–113-S39A. Totals of 20,286, 16,200, and 729 peaks were identified for Sox2, Sox2-S39A, and Sox21–113-S39A, respectively. The heights of the peaks were similar between Sox2 and Sox2-S39A but significantly reduced for Sox21–113-S39A (Fig. 6G). The comparatively lower number of peaks and peak height for Sox21–113-S39A were not surprising since we have shown above that this truncated form was much less retained on chromatin than full-length Sox2 (Fig. 5); hence, binding was expected to be weak. Nevertheless, the distributions of peaks across various genomic regions were comparable among the three data sets (Fig. 6H). The authenticity of the present ChIPseq was demonstrated by the significant concordance between our Sox2 and a published Sox2 ChIPseq (Fig. 6A) (61). However, the overlap is not perfect due to several experimental differences. First, our NSCs were directly isolated from embryonic day 13.5 (E13.5) mouse embryonic brains and expanded in culture as neurospheres, while the neural progenitor cell (NPC) line used by Lodato et al. was differentiated from ESCs in vitro (61). Second, we were pulling down myc-tagged, therefore exogenous Sox2, while Lodato et al. were studying the binding of endogenous Sox2 (61). In spite of these differences, our study picked up the canonical Sox2 motif as the topmost enriched motif for the Sox2 (P = 1 × 10−2,320) and Sox2-S39A (P = 1 × 10−2,478) peaks (Fig. 6I), suggesting that our approach is appropriate for evaluating the genome-wide occupancy of Sox2 and its variants.
FIG 6.


Nonphosphorylated Sox21–113 binds specifically to neurogenic genes. (A) Venn diagram displaying the extent of overlap between full-length Sox2 ChIPseq peaks obtained from the current study (red) and that from a published source (gray) (61). (B and C) Venn diagrams indicating the number of overlapping genes bound by full-length Sox2 (red) and Sox2-S39A (blue, B) or the fragment Sox21–113-S39A (green, C). (D) Gene ontology (GO) analysis of the target genes of Sox2 (red), Sox2-S39A (blue), and Sox21–113-S39A (green). The black dashed line indicates a P value of 0.01. (E) Gene lists were intersected and GO analysis was performed on genes that are regulated by Sox2 but not Sox2-S39A. Top hits are shown together with the GO identifier (ID) and P value. (F) Gene lists were intersected and GO analysis was performed on genes that are regulated by both Sox2 and Sox21–113-S39A. Top hits are shown together with the GO ID and P value. (G) Peak height distribution of the indicated libraries. Boxes represent the 25th to 75th percentile of peaks, with the median shown as an intersection. The y axis displays log 2-transformed peak scores. (H) Genome distribution of peaks obtained from the Sox2 (red), Sox2-S39A (blue), and Sox21–113-S39A (green) ChIPseq libraries. Peaks were assigned based on their proximity to promoters, 5′ untranslated region (UTR), 3′UTR, exons, introns, or intergenic regions. (I) Position weight matrices (PWMs) of motifs identified de novo in peaks bound by Sox2 or Sox2-S39A. The canonical Sox2 motif is the most enriched in both data sets. (J) Heat map to indicate reads from each peak of the indicated data sets (full-length Sox2 and Sox2-S39A) after it has been normalized to the average of merged reads from corresponding peaks of both data sets. Values would therefore range from 0 (black) to 1 (yellow), where values close to 1 represent peaks unique that given data set and a value of 0.5 indicating perfect concordance among the two data sets. Black horizontal lines indicate fold changes of more than 2 (upper) and 0.5 (lower). (K) Heat map as in panel J but comparing between Sox2 and Sox21–113-S39A. (L and M) Venn diagrams of the comparisons of our wild-type Sox2 ChIPseq peaks (red) with those described in references 79 (blue, L) and 80 (blue, M).
Using Sox2 (which we expect to be heavily phosphorylated at S39 in NSCs with high Cdk kinase activity) as the baseline, we proceeded to analyze how genomic binding sites may be altered by (i) the inhibition of phosphorylation (as in Sox2-S39A) and (ii) the inhibition of phosphorylation combined with cleavage (as in Sox21–113-S39A). Peaks from Sox2 and full-length Sox2-S39A largely overlapped, with only 7.1% of the peaks from Sox2 and 2.2% of the peaks from full-length Sox2-S39A displaying more than a 2-fold difference from each other (Fig. 6J). However, 96.1% of the peaks from Sox2 had more than a 2-fold change from Sox21–113-S39A (Fig. 6K). This huge variation is due to the massive reduction in the number of peaks obtained from Sox21–113-S39A. Nonetheless, within the Sox21–113-S39A data set, there were still 37.6% of the peaks that differed more than 2-fold compared to Sox2, suggesting that although the loss of phosphorylation did not majorly impact the genomic distribution of Sox2, the additional cleavage may have retargeted Sox2 to distinct loci or increased its affinity at existing bound regions.
Peaks from each data set were mapped to proximal genes, which were deemed potential targets. Target genes from Sox2-S39A and Sox21–113-S39A were compared to those from Sox2. Sox2 and Sox2-S39A continue to share a large number of target genes (Fig. 6B) and therefore control similar biological processes (Fig. 6D). However, when the gene ontology (GO) analysis was extended to the genes that were differentially regulated between Sox2 and Sox2-S39A, the set of 1,832 genes unique to Sox2 (Fig. 6B, red) were significantly enriched in processes that control the cell cycle and the negative regulation of apoptosis (Fig. 6E). Examples of such genes include those for Cdk1, Cdk6, Cpeb4, Itm2c, and Sh3glb1 (data not shown). This suggests that when the phosphorylation of Sox2 at S39 is prevented, NSCs may become more prone to cell cycle withdrawal or apoptosis. Next, GO analysis also revealed that among the genes regulated by Sox2, only those required for neurogenesis were retained by Sox21–113-S39A (Fig. 6D, right side). The other notable GO terms in Sox2 (cell cycle and regulation of apoptotic process) were specifically lost in Sox21–113-S39A (Fig. 6D). Detailed comparisons indicated that these neuron-associated functions (Fig. 6F) were especially enriched in the 405 genes of Sox21–113-S39A that overlapped with Sox2 (Fig. 6C, dark green), with no significant differences in astrocyte and oligodendrocyte development. Examples of such genes include those for Hexb, Pou4f1, and even Sox2 itself. In addition, there was a partial overlap with Sox11 binding sites for Sox21–113-S39A (data not shown). This implies that upon neuronal differentiation, when Sox2 is cleaved by serine proteases and phosphorylation is lost due to the concomitant drop in Cdk kinase activity, the binding of Sox2 becomes concentrated to a subset of loci that control neurogenesis and displaced from all other regions, such as those required for stem cell maintenance. Our ChIPseq results thus provide evidence for the involvement of Sox21–113-S39A in a transcriptional program unique for the specification of the neuronal lineage.
Nonphosphorylated cleaved Sox2 promotes neurogenic commitment in vitro and in vivo.
Considering that the Sox21–113-S39A fragment remained bound to chromatin while the phosphorylated counterparts were excluded (Fig. 5), it is reasonable to assume that Sox21–113-S39A may have retained some degree of transactivation activity that was relinquished in Sox21–113 and Sox21–113-S39D. Furthermore, the subset of genes controlled by Sox2 after it was cleaved and dephosphorylated seemed to have switched from those involved in stem cell maintenance to those required for neuron formation. To corroborate these statements, we expressed the Sox21–113 fragment or its mutants (S39D or S39A) in NSCs and examined the mRNA levels of selected target genes. Compared to full-length Sox2 (Fig. 2A), Sox21–113 was easier to overexpress in NSCs, with transcript levels typically reaching 4-fold those of endogenous Sox2 (Fig. 7A). As described above, Sox2 levels in stem cells are tightly regulated (48, 49). Therefore, this relative ease of overexpression could indicate that the loss of the C-terminal region in Sox21–113, which contains the activation domain required for partner interaction (62, 63), may have compromised its ability to function in the maintenance of self-renewal. Indeed, the induction of Nmyc seen with the overexpression of full-length Sox2 (Fig. 2B) was diminished with Sox21–113 (Fig. 7B). However, when we analyzed the transcript level of NeuroG2, a proneural basic helix-loop-helix transcription factor required for neurogenesis (60), we were surprised to find a significant upregulation specific to Sox21–113-S39A (Fig. 7C). This is noteworthy since the activation of a neurogenic gene occurred under nondifferentiating culture conditions dedicated to the support of stem cell proliferation.
FIG 7.

Nonphosphorylated Sox21–113 drives neurogenesis in vitro and in vivo. (A to C) qPCR analysis of the expression levels of Sox2 (A), Nmyc (B), and NeuroG2 (C) in NSCs overexpressing the fragment Sox21–113 or its mutants (S39D or S39A) under conditions where proliferation is favored. Each column represents the average fold change compared to vector control (n = 2 to 4) after normalization to β-actin, Hsp90, and cyclophilin A expression. *, P < 0.05. (D to F) qPCR analysis of the expression levels of Gfap (D), NeuroD1 (E), and NeuroG2 (F) in NSCs overexpressing the fragment Sox21–113 or its mutants (S39D or S39A) and differentiated with 10% FBS for 6 days. Each column represents the average fold change compared to vector control (n = 2 or 3) after normalization to β-actin, Hsp90, and cyclophilin A expression. *, P < 0.05. (G to I) Representative fluorescent images from in vivo mouse brain electroporation of control (G, RFP), Sox21–113-S39A (H), and Sox21–113-WT (I) expression vectors. An RFP construct (RFPnls) was coinjected to identify cells that have taken up the plasmids (red). Tbr2 staining (white) was included so that boundaries could be drawn between the ventricular zone (VZ), subventricular zone (SVZ), intermediate zone (IZ), and cortical plate (CP). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue) in the merged image. Scale bar represents 25 μm in all panels. (J) Quantification of RFPnls+ Tbr2− cells in the VZ (AP), RFPnls+ Tbr2+ cells in the VZ or SVZ (BP), and RFPnls+ Tbr2− cells in the SVZ, IZ, or CP (N), as in panels G to I. Vec-con, control. n = 3 or 4. AP, apical progenitors; BP, basal progenitors; N, neurons.
The consequences of these underlying gene expression changes were explored under differentiating culture conditions, similar to those for Fig. 2. NSCs overexpressing the phosphorylated equivalents Sox21–113 and Sox21–113-S39D responded similarly to the vector control (Fig. 7D to F), suggesting that the truncation eliminated the inhibitory effect full-length Sox2 has on differentiation (compare to Fig. 2D to F). This is consistent with the lack of induction of genes involved in self-renewal, such as the Nmyc gene (Fig. 7B). In contrast, NSCs infected with Sox21–113-S39A promptly generated neurons marked by the expression of NeuroD1 and NeuroG2 (Fig. 7E and F), while the production of astrocytes marked by the expression of Gfap was not affected (Fig. 7D). This is consistent with the seemingly inherent predisposition toward neuronal differentiation already detected in the stem cell state (Fig. 7C). Therefore, it appears that the cleavage of Sox2 not only diminishes its function in the maintenance of self-renewal but also favors neurogenic commitment depending on the phosphorylation status of S39.
To test whether truncated and dephosphorylated Sox2 is capable of inducing neuronal differentiation not only in vitro but also in vivo, we performed in utero electroporation of mouse embryos (36, 64) at E13.5 with Sox21–113-WT, Sox21–113-S39A, or control (RFP) expression vectors (Fig. 7G to I). Brains were collected 24 h later and the distribution of electroporated cells and their progeny was determined by the expression of a coelectroporated RFP vector harboring a nuclear localization signal (RFPnls) to reliably count individual cells.
To identify the boundaries between the ventricular zone (VZ), subventricular zone (SVZ), and intermediate zone (IZ), we used Tbr2 as a marker of intermediate/basal progenitors (65). This was important because electroporation targets primarily, if not exclusively, apical radial glial progenitors (AP) in the VZ (36, 64). Following electroporation, AP may switch from proliferative to differentiative divisions to generate basal progenitors (BP) that migrate to the SVZ, while, finally, BP divide to generate neurons (N) that transit through the IZ to form the cortical plate (CP). Hence, the redistribution of electroporated cells and their progeny from the VZ to the SVZ, IZ, and CP can be used as a direct readout of NSC differentiation during cortical development (36, 64).
Staining of Tbr2 provided us the possibility to distinguish AP from BP, in particular in the VZ, where these two cell types are intermingled. Specifically, RFPnls+ cells upon electroporation with Sox21–113-WT, Sox21–113-S39A, or control plasmids were divided into three groups: (i) Tbr2− cells in the VZ, (ii) Tbr2+ cells in the VZ or SVZ, and (iii) Tbr2− cells in the SVZ and cells in the IZ or CP irrespective of Tbr2 expression, representing AP, BP, and N, respectively (36, 64). Expression of Sox21–113-WT or Sox21–113-S39A did not affect the proportion of AP (Fig. 7J), but there was a slight decrease in BP as a result of Sox21–113-S39A expression. This decrease is not statistically significant but indicates differences between the WT and S39A Sox2. Nevertheless, there was a statistically significant increase in the population of neurons following forced Sox21–113-S39A expression compared to Sox21–113-WT or control, and this confirmed our previous observations in vitro (Fig. 7E and F) suggesting that Sox21–113-S39A promotes neurogenesis (Fig. 7J). It was also evident that ectopic expression of Sox21–113-S39A promoted the formation of neurons at the expense of the overall abundance of progenitors, although the statistical dispersion of individual AP and BP values did not allow us to assess the relative contribution of one specific progenitor pool relative to the other in the total neurogenic effect of Sox21–113-S39A (Fig. 7J). Overall, the net effect of Sox21–113-S39A expression is an increase in the formation of neurons. When we electroporated full-length Sox2, we detected an increase in AP, a decrease in BP, and no changes in neuron formation (Fig. 2L). These effects in the in vivo electroporations are comparable to what we have observed in NSCs under nondifferentiating conditions that favor self-renewal (Fig. 2A to C). Since we analyzed the results of the electroporations after 24 h, this is a phase where cells are still proliferating, which probably resembles more closely self-renewing rather than differentiating NSCs.
In conclusion, our data suggest that the truncated product of Sox2 is functionally active in vitro and in vivo and is likely to be involved in neurogenic commitment during embryogenesis, but only if S39 is dephosphorylated. Therefore, phosphorylation of Sox2 by Cdks at S39 (and other sites) regulates its functions during differentiation.
DISCUSSION
In this study, we uncovered that Cdk1/2 directly phosphorylate Sox2 on S39, and we propose that the levels of phospho-S39-Sox2 is used by NSCs as a measurement of the level of Cdk kinase activity to determine the mode of division (proliferative versus differentiative). In actively cycling NSCs with high Cdk kinase activity, phospho-S39-Sox2 accumulates, which drives the expression of genes required for cell cycle progression, cell survival, and stem cell maintenance. As neurodevelopment proceeds and Cdk kinase activity declines in committed progenitors, the shift toward nonphosphorylated Sox2 and the neurogenesis-specific cleavage of Sox2 results in the upregulation of proneural genes that drive neuron formation (Fig. 8). Our study therefore presents a novel mechanism through which components of the cell cycle machinery participate in the specification of neural cell fate via their activities on a master transcriptional regulator, Sox2, with functions in both self-renewal and differentiation.
FIG 8.

Phosphorylation of Sox2 regulates its function in the transition between cell states. A model of the functions of Sox2 in self-renewal of stem cells and during neurogenesis is shown. Dark blue ovals, full-length Sox2; light blue ovals with dotted outline, full-length Sox2 with reduced binding; cS, cleaved Sox2; empty halved oval with dashed outline, cS with no binding.
Phosphorylation of Sox2 by Cdks provides a missing molecular link to explain the role of cell cycle regulators in neuronal differentiation.
The cell cycle length hypothesis was proposed as a possible explanation for the observed correlation between cell cycle lengthening and neurogenesis (66). In this model, a prolonged G1 phase was suggested to allow cell fate determinants to accumulate to sufficient levels for the induction of differentiation. In fact, as recently proposed (67), cell cycle regulators and cell fate determinants did likely coevolve during speciation to play mutually interdependent roles. Yet this molecular coordination of cell cycle regulators and cell fate determinants was only inferred, with direct experimental evidence supporting this notion being limited.
A notable exception to this lack of evidence is a study demonstrating that Cdks phosphorylate the proneural transcription factor neurogenin 2 (Ngn2), thus inhibiting its binding to E box DNA and suppressing its neurogenic activity during proliferative divisions (68). When cell cycle length increases and Cdk kinase activity drops, hypophosphorylated Ngn2 with enhanced promoter occupancy accumulates to drive the expression of genes required for neuronal differentiation, including NeuroD. In the present study, we extended these findings by demonstrating that Cdks control the activity of a second critical cell fate determinant, Sox2, further supporting the hypothesis that cell cycle regulators and cell fate determinants play coordinated and mutually interdependent roles during somatic stem cell differentiation. By keeping neurogenic gene occupancy strictly limited to the dephosphorylated version of Sox2, we have uncovered an important failsafe mechanism that prevents commitment to the neuronal lineage until the cell is certain that Cdk kinase activity is sufficiently low, since cell cycle activity in neurons is known to cause apoptosis (69). Given that neurogenic gene induction is additionally coupled to the cleavage of Sox2 by serine proteases that are likely to be activated in response to external differentiative cues, we suggest that it is the synergistic cooperation between the control that Cdks have over the molecular activity of cell fate determinants and the G1 duration required for their accumulation that determines the acquisition of a neuronal cell fate.
Insights into the sensitivity of neurons to reduced levels of Sox2.
While we propose that the ability of Sox2 to modulate neurogenesis is mediated through the formation of a dephosphorylated cleavage product, it is important to note that this product is only detectable during the initial phases of neuron induction and the affinity for DNA is reduced compared to that of full-length Sox2. These findings may in part explain the phenotypes seen in hypomorphic mutants retaining less than one-third of Sox2 expression, where stem cell proliferation was not affected but neuronal differentiation was severely compromised (20). The residual levels of Sox2 may still be sufficient for the expression of genes required for stem cell maintenance, since full-length Sox2 is mainly localized on chromatin, but may be insufficient for the induction of neurogenic genes, since (i) cleavage occurs only transiently, and therefore, inadequate cleavage products are formed when the starting levels are reduced, and (ii) DNA binding capacity is less efficient than with the full-length version, which further diminishes the final amount targeted to neurogenic genes. Our study thus presents a possible explanation for the dose-dependency of Sox2 functions.
Conclusion.
In the present study, we demonstrated how phosphorylation of Sox2 at S39 is used as a sensor of Cdk kinase activity to balance neural progenitor maintenance and neuronal differentiation in coordination with cell cycle status. Phosphorylation of Sox2 enhances the expression of genes required for cell cycle progression and inhibition of apoptosis. Dephosphorylation and truncation of Sox2 refine binding to a subset of neurogenic genes. By modulating the levels of Sox2 phosphorylation, Cdks thus induce changes in gene expression patterns to specify cell fate. More importantly, S39 targeted by Cdks is exclusive to Sox2 and does not exist in any of the remaining 18 Sox family proteins analyzed, suggesting that it is a unique mechanism to regulate Sox2 activity without affecting other closely related family members, including those known to have roles in the CNS. Since Sox2, together with Oct4, Klf4, and c-Myc, reestablishes pluripotency in terminally differentiated cells (6–8) and its CNS partner, Brn2, together with other factors, converts fibroblasts into functional neurons (70–73), information acquired through deciphering how phosphorylation controls Sox2 activity may enable induced pluripotent stem (iPS) cells and induced neuronal (iN) cells to be produced with greater efficiency and bring us a step closer toward their prospective clinical application.
MATERIALS AND METHODS
Plasmid construction.
The full-length mouse Sox2 coding sequence (CDS) was PCR amplified from a cDNA library of NSCs using the primers PKO1704 (5′-GCG CGG ATC CGC CAC CAT GTA TAA CAT GAT-3′) and PKO1705 (5′-ATA TCT CGA GCA TGT GCG ACA GGG GCA GTG-3′) (underlining shows the beginning of the gene), gel purified, digested with BamHI/XhoI, and ligated into pMCs-puro retroviral vector (Cell Biolabs; RTV-041) containing a C-terminal myc tag between XhoI and NotI (PKB1219) to obtain pMCs-Sox2-myc-puro (PKB1221). At amino acid position 39 of Sox2, serine-to-aspartic acid (S39D; AGC-to-GAC) and serine-to-alanine (S39A; AGC-to-GCC) substitution mutations were introduced by PCR-based site-directed mutagenesis using the following primer sets: for S39D, PKO1742 (5′-GGC AAC CAG AAG AAC GAC CCG GAC CGC GTC A-3′) and PKO1743 (5′-TGA CGC GGT CCG GGT CGT TCT TCT GGT TGC C-3′), and for S39A, PKO1744 (5′-GCA ACC AGA AGA ACG CCC CGG ACC GCG TC-3′) and PKO1745 (5′-GAC GCG GTC CGG GGC GTT CTT CTG GTT GC-3′) (boldface indicates the site of mutation). PCR products were digested with DpnI before transformation of XL10-Gold ultracompetent bacterial cells (Stratagene; 200314) to obtain pMCs-Sox2-S39D-myc-puro (PKB1255) and pMCs-Sox2-S39A-myc-puro (PKB1256). Sox2 and Sox2-S39A were released from pMCs-myc-puro by BamHI/XhoI and transferred downstream of glutathione S-transferase (GST) in pGEX-6P-1 (GE Healthcare; 28-9546-48) to obtain pGEX-6P-1-Sox2 (PKB1257) and pGEX-6P-1-Sox2-S39A (PKB1258). Sox2 and Sox2-S39A were also subcloned into Δ13Tb (PKB1350 and PKB1352) by PCR amplification using PKO2270 (5′-GGG TCC ATG GAT AAC ATG ATG GAG ACG GAG CTG-3′) and PKO2271 (5′-ATA TGG ATC CTC ACA TGT GCG ACA GGG GCA GTG-3′), partial digestion with NcoI/BamHI (since Sox2 contains an internal NcoI-cut site), gel purification, and ligation of full-length inserts. Shorter versions of mouse Sox2, together with the mutant forms, were PCR amplified from full-length versions using the same forward primer, PKO1704 (5′-GCG CGG ATC CGC CAC CAT GTA TAA CAT GAT-3′), and the following reverse primers: for Sox21–235, PKO1848 (5′-ATA TCT CGA GGG GGG TGC CCT GCT GCG AGT-3′); for Sox21–159, PKO1849 (5′-ATA TCT CGA GCA TGC GCT GGT TCA CGC CCG-3′); and for Sox21–113, PKO1850 (5′-GCG CCT CGA GCC GGT ATT TAT AAT CCG GGT-3′). PCR products were digested with BamHI and XhoI and ligated into pMCs-myc-puro to obtain pMCs-Sox21–235-myc-puro (PKB1274), pMCs-Sox21–159-myc-puro (PKB1275), pMCs-Sox21–113-myc-puro (PKB1276), pMCs-Sox21–113-S39D-myc-puro (PKB1405), and pMCs-Sox21–113-S39A-myc-puro (PKB1406). Another truncated form of mouse Sox2 containing amino acids 33 to 141 (Sox233–141), which span the HMG domain, was cloned as previously described (58). S39D (PKB1402) and S39A (PKB1403) mutations of this plasmid were obtained as described above. All plasmids were validated through DNA sequencing using BigDye Terminator v3.1 and an Applied Biosystems 3730xl DNA analyzer.
Recombinant protein expression and purification.
BL21(DE3) competent bacterial cells (Stratagene; 200131) were transformed with pGEX-6P-1-Sox2 (PKB1257) or pGEX-6P-1-Sox2-S39A (PKB1258) for the expression of the respective GST fusion proteins. After cell growth to an optical density at 600 nm (OD600) of 0.6 in modified terrific broth containing 100 μg/ml of ampicillin, 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) was added to induce protein synthesis. Following another 4 h of growth at 25°C, bacterial cells were harvested and resuspended in lysis buffer (50 mM Tris-HCl [pH 7.5], 200 mM NaCl, 1% Triton X-100, 1 mM dithiothreitol [DTT], and 1:200 protease inhibitor cocktail set II [EMD Biosciences; 539132]). Resuspended cells were lysed in a French press at 18,000 lb/in2 and clarified by ultracentrifugation at 25,000 rpm for 45 min at 4°C. The supernatant was incubated with glutathione-Sepharose 4B (GE Healthcare; 17-0756-05) for 90 min at 4°C and washed twice with lysis buffer without protease inhibitors and twice with wash buffer (50 mM Tris-HCl [pH 7.5], 500 mM NaCl, 1% Triton X-100, and 1 mM DTT). GST fusion protein was eluted by competition with 10 mM reduced l-glutathione (Sigma; G4251), concentrated with an Amicon Ultra-15 centrifugal filter unit with an Ultracel-10 membrane (Millipore; UFC901024), quantified with bicinchoninic acid (BCA) protein assay (Thermo Scientific; 23225), and stored in 50 mM HEPES (pH 8), 10% glycerol, 150 mM NaCl, 1 mM DTT, and 1:200 protease inhibitor cocktail set II at −80°C. Expression and purification of Sox233–141, Sox233–141-S39D, and Sox233–141-S39A were performed as previously described (74).
In vitro phosphorylation assay.
Five micrograms of GST-Sox2, GST-Sox2-S39A, GST-Sox21–113, or GST-Sox21–113-S39A was incubated with 100 ng of Cdk/cyclin complexes, 30 μM ATP (Roche; 10127523001), and 5 μCi of [γ-32P]ATP (PerkinElmer; NEG502A) in buffer EB+++ (80 mM β-glycerophosphate [pH 7.3], 20 mM EGTA, 15 mM MgCl2, 1 mg/ml of ovalbumin, 1× protease inhibitors, and 10 mM DTT) for 30 min at room temperature. Expression, purification, and activation of Cdk2/cyclin A complexes were performed as previously described (75, 76). Other Cdk/cyclin complexes are commercially available: Cdk1/cyclin A (Cell Signaling; number 7477) and Cdk1/cyclin B (Cell Signaling; number 7518). In control samples, 3 μg of GST or 0.8 μg of histone H1 (Roche; 10223549001) was used as a substrate. Reactions were terminated with 1× SDS sample buffer, and products were boiled for 5 min and separated on a 12.5% polyacrylamide gel. The gel was subsequently fixed and stained in Coomassie blue and dried for 2 h at 80°C, and the levels of incorporated radioactivity were quantified with a PhosphorImager (Fujifilm; FLA-7000). For the anti-Sox2P-S39 antibody testing, 25 μg of GST-Sox2 or GST-Sox2-S39A was incubated with 100 ng of Cdk2/cyclin A complexes and 1 mM ATP in buffer EB+++ for 4 h at room temperature, with occasional vortexing. The reaction was terminated with 1× SDS sample buffer, boiled for 5 min, and separated on a 12.5% polyacrylamide gel.
Generation of anti-Sox2P-S39 antibody.
A phosphopeptide sequence containing an N-terminal cysteine followed by residues derived from Sox2 flanking phosphorylated serine 39 (CSAANNQKNS39PDRVKR) was synthesized by GL Biochem (Shanghai) Ltd. and coupled to Imject mariculture keyhole limpet hemocyanin (mcKLH; Thermo Scientific; 77600) with the cross-linker disuccinimidyl suberate (DSS; Thermo Scientific; 21555) at a ratio of 1 mg of phosphopeptide to 1.6 mg of mcKLH, according to the manufacturer's instructions. One microgram of mcKLH-conjugated phosphopeptide was mixed with Freud's complete adjuvant and injected subcutaneously into a rabbit at day 0. All subsequent immunizations at days 14, 35, 56, 77, 98, 133, 154, 175, 196, 217, and 238 were in the presence of Freud's incomplete adjuvant. The rabbit was bled at days 0, 49, 70, 91, 112, 147, 168, 189, 210, 231, and 252, and sera were stored at −80°C. The fourth bleed was affinity purified through a column prepared by covalently immobilizing 12.5 mg of phosphopeptide to 10 ml of SulfoLink coupling resin (Thermo Scientific; 20402) according to the manufacturer's instructions, and anti-phosphopeptide antibodies were eluted with 0.1 M glycine (pH 2.5) and 0.5 M NaCl. The eluate was neutralized with 1 M Tris (pH 12), concentrated with an Amicon Ultra-15 centrifugal filter unit with an Ultracel-10 membrane (Millipore; UFC901024), and quantified with a BCA protein assay (Thermo Scientific; 23225). Finally, antibodies were reconstituted to a final concentration of 3 mg/ml in phosphate-buffered saline (PBS) and 50% glycerol and stored at −20°C. Rabbits were treated humanely and according to the IACUC guidelines of the Biopolis Resource Center (number 150998).
Immunoblotting and immunocytochemistry.
Immunoblotting and immunocytochemistry were performed as previously described (35). For chromatin extraction, cells were incubated in ice-cold CSK buffer (10 mM HEPES [pH 7.4], 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 0.2% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.1 mM Na3VO4, and 10 μg/ml each of leupeptin, chymostatin, and pepstatin) for 5 min on ice and spun at 5,000 × g for 5 min at 4°C. The supernatant representing the soluble proteins was collected and stored at −80°C, and the pellet was washed twice with ice-cold CSK buffer. After the washes, the final pellet was lysed with ice-cold radioimmunoprecipitation assay (RIPA) buffer for 15 min to obtain the chromatin-bound proteins. Antibodies used in the present study include mouse anti-Sox2 (Cell Signaling; number 4195), rabbit anti-Sox2 (Cell Signaling; number 2748), rabbit anti-Sox2 (Abcam; ab59776), mouse anti-Hsp90 (BD Transduction; 610419), rabbit anti-Cdk1 (Santa Cruz; sc-954), mouse anti-Cdk2 (Santa Cruz; sc-6248), mouse anti-HA tag (Cell Signaling; number 2367), rabbit anti-HA tag (Cell Signaling; number 3724), mouse anti-Myc tag (Cell Signaling; number 2276), rabbit anti-Myc tag (Cell Signaling; number 2278), rabbit anti-NeuroD (Cell Signaling; number 4373), rabbit anti-Tbr2 (Abcam; ab23345), rabbit anti-histone H3 (Cell Signaling; number 4499), rabbit anti-cyclin A2 (Santa Cruz; sc-596), and mouse anti-p27 (BD Transduction; 610242).
Isolation and culture of MEFs and NSCs.
Isolation and culture of mouse embryonic fibroblasts (MEFs) and NSCs were performed as previously described (35). For the serum starvation experiment, MEFs were plated at high densities and allowed to grow to full confluence. The cells were then washed twice with PBS and serum starved in 0.1% FBS–DMEM for 72 h before collection for Western blot analysis. For the Cre-mediated recombination experiment, NSCs containing Cdk1 floxed alleles and ROSA26-CreERT2 were mechanically dissociated into single cells, grown in culture for 3 days, and treated with 20 ng/ml of 4-hydroxy-tamoxifen (Sigma; T5648) for another 3 days before collection for Western blot analysis. For the differentiation experiment, NSCs were mechanically dissociated into single cells and seeded onto poly-d-lysine coated 100-mm culture dishes (BD Biosciences; 356469) in NSC culture medium. The next day, 10% FBS–DMEM was added, and it was replaced every other day until the cells were collected for qPCR analysis 6 days postinduction. The directed neuronal differentiation experiment was performed as previously described (35). Mice were treated humanely and according to the IACUC guidelines of the Biopolis Resource Center (number 140927).
Retrovirus production and infection.
To prepare retroviruses for the infection of primary MEFs and NIH 3T3 cells, plasmids were transfected into the ecotrophic packaging cell line Platinum-E (Plat-E; Cell Biolabs; RV-101) using FuGENE6 (Roche; 11 988 387 001) at a transfection reagent/DNA ratio of 3:1 in Opti-MEM (Gibco; 31985). Six hours later, the transfection medium was replaced with 10% FBS–DMEM. Two days later, viral supernatant was harvested, passed through a 0.45-μm syringe filter, and added to MEFs or NIH 3T3 cells in the presence of 4 μg/ml of Polybrene (Sigma; H9268). Infection medium was removed 24 h postinfection, and after an additional 24 h of incubation, infected cells were selected with 5 μg/ml of puromycin (Calbiochem; 540411) for 3 days. Infected cells were subsequently maintained in 1 μg/ml of puromycin. For the infection of NSCs, Plat-E cells were transfected as before and retroviruses were harvested twice, at 48 h and 72 h posttransfection. Pooled viral supernatant was filtered, centrifuged at 25,000 rpm for 2 h at 4°C, and resuspended in NSC culture medium. Infection of NSCs was conducted through spinoculation in 6-well plates at 1,200 × g for 2 h at 15°C (77) in the presence of 4 μg/ml of Polybrene. The same procedure was repeated again the next day. Forty-eight hours postinfection, NSCs were trypsinized and transferred to NSC culture medium. Once neurospheres reformed, they were mechanically dissociated into single cells and treated with 0.2 μg/ml of puromycin for the selection of cells that have acquired the puromycin plasmid.
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed using a 5′-, Cy5-labeled double-stranded DNA (dsDNA) probe synthesized by Sigma Proligo, Singapore, and annealed through heating an equimolar mixture of complementary strands to 95°C in 50 mM MgCl2, 50 mM KCl, and 20 mM Tris-HCl, followed by gradual cooling to ambient temperature over at least 5 h (58). The sequence of the forward oligonucleotide is as follows: 5′-GGC GCG GCA TTG TCA TGC AAA TCG GCG GCG-3′; the sequence contains both optimal Sox and Oct binding sites, as has been described previously (59). In the binding assays, 1 nM dsDNA probe was mixed with various concentrations of protein in a buffer containing 10 mM Tris-HCl (pH 8), 0.1 mg/ml of bovine serum albumin (BSA), 50 μM ZnCl2 100 mM KCl, 10% glycerol, 0.1% NP-40, and 2 mM β-mercaptoethanol and incubated for 1 h at 4°C in the dark. Bound and unbound probes were subsequently separated at 4°C on a prerun 12% 1× Tris-glycine polyacrylamide gel in 1× Tris-glycine buffer for 30 min at 200 V. Fluorescence was detected using a Typhoon 9140 PhosphorImager (Amersham Biosciences).
RNA analysis.
RNA extraction, first-strand cDNA synthesis, and qPCR were performed as previously described (35). Primer sequences are as follows: Sox2 set 1, PKR99 (forward [F], CAT GAA CGG CTG GAG CAA CG; reverse [R], GCG AGC TGG TCA TGG AGT TG); Sox2 set 2, PKR234 (F, ACC GGC GGC AAC CAG AAG AA; R, AAG TTT CCA CTC CGC GCC CA); N-myc, PKR227 (F, GTG TCT GTT CCA GCT ACT GC; R, CCT CAT CAT CTG AGT CGC TC); Neurog2, PKR159 (F, GCA TCA AGA AGA CCC GCA GG; R, CAG CGC GGC GTT TAG GTT GT); GFAP, PKR103 (F, AGG GCC AAA GCC TCA AGG AG; R, TGC GGT TTT CTT CGC CCT CC); NeuroD1 set 1, PKR137 (F, CCC TCG GAC TTT CTT GCC TG; R, GGG ACT GGT AGG AGT AGG GA); NeuroD1 set 2, PKR205 (F, ATA GAG ACA CTG CGC TTG GC; R, ATT GGT AGT GGG CTG GGA CA); NeuroD1 set 3, PKR206 (F, TGG ACA GCT CCC ACG TCT TC; R, CGT CAA AGG AAG GGC TGG TG); Rbpj set 1, PKR216 (F, ATC GGC GGG GAA GTT TGG TGA; R, GAG CAC TGT TTG ATC CCC TCG); Rbpj set 2, PKR219 (F, GCA AAA GTT GCA CAG AAG TC; R, CCT ATT CCA ATA AAC GCA CA); Gli2 set 1, PKR220 (F, CAT CTG AAA GAG AGG GGA CT; R, GGT CAC ACG TGG ACT AGA GA); Gli2 set 2, PKR224 (F, AAC CCT GTG GAT GCA TCA CG; R, TCA TGC GTT GTA GGT CGA GG); Gli3 set 1, PKR221 (F, CTT GCC CTT CAT TAG GAT CT; R, CAG AGC CAT CTG GTG ATA GT); Gli3 set 2, PKR225 (F, TGG AGC CTC ACT ACC ACC CT; R, CAA GTC TGG ATA CGT CGG GC); Jag1 set 1, PKR217 (F, TTG GCT GCA ATA AGT TCT GT; R, TGC AGT CAC CTG GAA GTT TA); Jag1 set 2, PKR223 (F, CCC CAC GTG TTC CAC AAA CA; R, GCC TCG CAC TCA TTT GCA TC); Tulp3 set 1, PKR222 (F, AAG CCT CAG GTT CTC TCT GT; R, GCT CCT CGT CAT AGT TCA CA); and Tulp3 set 2, PKR226 (F, AGC GAC GTC ATC CTA CAC GG; R, CCT TCC GTG CCT TCT TCT GG). All data were normalized to the expression levels of three housekeeping genes, the genes for the following: β-actin, PKR143 (F, ACG GCT CCG GCA TGT GCA AA; R, TTC CCA CCA TCA CAC CCT GG); Hsp90, PKR144 (F, ACT GCT CTG CTC TCC TCT GG; R, CAT CGA TGC CCA GGC CTA GT); and cyclophilin A, PKR195 (F, CCT TGG GCC GCG TCT CCT T; R, CAC CCT GGC ACA TGA ATC CTG).
ChIPseq.
Chromatin immunoprecipitation was performed as previously described (78). Myc-tagged proteins were immunoprecipitated using anti-myc-tag antibodies (Cell Signaling; number 2276). Statistical significance was estimated by the hypergeometric distribution function in R and further corrected using the Benjamini-Hochberg method.
In utero electroporation.
Pregnant mice were isoflurane anesthetized at E13.5, and 1 to 3 μl of PBS containing an equimolar mixture of Sox21–113-S39A and RFPnls vectors (ca. 1 to 3 mg/ml each) was coinjected into the lumen of the telencephalon, followed by 6 pulses of 30 V for 50 ms each at 1-s intervals delivered through platinum electrodes using a BTX-830 electroporator (Genetronics) as previously described (36, 64). Statistical significance was estimated by a two-tailed t test with unequal variance. Mice were treated humanely and according to the IACUC guidelines of TU-Dresden (24D-9168.11-1/2008-16 and 2011-41).
Accession number(s).
The raw data for the chromatin immunoprecipitation-sequencing (ChIPseq) experiments have been deposited at NCBI and can be accessed at https://www.ncbi.nlm.nih.gov/bioproject with accession number PRJNA371527.
ACKNOWLEDGMENTS
We deeply appreciate the support and encouragement provided by all members of the Kaldis lab. We thank June Wang, Vithya Anantaraja, and Chloe Sim for help with animal care, Sheena Wee and Jayantha Gunaratne for mass spectrometry, Jiaxuan Chen for ChIP-sequencing, Calista K. L. Ng, Ralf Jauch, and Prasanna R. Kolatkar for EMSA, Araki Marito for the in vitro ubiquitylation assay, Xavier Bisteau for help with the preparation of the final figures, and Wei Theng Poh for chromatin extraction. We acknowledge the technical expertise provided by the Advanced Molecular Pathology Laboratory at IMCB.
The authors contributed to this article as follows: S.L., conception and design, collection and/or assembly of data, data analysis and interpretation, and manuscript writing; A.B., I.A., and L.W.S., data collection, data analysis, and interpretation for ChIP-sequencing; J.A., S.B.A., and F.C., data collection, data analysis and interpretation, and manuscript writing for in vivo mouse brain electroporation; C.F.C., discussion; P.K., conception and design, data analysis and interpretation, manuscript writing, financial support, and direction of the study.
This work was supported by the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research), Singapore.
We declare that we have no competing interests.
All data needed to evaluate the conclusions in this paper are presented here. Additional data related to this work may be requested from the authors.
REFERENCES
- 1.Wegner M. 1999. From head to toes: the multiple facets of Sox proteins. Nucleic Acids Res 27:1409–1420. doi: 10.1093/nar/27.6.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. 2003. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan H, Corbi N, Basilico C, Dailey L. 1995. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev 9:2635–2645. doi: 10.1101/gad.9.21.2635. [DOI] [PubMed] [Google Scholar]
- 4.Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. 2006. Dissecting self-renewal in stem cells with RNA interference. Nature 442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 5.Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, Okochi H, Okuda A, Matoba R, Sharov AA, Ko MSH, Niwa H. 2007. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 9:625–635. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- 6.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 9.Rex M, Orme A, Uwanogho D, Tointon K, Wigmore PM, Sharpe PT, Scotting PJ. 1997. Dynamic expression of chicken Sox2 and Sox3 genes in ectoderm induced to form neural tissue. Dev Dyn 209:323–332. doi:. [DOI] [PubMed] [Google Scholar]
- 10.Pevny L, Rao MS. 2003. The stem-cell menagerie. Trends Neurosci 26:351–359. doi: 10.1016/S0166-2236(03)00169-3. [DOI] [PubMed] [Google Scholar]
- 11.Zappone MV, Galli R, Catena R, Meani N, De Biasi S, Mattei E, Tiveron C, Vescovi AL, Lovell-Badge R, Ottolenghi S, Nicolis SK. 2000. Sox2 regulatory sequences direct expression of a (beta)-geo transgene to telencephalic neural stem cells and precursors of the mouse embryo, revealing regionalization of gene expression in CNS stem cells. Development 127:2367–2382. [DOI] [PubMed] [Google Scholar]
- 12.Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, McMahon A, Rao M, Pevny L. 2004. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci 26:148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- 13.Graham V, Khudyakov J, Ellis P, Pevny L. 2003. SOX2 functions to maintain neural progenitor identity. Neuron 39:749–765. doi: 10.1016/S0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 14.Suh H, Consiglio A, Ray J, Sawai T, D'Amour KA, Gage FH. 2007. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell 1:515–528. doi: 10.1016/j.stem.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Archer TC, Jin J, Casey ES. 2011. Interaction of Sox1, Sox2, Sox3 and Oct4 during primary neurogenesis. Dev Biol 350:429–440. doi: 10.1016/j.ydbio.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bylund M, Andersson E, Novitch BG, Muhr J. 2003. Vertebrate neurogenesis is counteracted by Sox1-3 activity. Nat Neurosci 6:1162–1168. doi: 10.1038/nn1131. [DOI] [PubMed] [Google Scholar]
- 17.Collignon J, Sockanathan S, Hacker A, Cohen-Tannoudji M, Norris D, Rastan S, Stevanovic M, Goodfellow PN, Lovell-Badge R. 1996. A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development 122:509–520. [DOI] [PubMed] [Google Scholar]
- 18.Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. 2006. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev 20:1187–1202. doi: 10.1101/gad.1407906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyagi S, Masui S, Niwa H, Saito T, Shimazaki T, Okano H, Nishimoto M, Muramatsu M, Iwama A, Okuda A. 2008. Consequence of the loss of Sox2 in the developing brain of the mouse. FEBS Lett 582:2811–2815. doi: 10.1016/j.febslet.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Cavallaro M, Mariani J, Lancini C, Latorre E, Caccia R, Gullo F, Valotta M, DeBiasi S, Spinardi L, Ronchi A, Wanke E, Brunelli S, Favaro R, Ottolenghi S, Nicolis SK. 2008. Impaired generation of mature neurons by neural stem cells from hypomorphic Sox2 mutants. Development 135:541–557. doi: 10.1242/dev.010801. [DOI] [PubMed] [Google Scholar]
- 21.Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S, Pandolfi PP, Sala M, DeBiasi S, Nicolis SK. 2004. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- 22.Heinrich C, Bergami M, Gascon S, Lepier A, Vigano F, Dimou L, Sutor B, Berninger B, Gotz M. 2014. Sox2-mediated conversion of NG2 glia into induced neurons in the injured adult cerebral cortex. Stem Cell Rep 3:1000–1014. doi: 10.1016/j.stemcr.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuruzoe S, Ishihara K, Uchimura Y, Watanabe S, Sekita Y, Aoto T, Saitoh H, Yuasa Y, Niwa H, Kawasuji M, Baba H, Nakao M. 2006. Inhibition of DNA binding of Sox2 by the SUMO conjugation. Biochem Biophys Res Commun 351:920–926. doi: 10.1016/j.bbrc.2006.10.130. [DOI] [PubMed] [Google Scholar]
- 24.Van Hoof D, Munoz J, Braam SR, Pinkse MW, Linding R, Heck AJ, Mummery CL, Krijgsveld J. 2009. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell 5:214–226. doi: 10.1016/j.stem.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Savare J, Bonneaud N, Girard F. 2005. SUMO represses transcriptional activity of the Drosophila SoxNeuro and human Sox3 central nervous system-specific transcription factors. Mol Biol Cell 16:2660–2669. doi: 10.1091/mbc.E04-12-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernández-Lloris R, Osses N, Jaffray E, Shen LN, Vaughan OA, Girwood D, Bartrons R, Rosa JL, Hay RT, Ventura F. 2006. Repression of SOX6 transcriptional activity by SUMO modification. FEBS Lett 580:1215–1221. doi: 10.1016/j.febslet.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 27.Girard M, Goossens M. 2006. Sumoylation of the SOX10 transcription factor regulates its transcriptional activity. FEBS Lett 580:1635–1641. doi: 10.1016/j.febslet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 28.Baltus GA, Kowalski MP, Zhai H, Tutter AV, Quinn D, Wall D, Kadam S. 2009. Acetylation of Sox2 induces its nuclear export in embryonic stem cells. Stem Cells 27:2175–2184. doi: 10.1002/stem.168. [DOI] [PubMed] [Google Scholar]
- 29.Jeong CH, Cho YY, Kim MO, Kim SH, Cho EJ, Lee SY, Jeon YJ, Lee KY, Yao K, Keum YS, Bode AM, Dong Z. 2010. Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells 28:2141–2150. doi: 10.1002/stem.540. [DOI] [PubMed] [Google Scholar]
- 30.Fang L, Zhang L, Wei W, Jin X, Wang P, Tong Y, Li J, Du JX, Wong J. 2014. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol Cell 55:537–551. doi: 10.1016/j.molcel.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 31.Lim S, Kaldis P. 2013. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140:3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi T, Nowakowski RS, Caviness VS. 1995. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci 15:6046–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calegari F, Haubensak W, Haffner C, Huttner WB. 2005. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci 25:6533–6538. doi: 10.1523/JNEUROSCI.0778-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calegari F, Huttner WB. 2003. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci 116:4947–4955. doi: 10.1242/jcs.00825. [DOI] [PubMed] [Google Scholar]
- 35.Lim S, Kaldis P. 2012. Loss of Cdk2 and Cdk4 induces a switch from proliferation to differentiation in neural stem cells. Stem Cells 30:1509–1520. doi: 10.1002/stem.1114. [DOI] [PubMed] [Google Scholar]
- 36.Lange C, Huttner WB, Calegari F. 2009. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5:320–331. doi: 10.1016/j.stem.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 37.Hindley C, Ali F, McDowell G, Cheng K, Jones A, Guillemot F, Philpott A. 2012. Post-translational modification of Ngn2 differentially affects transcription of distinct targets to regulate the balance between progenitor maintenance and differentiation. Development 139:1718–1723. doi: 10.1242/dev.077552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalton S. 2015. Linking the cell cycle to cell fate decisions. Trends Cell Biol 25:592–600. doi: 10.1016/j.tcb.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dinkel H, Van Roey K, Michael S, Kumar M, Uyar B, Altenberg B, Milchevskaya V, Schneider M, Kühn H, Behrendt A, Dahl SL, Damerell V, Diebel S, Kalman S, Klein S, Knudsen AC, Mäder C, Merrill S, Staudt A, Thiel V, Welti L, Davey NE, Diella F, Gibson TJ. 2016. ELM 2016—data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44:D294–D300. doi: 10.1093/nar/gkv1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nigg EA. 1993. Cellular substrates of p34cdc2 and its companion cyclin-dependent kinases. Trends Cell Biol 3:296–301. doi: 10.1016/0962-8924(93)90011-O. [DOI] [PubMed] [Google Scholar]
- 41.Holmes JK, Solomon MJ. 1996. A predictive scale for evaluating cyclin-dependent kinase substrates. A comparison of p34cdc2 and p33cdk2. J Biol Chem 271:25240–25246. [DOI] [PubMed] [Google Scholar]
- 42.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. 2009. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct Biol 9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang J, Yu W, Liu J, Zhang N, Florens L, Chen J, Liu H, Washburn M, Pei D, Xie T. 2015. Cyclin-dependent kinase-mediated Sox2 phosphorylation enhances the ability of Sox2 to establish the pluripotent state. J Biol Chem 290:22782–22794. doi: 10.1074/jbc.M115.658195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, Kaldis P. 2012. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci U S A 109:3826–3831. doi: 10.1073/pnas.1115201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vooijs M, Jonkers J, Berns A. 2001. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep 2:292–297. doi: 10.1093/embo-reports/kve064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kopp JL, Ormsbee BD, Desler M, Rizzino A. 2008. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 26:903–911. doi: 10.1634/stemcells.2007-0951. [DOI] [PubMed] [Google Scholar]
- 47.Boer B, Kopp J, Mallanna S, Desler M, Chakravarthy H, Wilder PJ, Bernadt C, Rizzino A. 2007. Elevating the levels of Sox2 in embryonal carcinoma cells and embryonic stem cells inhibits the expression of Sox2:Oct-3/4 target genes. Nucleic Acids Res 35:1773–1786. doi: 10.1093/nar/gkm059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hagey DW, Muhr J. 2014. Sox2 acts in a dose-dependent fashion to regulate proliferation of cortical progenitors. Cell Rep 9:1908–1920. doi: 10.1016/j.celrep.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 49.Hutton SR, Pevny LH. 2011. SOX2 expression levels distinguish between neural progenitor populations of the developing dorsal telencephalon. Dev Biol 352:40–47. doi: 10.1016/j.ydbio.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 50.Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, Szumska D, Kockx C, van Ijcken W, Dekkers DH, Demmers J, Rijkers EJ, Bhattacharya S, Philipsen S, Pevny LH, Grosveld FG, Rottier RJ, Lenhard B, Poot RA. 2011. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 43:607–611. doi: 10.1038/ng.825. [DOI] [PubMed] [Google Scholar]
- 51.Peltier J, Conway A, Keung AJ, Schaffer DV. 2011. Akt increases sox2 expression in adult hippocampal neural progenitor cells, but increased sox2 does not promote proliferation. Stem Cells Dev 20:1153–1161. doi: 10.1089/scd.2010.0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D. 1999. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 208:166–188. doi: 10.1006/dbio.1998.9192. [DOI] [PubMed] [Google Scholar]
- 53.Bani-Yaghoub M, Tremblay RG, Lei JX, Zhang D, Zurakowski B, Sandhu JK, Smith B, Ribecco-Lutkiewicz M, Kennedy J, Walker PR, Sikorska M. 2006. Role of Sox2 in the development of the mouse neocortex. Dev Biol 295:52–66. doi: 10.1016/j.ydbio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 54.Liu L, Michowski W, Inuzuka H, Shimizu K, Nihira NT, Chick JM, Li N, Geng Y, Meng AY, Ordureau A, Kolodziejczyk A, Ligon KL, Bronson RT, Polyak K, Harper JW, Gygi SP, Wei W, Sicinski P. 2017. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat Cell Biol 19:177–188. doi: 10.1038/ncb3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inoue M, Kamachi Y, Matsunami H, Imada K, Uchikawa M, Kondoh H. 2007. PAX6 and SOX2-dependent regulation of the Sox2 enhancer N-3 involved in embryonic visual system development. Genes Cells 12:1049–1061. doi: 10.1111/j.1365-2443.2007.01114.x. [DOI] [PubMed] [Google Scholar]
- 56.Miyagi S, Nishimoto M, Saito T, Ninomiya M, Sawamoto K, Okano H, Muramatsu M, Oguro H, Iwama A, Okuda A. 2006. The Sox2 regulatory region 2 functions as a neural stem cell-specific enhancer in the telencephalon. J Biol Chem 281:13374–13381. doi: 10.1074/jbc.M512669200. [DOI] [PubMed] [Google Scholar]
- 57.Tomioka M, Nishimoto M, Miyagi S, Katayanagi T, Fukui N, Niwa H, Muramatsu M, Okuda A. 2002. Identification of Sox-2 regulatory region which is under the control of Oct-3/4-Sox-2 complex. Nucleic Acids Res 30:3202–3213. doi: 10.1093/nar/gkf435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jauch R, Aksoy I, Hutchins AP, Ng CK, Tian XF, Chen J, Palasingam P, Robson P, Stanton LW, Kolatkar PR. 2011. Conversion of Sox17 into a pluripotency reprogramming factor by reengineering its association with Oct4 on DNA. Stem Cells 29:940–951. doi: 10.1002/stem.639. [DOI] [PubMed] [Google Scholar]
- 59.Ng CK, Li NX, Chee S, Prabhakar S, Kolatkar PR, Jauch R. 2012. Deciphering the Sox-Oct partner code by quantitative cooperativity measurements. Nucleic Acids Res 40:4933–4941. doi: 10.1093/nar/gks153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertrand N, Castro DS, Guillemot F. 2002. Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- 61.Lodato MA, Ng CW, Wamstad JA, Cheng AW, Thai KK, Fraenkel E, Jaenisch R, Boyer LA. 2013. SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet 9:e1003288. doi: 10.1371/journal.pgen.1003288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ambrosetti DC, Scholer HR, Dailey L, Basilico C. 2000. Modulation of the activity of multiple transcriptional activation domains by the DNA binding domains mediates the synergistic action of Sox2 and Oct-3 on the fibroblast growth factor-4 enhancer. J Biol Chem 275:23387–23397. doi: 10.1074/jbc.M000932200. [DOI] [PubMed] [Google Scholar]
- 63.Nowling TK, Johnson LR, Wiebe MS, Rizzino A. 2000. Identification of the transactivation domain of the transcription factor Sox-2 and an associated co-activator. J Biol Chem 275:3810–3818. doi: 10.1074/jbc.275.6.3810. [DOI] [PubMed] [Google Scholar]
- 64.Artegiani B, Lange C, Calegari F. 2012. Expansion of embryonic and adult neural stem cells by in utero electroporation or viral stereotaxic injection. J Vis Exp 2012(68):4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. 2005. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salomoni P, Calegari F. 2010. Cell cycle control of mammalian neural stem cells: putting a speed limit on G1. Trends Cell Biol 20:233–243. [DOI] [PubMed] [Google Scholar]
- 67.Borrell V, Calegari F. 2014. Mechanisms of brain evolution: regulation of neural progenitor cell diversity and cell cycle length. Neurosci Res 86:14–24. doi: 10.1016/j.neures.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 68.Ali F, Hindley C, McDowell G, Deibler R, Jones A, Kirschner M, Guillemot F, Philpott A. 2011. Cell cycle-regulated multi-site phosphorylation of Neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. Development 138:4267–4277. doi: 10.1242/dev.067900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Herrup K, Yang Y. 2007. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 8:368–378. doi: 10.1038/nrn2124. [DOI] [PubMed] [Google Scholar]
- 70.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. 2010. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, Lipton SA, Ding S. 2011. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9:113–118. doi: 10.1016/j.stem.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, Wernig M. 2011. Induction of human neuronal cells by defined transcription factors. Nature 476:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, Dufour A, Bjorklund A, Lindvall O, Jakobsson J, Parmar M. 2011. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A 108:10343–10348. doi: 10.1073/pnas.1105135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ng CK, Palasingam P, Venkatachalam R, Baburajendran N, Cheng J, Jauch R, Kolatkar PR. 2008. Purification, crystallization and preliminary X-ray diffraction analysis of the HMG domain of Sox17 in complex with DNA. Acta Crystallogr Sect F Struct Biol Cryst Commun 64:1184–1187. doi: 10.1107/S1744309108038724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaldis P, Ojala PM, Tong L, Makela TP, Solomon MJ. 2001. CAK-independent activation of CDK6 by a viral cyclin. Mol Biol Cell 12:3987–3999. doi: 10.1091/mbc.12.12.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaldis P, Russo AA, Chou HS, Pavletich NP, Solomon MJ. 1998. Human and yeast cdk-activating kinases (CAKs) display distinct substrate specificities. Mol Biol Cell 9:2545–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu Y, Melton DW, Zhang Y, Hornsby PJ. 2009. Improved coinfection with amphotropic pseudotyped retroviral vectors. J Biomed Biotechnol 2009:901079. doi: 10.1155/2009/901079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhinge A, Poschmann J, Namboori SC, Tian X, Jia Hui Loh S, Traczyk A, Prabhakar S, Stanton LW. 2014. MiR-135b is a direct PAX6 target and specifies human neuroectoderm by inhibiting TGF-beta/BMP signaling. EMBO J 33:1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bergsland M, Ramskold D, Zaouter C, Klum S, Sandberg R, Muhr J. 2011. Sequentially acting Sox transcription factors in neural lineage development. Genes Dev 25:2453–2464. doi: 10.1101/gad.176008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hagey DW, Zaouter C, Combeau G, Lendahl MA, Andersson O, Huss M, Muhr J. 2016. Distinct transcription factor complexes act on a permissive chromatin landscape to establish regionalized gene expression in CNS stem cells. Genome Res 26:908–917. doi: 10.1101/gr.203513.115. [DOI] [PMC free article] [PubMed] [Google Scholar]