

ABSTRACT
Marburg virus (MARV) encodes a nucleoprotein (NP) to encapsidate its genome by oligomerization and form a ribonucleoprotein complex (RNP). According to previous investigation on nonsegmented negative-sense RNA viruses (nsNSV), the newly synthesized NPs must be prevented from indiscriminately binding to noncognate RNAs. During the viral RNA synthesis process, the RNPs undergo a transition from an RNA-bound form to a template-free form, to open access for the interaction between the viral polymerase and the RNA template. In filoviruses, this transition is regulated by VP35 peptide and other viral components. To further understand the dynamic process of filovirus RNP formation, we report here the structure of MARV NPcore, both in the apo form and in the VP35 peptide-chaperoned form. These structures reveal a typical bilobed structure, with a positive-charged RNA binding groove between two lobes. In the apo form, the MARV NP exists in an interesting hexameric state formed by the hydrophobic interaction within the long helix of the NPcore C-terminal region, which shows high structural flexibility among filoviruses and may imply critical function during RNP formation. Moreover, the VP35 peptide-chaperoned NPcore remains in a monomeric state and completely loses its affinity for single-stranded RNA (ssRNA). The structural comparison reveals that the RNA binding groove undergoes a transition from closed state to open state, chaperoned by VP35 peptide, thus preventing the interaction for viral RNA. Our investigation provides considerable structural insight into the filovirus RNP working mechanism and may support the development of antiviral therapies targeting the RNP formation of filovirus.
IMPORTANCE Marburg virus is one of the most dangerous viruses, with high morbidity and mortality. A recent outbreak in Angola in 2005 caused the deaths of 272 persons. NP is one of the most essential proteins, as it encapsidates and protects the whole virus genome simultaneously with self-assembly oligomerization. Here we report the structures of MARV NPcore in two different forms. In the MARV NP apo form, we identify an interesting hexamer formed by hydrophobic interaction within a long helix, which is highly conserved and flexible among filoviruses and may indicate its critical function during the virus RNP formation. Moreover, the structural comparison with the NP-VP35 peptide complex reveals a structural transition chaperoned by VP35, in which the RNA binding groove undergoes a transition from closed state to open state. Finally, we discussed the high conservation and critical role of the VP35 binding pocket and its potential use for therapeutic development.
KEYWORDS: Marburg virus, nucleoprotein, crystal structure, filovirus, assembly mechanism
INTRODUCTION
Marburg virus (MARV), as one of the most dangerous viruses in the world, causes convulsions and bleeding from the mucous membranes, skin, and organs with a fatality rate higher than 90%, similar to that of Ebola virus (EBOV) (1–3). Recently, MARV and EBOV have been listed by the WHO in Geneva, Switzerland, among the top 5 to 10 emerging pathogens likely to cause severe outbreaks in the near future, along with other viruses. The genera Ebolavirus, Marburgvirus, and Cuevavirus constitute the Filoviridae family (4), a typical family of the Mononegavirales order. Similar to other nonsegmented negative-strand RNA viruses (nsNSVs), filoviruses contain a single-stranded RNA (ssRNA) genome encoding a glycoprotein (GP), viral protein 30 (VP30) and VP35, a nucleocapsid protein (NP), an RNA-dependent RNA polymerase (RdRp), and a matrix protein (VP40) in the order of 3′-NP-VP-matrix-GP-RdRp-5′ (5). In the virus replication process, NP recognizes and encapsidates the virus RNA genome to form the ribonucleoprotein (RNP), which can help RNA escape from the cellular innate immune response, making it resistant to ribonucleases (6, 7), reduces potential double-stranded RNA (dsRNA) accumulation, and mediates the interaction between the virus genome and RdRp (8). RNP is relatively stable and can serve as a direct template for replication (9). Moreover, during the viral RNA synthesis, the RNPs undergo a transition from an RNA-bound form to a template-free form, to open access for the interaction between viral RdRp and the viral RNA template. Therefore, as NSVs evolve a mechanism for modulating this dynamic RNA-binding process, the multifunctional NP plays an essential role for the viral RNA synthesis machinery.
After years of NP structural investigation, several NSV nucleoprotein structures have been reported, such as Lassa fever virus (LAFV) (10, 11), Crimean-Congo hemorrhagic fever virus (CCHFV) (12, 13), influenza A virus (14), hantavirus (15), and SFTS virus (16) among segmented NSVs (sNSVs) and Borna disease virus (BDV) (17), vesicular stomatitis virus (VSV) (18, 19), respiratory syncytial virus (RSV) (20), parainfluenza virus 5 (PIV-5) (21), and Nipah virus (NiV) (22) among nonsegmented NSVs. Despite the structural differences among these nucleoproteins, most of the structures share a typical bilobe fold consisting of N- and C-terminal domains, with a positive-charged RNA binding groove between the two lobes. For some available oligomeric nucleoproteins, the N- and C-terminal arms (consisting mostly of helix or sheet) from each protomer extend and interact with an adjacent nucleoprotein for oligomerization. In addition, recent results have clearly demonstrated that viral NP could be directly used as the target for antiviral development (23, 24), raising great potential to find new antiviral agents with a novel mechanism against the drug resistance occurring in traditional antiviral drugs targeting highly mutable surface proteins.
Typically, a filovirus nucleocapsid consists of NP, RdRp, VP24, VP30, and VP35 (25–27). Besides the crucial function of VP35 in immune suppression (28, 29), another major function is mediating the interaction of NP and RdRp during virus replication and transcription (5, 25, 30). The VP35 homo-oligomerization mechanism, which is essential for the interaction with RdRp for its function, appears to be dispensable to NP function and interactions with other components (31). Investigations of the VP35 homolog phosphoprotein (P) showed a chaperoning function of nascent NP into a monomer and RNA-free state (N0) before encapsidating RNA and self-assembly multimerization, preventing NP from nonspecific aggregation (32, 33). The crystal structure of the NiV and VSV N-P complex also reveals the mechanism of the interaction between N and P N-terminal peptides (18, 19, 22), which is similar to the function of VP35 in the EBOV life cycle (34, 35).
During the 2014-2016 Ebola outbreak in West Africa, three independent research groups reported the crystal structures of EBOV NP in the apo form (36) and in the VP35 peptide-bound form (34, 35). The EBOV NP core domain possesses an N lobe and C lobe to clamp an RNA binding groove, presenting similarities with the structures of the other reported viral NPs encoded by the members of Mononegavirales order. In the VP35 peptide-bound form reported by two research groups, the differences in RNA binding affinity and oligomeric functions of respective constructs were examined. The most significant difference for these structures, located at the C-terminal region, compared with the structure of the EBOV VP35-NP34–367 fusion protein (34) is that the complex structure of EBOV VP35-NP25–457 presents a long U-turned helix from E338 to N384 (35), which mediates crystal contacts and may represent a physiological oligomerization interaction between NP protomers.
Despite previous efforts and investigations on the MARV NP working mechanism, it still remains elusive. Our interest in understanding the structures and functions of filovirus virus-encoded NPs, as well as the attempt to find a novel druggable target, prompted us to initiate this investigation.
RESULTS
Purification and characterization of MARV NPcore.
Our previous studies on EBOV NP have suggested that the first 450 amino acids contain the most stable core domain of MARV NP. We therefore studied this segment after the initial full-length trial failed. During production, we found that the N-terminal 19-amino-acid segment causes NP to oligomerize and precipitate, even under ultrahigh ionic strength conditions, which is consistent with previous work on EBOV (34–36). After testing a series of truncations (Fig. 1A), we found the best construct for MARV NP, which was NP19–370 (here named NPcore), with a high production yield and proper biochemical features. The MARV NPcore was produced in Escherichia coli and subsequently purified as a mixture of oligomer and monomer with the ratio of 3:1 (Fig. 1B). The A280/A260 UV absorption ratio of 1.67 demonstrated some nucleic acid binding with MARV NPcore. For understanding of the molecular mechanism for the conformational shifting of NP from a monomer to a high-ordered oligomer, we studied both fractions by crystallography.
FIG 1.

Purification and crystal structure of MARV nucleoprotein core domain. (A) Construction of MARV nucleoprotein. Representative truncations are presented, including a full-length construct of NPFL. The best truncation of MARV NP is NP19–370 (NPcore). (B) Purification of MARV nucleoprotein. The sample containing MARV NPcore was injected into a Superdex-200 column for size exclusion chromatography (SEC). The molecular masses of standard protein markers are shown on the top. The blue and red lines indicate A280 and A260, respectively. SDS-PAGE analysis of the peak fractions is shown in the inset. (C) Cartoon representation of the overall structure of MARV NPcore. Missing residues are linked by dotted lines. Four positive residues, K142A, K153A, R156A, and K230 (shown as sticks in yellow), are labeled on NP. The protruding last α-helix (α20) is labeled. (D) EMSA of MARV NPcore WT and four mutations with ssRNA. Site-directed K142A, K153A, R156A, and K230A mutation proteins are sample loaded along with the wild type (WT) after incubation with an ssRNA probe. The dosages of NP and ssRNA probe are all set at 0.2 nM and 0.1 nM, respectively, as final concentrations. The free and combined probes are labeled. SDS-PAGE analysis of the MARV NPcore WT and mutation fractions are shown in the inset. The fraction binding for each mutation (black bars) is indicated in a chart made by GraphPad Prism after intensity quantification of the film by ImageJ and normalization to WT (gray bar). Error bars represent the standard deviations (SD) from three independent replicates.
Overall structure of MARV NPcore.
The MARV NPcore oligomer produced a better crystal than that in the monomeric state. The oligomer crystal structure was finally solved, while the monomeric structure was limited to a low resolution (the best crystals diffract lower than 8 Å). The MARV NPcore structure was solved by molecular replacement methods using EBOV NPcore (PDB 4Z9P) as the initial search model, and ultimately, the structure was refined to a resolution of 2.9 Å (Table 1) (Fig. 1C). Most residues in the MARV NP polypeptide were built into the final model, except some short gaps (residues E120, G206, A314, G315, V318, and G319).
TABLE 1.
Data collection and refinement statisticsc
| Parameter | Native NPcore | VP35 complex |
|---|---|---|
| Data collection statistics | ||
| Cell parameters | ||
| a (Å) | 147.96 | 98.396 |
| b (Å) | 147.96 | 98.396 |
| c (Å) | 58.98 | 95.753 |
| γ (°) | α = β = 90°, γ = 120° | α = β = 90°, γ = 120° |
| Space group | P321 | P32 |
| Wavelength used (Å) | 0.9785 | 0.9785 |
| Resolution (Å) | 50.00–2.90 (2.95–2.90) | 50.00–2.20 (2.24–2.20) |
| No. of all reflections | 477,991 | 1,008,672 |
| No. of unique reflections | 16,663 (833) | 52,682 (2,624) |
| Completeness (%) | 99.9 (99.6) | 100 (100) |
| Average I/σ(I) | 27.75 (4.25) | 36.78 (4.20) |
| Redundancy | 10.8 (10.6) | 10.3 (8.9) |
| Rmergea (%) | 6.3 (66.9) | 4.3 (53.5) |
| Rpim (%) | 2.7 (21.9) | 2.1 (22.0) |
| CC 1/2 | 0.996 (0.894) | 0.998 (0.926) |
| Wilson B factor | 43.44 | 24.88 |
| Refinement statistics | ||
| No. of reflections used (σ(F) > 0) | 16,599 | 47,813 |
| Rworkb (%) | 21.22 | 21.14 |
| Rfreeb (%) | 26.91 | 27.73 |
| RMSD bond distance (Å) | 0.003 | 0.015 |
| RMSD bond angle (°) | 0.596 | 1.847 |
| Average B value (Å2) | 42.1 | 32.8 |
| No. of protein atoms | 5,361 | 8,250 |
| No. of ligand atoms | 0 | 0 |
| No. of solvent atoms | 3 | 0 |
| Ramachandran plot resolution (%) | ||
| In favored regions | 95.11 | 95.27 |
| In generously allowed regions | 4.89 | 4.73 |
| In disallowed regions | 0 | 0 |
Rmerge = Σh Σl | Iih − <Ih> |/Σh ΣI <Ih>, where <Ih> is the mean of the observations Iih of reflection h.
Rwork = Σ(||Fp(obs)|−|Fp(calc)||)/Σ|Fp(obs)|; Rfree is an R factor for a preselected subset (5%) of reflections that was not included in refinement.
Numbers in parentheses are corresponding values for the highest-resolution shell.
Similar to EBOV, MARV shares the same topology in two separate portions, an N lobe and a C lobe, which are connected by a single interflexible loop domain. Both domains are predominantly composed of α-helices, with 13 helices in the N lobe (α1 to α13) and seven helices in the C lobe (α14 to α20). There are two additional β-strands in the N lobe (β1 and β2) and an additional β-hairpin in the C lobe (β3 and β4) (Fig. 1C). Alignment of MARV and EBOV NPcore indicates that the overall structure and topology are similar, with a root mean square deviation (RMSD) of 1.080 Å for the Cα atoms of 230 residues, revealing structural conservation among filovirus NPs. However, the C lobe shows some variation: the C lobe also contains a significantly extraordinary long α-helix (α20, Q321 to E367), which is directly protruding outwards (Fig. 1C).
RNA binding groove of MARV NPcore.
NP encapsidates the viral genome to perform its natural function. However, the A280/A260 ratio was approximately 1.67 during the purification of MARV NPcore (Fig. 1B), which revealed very weak binding with nucleic acid (Fig. 1B). We speculate that this likely owes to the truncated construct without the N-terminal and C-terminal portions. Consistently, no continuous or discontinuous electron density for nucleic acid can be observed in the structure of MARV NPcore. However, the molecular surface of MARV NPcore showed a positively charged groove (Fig. 1C) without any obstructions hindering it and four positive-charged residues potentially for RNA binding, K142, K153, R156, and K230 (Fig. 1C).
It is well established that negative-strand RNA virus NP can specifically bind only with ssRNA, not dsRNA or DNA (5, 27, 37–40). To further clarify the RNA binding feature of MARV NP, we performed an electrophoretic mobility shift assay (EMSA), using a biotin-labeled single-strand RNA derived from the 3′ end of Marburg virus S segment as a probe binding to NP. In the sample preparation process, the probe and NP were incubated together at an increasing ratio, and samples were loaded according to the directions of the manufacturer. The results show that MARV NPcore was able to bind ssRNA, resulting in a significant shift of ssRNA probe on a native PAGE gel, which is consistent with EBOV and other NSV nucleoproteins.
Subsequently, a series of site-directed mutations of putative RNA binding residues were assessed in the EMSA and showed different binding ability for ssRNA binding. K142A, K153A, and R156A mutants almost completely lost their ability for ssRNA binding, while the K230A mutant still retained some binding ability (Fig. 1D). Taken together, this recombinant MARV NPcore shows the ability to bind ssRNA in vitro, and the residues K142, K153, and R156 are proved to be essential for the NP-RNA interaction.
MARV NPcore hexamerization.
Oligomerization is one of the key features of viral NP function. Although we observe an interesting hexamer formation of MARV NPcore, it is unlikely to be the authentic oligomerization state during the RNP formation, due to the inconsistency with the previously reported filovirus particle within infected cells in assays using cryo-electron tomography (26, 27). However, this oligomerization state may also provide an insight into the intermolecular interaction of MARV NPcore. Gel filtration chromatography during protein purification indicates the molecular size of possible hexameric oligomerization, and the structure of this fraction shows that MARV NPcore is in a hexameric state in the crystal (Fig. 2A). Two NPcore protomers form a dimer with their last C-terminal α-helices (α20) overlapping in an antiparallel orientation (Fig. 2B), and then three dimers constitute a hexamer with the dimers overlapping, forming a triangle-like hexamer (Fig. 2A). There are three pairs of interactions on the inner-dimer interface: E350-R339, I357-L336, and F361-A328 interactions (Fig. 2B). On the dimer contact surface, each dimer contacts another two adjacent dimers by interactions between I345-L233 and I348-L226 (Fig. 2B).
FIG 2.
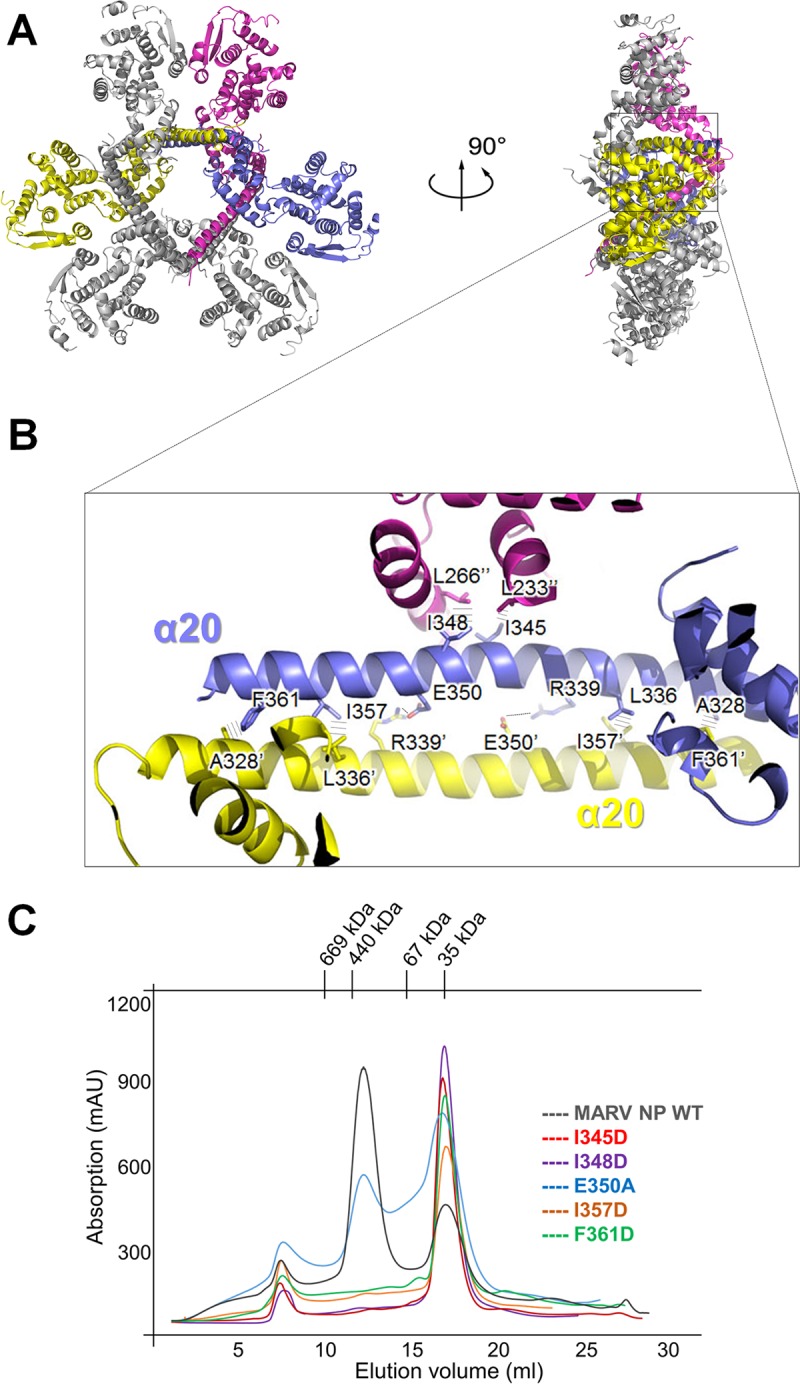
Recombinant MARV NPcore exists as a hexamer both in crystals and in solution. (A) The hexamer of MARV NPcore in a cartoon representation (left) and in a 90°-rotated configuration (right). Three protomers representing a pair of the interactions inside the hexamer are colored blue, yellow, and purple, respectively. (B) A representative of the interactions inner the hexamer on α20 helices. The key residues are shown as sticks and labeled. The interactions between residues are depicted as dashed lines. (C) Mutational analysis of the interprotomer interface. SEC (Superdex-200 column) of MARV NPcore site-directed mutations I345D (red), I348D (purple), E350A (blue), I357D (orange), and F361D (green) are merged together with WT (gray) as the control.
To further investigate the formation of this hexameric form, a series of site-specific (I345D, I348D, E350A, I357D, and F361D) mutants were assessed with size exclusion chromatography (SEC). Most of these mutations, i.e., I345D, I348D, I357D, and F361D, result in nearly complete disruption of the hexamer (Fig. 1B and 2C), which reveals that these hydrophobic interactions are crucial for hexamer formation. In contrast, the E350A mutant retained some hexamer fraction, which suggests that E350-R339 is not a primary factor for the formation of the hexamer. Thus, we confirm that the hydrophobic bond interaction by I345D, I348D, I357D, and F361D mutants is the major driving force for this hexameric form. All of these key residues are located on helix α20 (Fig. 2B), suggesting an important role for this long protruding helix for NP oligomerization.
VP35 interacts with NP as a chaperone.
Newly synthesized NP must be prevented from indiscriminately binding to cellular RNA. Once released from the host ribosome, NPs will be chaperoned by viral components and remain in an RNA-free (“N0”) state before self-assembly oligomerization and encapsidation of RNA to form the RNP (32, 41). Previous studies showed that phosphoprotein (P) homolog VP35 is essential to mediate virus transcription and replication (5, 25). Additionally, studies on EBOV confirmed that VP35 chaperones NP and keeps it in the “N0” state (34, 35). The mechanism of MARV VP35 chaperoning NP remains unclear and needs more investigation.
The MARV VP35 N-terminal peptide (M1-P29) was synthesized and assessed for its interaction with NP. The purified NPcore hexamer was incubated with VP35 peptide at 4°C for 1 h before SEC purification. During the SEC purification, we found that the NPcore hexamer was totally disrupted into a monomer by the VP35 peptide (Fig. 1B and 3A), suggesting a strong interaction between NP and VP35 peptide. To measure the affinity of VP35 peptide for NPcore, the isothermal titration calorimetry (ITC) assay was conducted, and the results indicate that the VP35 peptide has a strong affinity for binding NPcore, with an equilibrium dissociation constant (KD) of up to 16.2 ± 3.0E−8 M (Fig. 3B).
FIG 3.

MARV VP35 peptide chaperones NPcore. (A) MARV NPcore oligomerization is disrupted by VP35 peptide. SEC (Superdex-200 column) of MARV NPcore oligomer with VP35 peptide complex (red), merged together with MARV NPcore WT (blue). (B) ITC assay of the interaction between MARV NPcore and VP35 peptide. Data shown on the right side were calculated by Origin. Experiments were repeated more than three times. (C) Cartoon representation of MARV VP35 peptide rotating the C lobe of MARV NPcore. The conformation changes between the MARV NPcore apo form (blue) and its complex form (gray) are calculated by RMSD. The VP35 peptide (purple) induced a ∼25° lobe rotation, causing a potential opening of the RNA binding groove (blue bubble) and a potential RNA wrapping loop region (G310 to G319, dotted lines) movement. (D) Comparison of MARV and EBOV NPcore with or without VP35 peptides presented as a cartoon. The MARV NPcore apo form is colored in blue, and the EBOV NP apo form is colored in red. The two different EBOV NP (green for 4YPI and yellow for 4ZTG)-VP35 peptide (cyan for 4YPI and red for 4ZTG) complex structures are put into alignment with the MARV NP (gray)-VP35 peptide (purple) complex. The α20 and α21 helixes are labeled.
The hexamer became a monomer during the purification process, with an A280/A260 ratio increase to 1.82, indicating that there was barely any nucleic acid binding. Benefiting from the chaperoning effect of the VP35 peptide, the NP-VP35 peptide complex was crystallized and solved to a higher resolution of 2.2 Å (Table 1). The structure of the NP-VP35 complex gives a lower Wilson B factor, from 43.44 to 24.88 (Table 1), and most gaps lost in the NPcore apo form could be remodeled, such as the short gap (L203 to H207) and the loop region (G310 to G319). There are three molecules in one crystallographic asymmetric unit without any crystal packing interactions, and the alignment of these three molecules gives an average RMSD of 0.877 Å for 342 residues (0.52 Å, 1.119 Å, and 0.991 Å).
There are several points of conformation changes in these two structures. The structural alignment gives an overall RMSD of 0.813 and a C lobe RMSD of 2.198, leading to the conclusion that the conformation change lies mostly in the C lobe, which undergoes an ∼25° relative domain rotation (Fig. 3C). The relative rotation enlarged the RNA binding groove, leaving an “open” state instead of the “closed” state in the apo form of NPcore. Moreover, in the VP35-chaperoned structure, one mostly missing loop region (G310 to G319, GSTLAGVNVG, dotted lines in Fig. 1C) in the apo form can be remodeled; however, this loop region moved backward to the RNA binding groove at a distance of around 9 Å (Fig. 3C), and this movement may be another proof that the RNA binding groove undergoes the transition to the “open” state. Finally, the long protruding α-helix (α20) can build only to R338; however, R339-H370 was invisible in the VP35-chaperoned structure (Fig. 1C and 3C), due to the loss of continuous electron density.
We further superimpose the structures with EBOV NP, both in the apo form and the VP35 peptide bond form. The N lobes are mostly identical, while the C lobes are variable (Fig. 3D). Benefiting from the structures of the EBOV NP-VP35 complex reported by two independent groups (34, 35), we can compare the NP conformation difference within filoviruses: the C lobes of the two EBOVs (PDB codes 4ZTG and 4YPI) and MARV NP share a similar conformation, except for a significant difference at the end of the C lobe (Fig. 3D). Unlike the long protruding helix α20 in MARV NP (Fig. 1C), this region is split into U-turned α20 and α21 in EBOV NP (PDB code 4YPI) (Fig. 3D), which mediates crystal contacts and may represent a physiological oligomerization interaction between C lobes (34).
NP-VP35 peptide interaction is conserved in filoviruses.
Although the length of the VP35-encoding sequence varies among filoviruses, the primary sequence alignment of filovirus VP35 showed a conserved hydrophobic N-terminal region (Fig. 4A), with the potential key residues V10, L14, M15, I21, V24, and F25; thus, VP35 peptide may share a conserved mechanism for binding NP in filovirus.
FIG 4.

VP35 binds NPcore through hydrophobic interactions. (A) Sequence alignment of the conserved VP35 N-terminal peptide. The amino acids sharing identity are background-shaded red in blue squares, and similar amino acids are red in blue squares. The speculated potential key hydrophobic residues are indicated by arrowheads. (B) Representation of the hydrophobic interaction between MARV NPcore (surface in gray) and VP35 peptide (cartoon in purple). The potential key residues V10, S11, L14, and M15 are emphasized as sticks and colored in white. (C) Summary of ITC binding measurements between VP35 mutation derivatives and MARV NPcore. The experiments were conducted in the same manner as described for Fig. 3B, with at least three repeats.
The structure of the MARV NPcore-VP35 peptide complex reveals that the interactions between NPcore and VP35 peptide are mainly hydrophobic interactions. The C lobe of the NPcore constitutes a hydrophobic pocket occupied by the VP35 peptide (Fig. 4B). Using the protein surface charge and hydrophobicity as a guide, we hypothesize that the VP35 residues S11, E12, M15, and L14 are essential for its interaction with NPcore (Fig. 4A and B). The mutations M7P, Q9A, E12A, and V19D had little impact on protein binding interactions; however, the mutations V10D, S11P, I21D, V24D, and F25A remarkably changed the KD value by 1 order of magnitude (Fig. 3B and 4C). Most strikingly, the hydrophobic interaction amino acid mutations L14D and M15P significant abolished the interaction of MARV NPcore with VP35 peptide (Fig. 4C).
VP35 peptide binding stabilizes NP and competes with NP-RNA binding.
Considering the high affinity of VP35 peptide with MARV NPcore, the peptide binds with NP tightly and might stabilize this complex as an integrated component. Meanwhile, during the structure determination process for the complex structure, the last amino acid can be traced only to R338, and most of the α20 helices were missing in the complex. To further clarify the consequence of VP35 peptide binding to MARV NPcore, we conducted a protein self-degradation experiment in vitro. As a control, MARV NPcore completely degraded within 5 days at 16°C (Fig. 5A); however, the VP35-chaperoned MARV NPcore complex degraded to a certain state (the accurate sequence is located as R19 to R338, confirmed by mass spectrum results) and remained stable for more than a week (Fig. 5A).
FIG 5.

The chaperon effect of VP35 on NP. (A) SDS-PAGE analysis of MARV NPcore (left lines) and NPcore-VP35 peptide complex (right lines) self-degradation. Each line contains the same quantity of original proteins. Time is given in hours (h) or days (d). The speculated region of MARV NPcore is boxed in red. Experiments were conducted in three individual repeats. (B) EMSA of the affinity competitions of VP35 peptide and ssRNA for NP. NP was pretreated with ssRNA and then incubated with VP35 peptide or in reverse order before sample loading. The dosages (final concentrations) are listed on the top. The free and combined ssRNAs are labeled on the side. Experiments were repeated more than three times. (C) Electrostatic surface potential (calcluated with APBS tools, with limits of ±5 kbT/ec) and cartoon representation for structure comparison of P/VP35 peptide or Ni-1 arm effect on NP (N). The P (green)/VP35 (pink) peptides, Ni-1 arms (orange), and α20 helices (cyan) are shown as a cartoon and labeled.
The VP35 peptide of EBOV was previously shown to be sufficient to disassociate RNA binding with NP (34, 35). To further clarify the impact of RNA binding after VP35 was chaperoned, we conducted a competition experiment to investigate the impact of VP35 on RNA binding in EMSA. In a positive competition experiment, NPcore and the VP35 peptide were pretreated at an increasing ratio before ssRNA was added, and the dose dependency chemiluminescence results reflect that ssRNA binding ability greatly decreased when the concentration of VP35 peptide increased (Fig. 5B). The same impact was also observed in a reverse competition experiment, when NP and ssRNA were pretreated at an increasing ratio before the VP35 peptide was added (Fig. 5B). In both situations, VP35 peptide was able to prevent ssRNA binding to NPcore and even dissociated ssRNA from the NP-RNA complex.
DISCUSSION
The structural investigation of negative single-strand RNA (−ssRNA) virus-encoded NP has achieved great progress in the past years. Although NPs in different virus families show large structural variations in detail, their overall structures share a conserved topology. In general, their structure features can be divided into two major groups. The NPs in the first group exhibit a typical bilobed structure to form a positively charged RNA binding groove between the N and C lobes and adopt diverse structural components for the formation of virus RNP, mostly by the interaction within protomers by the adjacent N-terminal arm or C-terminal tail. The first group includes most of the nonsegmented or segmented −ssRNA viruses, such as BDV (17) from the Bornaviridae family, VSV (18, 19, 42) and rabies virus (43) from the Rhabdoviridae family, RSV (20) from the Paramyxoviridae family, influenza virus (44) from the Orthomyxoviridae family, Rift Valley fever virus (RVFV) (45) from the Phenuiviridae family, Sin Nombre orthohantavirus (15) from the Hantaviridae family, and Bunyamwera virus (BUNV) (46) from the Peribunyaviridae family. The structural feature of the second group was discovered after 2010; this group includes LAFV (10, 11) and Junin virus (47) in the Arenaviridae family and CCHFV (12) and Hazara orthonairovirus (48, 49) in the Nairoviridae family. In this group, NPs do not exhibit as the typical divided two lobes; however, the positive-charged RNA binding groove is located at the center of one compact domain, which adopts an interesting gating mechanism for virus genome encapsidation; moreover, these NPs also show unanticipated enzymatic activities.
In this work, we reported the Marburg virus nucleoprotein structures, both in apo form and VP35 peptide-bound form. The structure of MARV NPcore is categorized into the first group. However, it is worth noting that the whole encoding sequence of MARV nucleoprotein contains nearly 700 amino acids, and only the region encompassing the first 450 amino acids accounts for the typical function of RNA encapsidation and homotypic NP interaction. The precise biological function of the C-terminal extension part warrants further investigations.
Here, we delineated a conformation change of MARV NP chaperoned by VP35 (Fig. 1C and 3C). The reconstituted structure unveiled the flexibility of MARV NP, revealing the transition of NP during virus proliferation. As previously reported, dynamic binding with viral RNA is the key function of NP in −ssRNA viruses. In the purification process, N-terminally truncated NPcore shows low ability for binding RNA, whereas the constructs containing the N-terminal region can bind cellular nucleic acid tightly. NP oligomerization recruits nucleic acids during RNP formation, and the RNA binding to NP will further stabilize oligomerization (34), which is consistent with the so-called “RNA-stabilized oligomerization” process (12). For EBOV, the modulation of dynamic RNA binding ability is regulated by the chaperoning of VP35; however, neither the monomeric NPcore nor the NP-VP35 peptide complex binds to RNA with any appreciable affinity (34, 35). Since three research groups separately reported the EBOV NP structure in the apo form (36) or the VP35 peptide-bound form (34, 35), it is hard to delineate the whole picture of the conformational change during the VP35-chaperoned process, especially for the modulation of RNA binding ability. Here, our investigation of MARV NP both in apo form and VP35 peptide-bound form leads us to propose a working mechanism of this VP35 modulation process. The most significant difference between these two structures is the rotation of the C lobe after it has been chaperoned by VP35 peptide. Additionally, a missing loop region (G310 to G319), which is mostly invisible in the apo form (Fig. 1C), can be remodeled in the VP35 peptide-chaperoned complex structure (Fig. 3C); these clues encourage us to propose the hypothesis that VP35 prevents or disassociates the interaction of NP and RNA by inducing the RNA binding groove transit from a “closed” state to an “open” state. (Fig. 3C); in addition, a homologous modulation mechanism is also reported in NiV (22) and is consistent with the result of EBOV in apo form (36) and VP35-chaperoned form (34, 35).
Besides the modulation of the NP protomer, the abolishment of oligomerization by VP35 (Fig. 3A) is also critical for the low RNA binding affinity. Once the oligomer was disrupted into the monomer by the VP35 peptide, the structure of the continuous positive-charged groove was damaged; hence, NP lost its function to bind nonspecific RNA with high affinity. This mechanism prevents newly synthesized NP from nonspecifically binding with cytosolic RNA. When the NP-VP35 complex reaches the RNA replication machinery, it is very likely that VP35 dissociates from NP concurrently with RNP reconstitution by an unknown mechanism, recovering the oligomerization ability of NP, followed by NP self-assembly and RNA encapsidation.
Another significant structural feature of MARV NP is the last α-helix in the C lobe, α20, which is protruding in the apo form (Fig. 1C) but mostly invisible in the VP35-chaperoned form (Fig. 3C). As we have discussed above, α20 is composed mainly of hydrophobic residues and responsible for the C-terminal interaction to form a hexamer (Fig. 2B). Meanwhile, α20 may be highly flexible during the RNP formation process. In EBOV NP apo form, α20 is located at the highly conserved hydrophobic pocket (36) (Fig. 3D and 5C). However, in the structure of EBOV NP bound with VP35, the correspondent long helix is either degraded (34) (PDB code 4ZTG) or split into two α-helices, α20 and α21 (35) (PDB code 4YPI) (Fig. 3D), responsible for the hydrophobic interaction within protomers in an asymmetry unit. All these differences suggest the high flexibility of this region, implying its critical role during authentic RNP formation. This warrants further structural and biological validations, such as determination of the whole sequence structure of filovirus nucleoprotein or RNP bound with RNA.
Interestingly, the highly conserved hydrophobic pocket may also show multifunctional interactions for binding with the NPi-1 N-terminal arm, P protein, and VP35 protein, or NP C-terminal helix as well. In RSV (19) and PIV-5 (20), each N-terminal arm of Ni-1 interacts with Ni by interacting with this pocket as the oligomerization bridge (Fig. 5C). In NiV (22) and VSV (18, 19), the P peptide binds with the hydrophobic pocket, locking the “open” conformation (NiV [22]) or occupying the RNA binding groove (VSV [18, 19]) (Fig. 5C). In addition, recent works on measles virus (MeV) (50) also delineated the structure of the N-P fusion protein, stretching the hydrophobic interaction of P peptide and collapsing the RNA binding groove. Except for the VSV-P complex with exact steric hindrance for RNA binding (19), the VP35 (P) peptide does not directly block the RNA binding groove under most conditions. In our previous work on EBOV NPcore, the C lobe helix α20 was clearly located in the hydrophobic pocket (Fig. 3D and 5C). An expected result is that when we purified MARV and EBOV NP without VP35 peptide, the presence of N-terminal 19 residues caused severe high-ordered oligomerization and thus resulted in high-level precipitation, and the release of VP35 peptide caused NP to reconstitute into a high-ordered oligomer in vitro (34); we thus hypothesize that the hydrophobic pocket may adapt the binding with both the adjacent Ni-1 N-terminal arm, P protein, or VP35 N-terminal sequence and the C-terminal helix in NP, to modulate the transition from monomeric and oligomeric states.
Considering the critical role of correct RNP formation and function for virus replication, transcription, and assembly, it is reasonable to design inhibitors to block this process for antiviral development. In the last few years, considerable progress has been achieved based on this new antiviral strategy, particularly for the influenza virus (23, 24). These results demonstrated that viral RNP is a valid druggable target for the development of small-molecule therapies, which is greatly supportive in counteracting the current severe situation of resistance to drugs targeting viral surface proteins. Here, we discuss a distinct hydrophobic pocket located in the core domain of MARV NP, which is highly conserved among all viruses within Filoviridae and even Mononegavirales. Recently, two independent teams have screened natural compounds targeting the RNA binding groove (51) or FDA-approved molecular drugs that target the EBOV NP-VP35 interaction surface (52); the high conservation of the NP-VP35 interface among filoviruses suggests that these assays based on the filovirus nucleoprotein have great potential to identify panfiloviral inhibitors. Therefore, the structure of MARV NP not only aids in understanding the structural and functional differences among NPs encoded by −ssRNA viruses but also benefits the development of antiviral therapies against filovirus infection.
MATERIALS AND METHODS
Protein production.
The wild-type cDNA of MARV NPcore (residues 19 to 370) was synthesized by Genewiz. MARV NPcore was cloned into the pET-28a (Novagen) vector using the NdeI and HindIII restriction sites within the cloning primers. The sequences of the primers were as follows: forward, 5′-CGCCATATGCGTAATAAGAAAGTG-3′; reverse, 5′-CCCAAGCTTTTAGTGTGTGATTTCAG-3′. Gene sequencing was applied to verify the accuracy of the vector.
The recombinant plasmid of MARV NPcore was transformed into E. coli strain BL21(DE3) (TransGen Biotech, Beijing, China) and overexpressed. The cells were cultured at 37°C in 800 ml of LB medium containing 50 μg/ml kanamycin. Once the optical density at 600 nm (OD600) reached approximately 0.6, the culture was transferred to 16°C. Then, protein expression was induced by the addition of 0.25 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for an additional 16 h. The harvested cells were resuspended in lysis buffer containing 20 mM HEPES (pH 7.5), 500 mM NaCl, and 5% (vol/vol) glycerol and homogenized with a low-temperature ultrahigh pressure cell disrupter (Jnbio). The lysate was centrifuged at 25,000 × g for 30 min at 4°C to remove cell debris. The supernatant was then loaded twice onto an Ni-nitrilotriacetic acid (Ni-NTA) column (GE Healthcare) preequilibrated with lysis buffer. After washing the resin with wash buffer containing 20 mM HEPES (pH 7.5), 500 mM NaCl, 5% (vol/vol) glycerol, and 50 mM imidazole, the target protein was eluted with elution buffer containing 20 mM HEPES (pH 7.5), 500 mM NaCl, 5% (vol/vol) glycerol, and 300 mM imidazole. The protein was further purified by Superdex 200 gel filtration chromatography (GE Healthcare) with buffer containing 20 mM HEPES (pH 7.5) and 200 mM NaCl. SDS-PAGE analysis revealed over 97% purity for the hexamer protein and over 95% purity for the monomer protein. The two purified proteins were then concentrated separately to 8 mg/ml in storage buffer containing 20 mM HEPES (pH 7.5) and 200 mM NaCl. The complex of MARV NPcore oligomer protein and the VP35 peptide (MWDSSYMQQVSEGLMTGKVPIDQVFGANP) [GL Biochem (Shanghai) Ltd.] were mixed in a ratio of 1:1.3 and purified by Superdex 200 gel filtration chromatography (GE Healthcare) after 1 h of incubation at 4°C.
Crystallization.
Commercial crystal screening kits, including the Index, Crystal Screen, PEG/Ion, Salt/RX, and Crystal Screen Cryo from Hampton Research, were used for screening the crystallization conditions by the hanging-drop vapor diffusion method at 16°C. The protein and reservoir solutions were mixed in a ratio of 1:1, and all conditions were equilibrated against 80 μl of reservoir solution in a 96-well format.
Tiny crystals of MARV NPcore first appeared after 1 day in 200 mM calcium chloride dihydrate, 100 mM HEPES sodium salt (pH 7.5), and 28% (vol/vol) polyethylene glycol 400. Further optimization was performed, and the final optimized crystals were grown in 200 mM calcium chloride dihydrate, 100 mM HEPES sodium salt (pH 7.5), and 15% (vol/vol) polyethylene glycol 400. Rod-like crystals grew to a final size of 40 by 40 by 300 μm within 1 week at 16°C. The crystals were harvested, cryoprotected in the well solution supplemented with 10% (vol/vol) glycerol, and cooled in a dry nitrogen stream at 100 K for X-ray data collection.
Tiny crystals of the MARV NPcore-VP35 peptide complex first appeared after 1 day in 200 mM lithium sulfate monohydrate, 100 mM Tris (pH 8.5), and 25% (wt/vol) polyethylene glycol 3,350. Further optimization was performed, and the final optimized crystals were grown in 200 mM lithium sulfate monohydrate, 100 mM Tris (pH 8.5), and 22% (wt/vol) polyethylene glycol 3,350. Prism-like crystals grew to a final size of 30 by 30 by 180 μm within 4 days at 16°C. The crystals were harvested and cooled in liquid nitrogen for X-ray data collection.
X-ray data collection, processing, and structure determination.
The data were collected at beamline BL19U (SSRF, China) under cryogenic conditions at 100 K. The data sets were processed using the HKL3000 package (53). The initial phases were calculated using the Molecular Replacement program in the Phenix software suite (54) with EBOV NPcore (PDB code 4Z9P) as the start model. All the models were then manually built into the modified experimental electron density using Coot (55) and further refined in Phenix (56). Model geometry was verified using the program MolProbity (57). Structural figures were drawn using the program PyMOL (58) and the APBS plugin (59). The primary sequence alignment was performed using ClustalW (60) and rescripted by ESPript3.0 (61). The coordinates and structure factors were deposited in the RCSB Protein Data Bank (see below).
Electrophoretic mobility shift assays.
EMSAs were performed to evaluate the potential interactions between the MARV-encoded NP and RNA probe and the effect of the presence of the MARV VP35 peptide. Experiments were performed according to the instructions of a commercial EMSA kit (Chemiluminescent EMSA kit; Beyotime Institute of Biotechnology, Haimen, China). Biotin-labeled single-strand RNA (ssRNA) containing the sequence (5′-UCUCAAAGAAAGUUG-3′) derived from the 3′ end of Marburg virus S segment was synthesized by TaKaRa Biotechnology (Dalian, China) as a probe. The probe and NP were mixed together at a ratio of 1:2 with 0.1 nM probe and 0.2 nM NP as final concentrations. The dosage of VP35 peptide was set at an increasing ratio with NP, from 1:4 to 1:1. After being incubated for 15 min at room temperature in the binding buffer, the mixtures were loaded onto a 10% native polyacrylamide gel in 0.5% Tris-borate-EDTA for electrophoresis. Then, the mixtures were transferred and UV cross-linked onto nylon membranes (FFN12; Beyotime Institute of Biotechnology). The bands were visualized and exposed to a film using a probe biotin-labeling kit (BeyoECL Plus; Beyotime Institute of Biotechnology).
ITC.
The binding of NPcore and VP35 was analyzed by ITC200 (MicroCal). Fresh NPcore protein and VP35 peptide samples were diluted or dissolved in storage buffer containing 20 mM HEPES (pH 7.5) and 200 mM NaCl. NPcore samples were loaded into the isothermal titration calorimetry (ITC) cell, and the VP35 peptide samples were loaded into the syringe. The reactions were run at 25°C with 17 or 20 injections. The raw ITC data were processed using Origin (OriginLab).
Accession number(s).
The coordinates and structure factors were deposited in the RCSB PDB under the accession codes 5F5M (MARV NPcore) and 5F5O (MARV NPcore and VP35 peptide complex).
ACKNOWLEDGMENTS
We gratefully acknowledge the staff members of BL18U1 and BL19U1, SSRF, China, for their assistance with the diffraction data collection.
This work was supported by the National Natural Science Foundation of China (grant numbers 31670731, 813300237, 81520108019), Ministry of Science and Technology of China “973” Project (grant numbers 2013CB911103, 2014CB542800, 2014CBA02003), Tianjin Municipal Natural Science Foundation (grant numbers 13ZCZDSY04200 and 14ZCZDSY00039), Strategic Priority Research Program of the Chinese Academy of Sciences (grant number XDB08000000), and the Fundamental Research Funds for the Central Universities.
We declare that we have no competing financial interests.
REFERENCES
- 1.Bausch DG, Nichol ST, Muyembe-Tamfum JJ, Borchert M, Rollin PE, Sleurs H, Campbell P, Tshioko FK, Roth C, Colebunders R, Pirard P, Mardel S, Olinda LA, Zeller H, Tshomba A, Kulidri A, Libande ML, Mulangu S, Formenty P, Grein T, Leirs H, Braack L, Ksiazek T, Zaki S, Bowen MD, Smit SB, Leman PA, Burt FJ, Kemp A, Swanepoel R, International Scientific and Technical Committee for Marburg Hemorrhagic Fever Control in the Democratic Republic of the Congo. 2006. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N Engl J Med 355:909–919. doi: 10.1056/NEJMoa051465. [DOI] [PubMed] [Google Scholar]
- 2.Towner JS, Khristova ML, Sealy TK, Vincent MJ, Erickson BR, Bawiec DA, Hartman AL, Comer JA, Zaki SR, Stroher U, Gomes da Silva F, del Castillo F, Rollin PE, Ksiazek TG, Nichol ST. 2006. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J Virol 80:6497–6516. doi: 10.1128/JVI.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Messaoudi I, Amarasinghe GK, Basler CF. 2015. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol 13:663–676. doi: 10.1038/nrmicro3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhn JH, Becker S, Ebihara H, Geisbert TW, Johnson KM, Kawaoka Y, Lipkin WI, Negredo AI, Netesov SV, Nichol ST, Palacios G, Peters CJ, Tenorio A, Volchkov VE, Jahrling PB. 2010. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch Virol 155:2083–2103. doi: 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muhlberger E, Weik M, Volchkov VE, Klenk H-D, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Y, Guo Y, Lou Z. 2012. A versatile building block: the structures and functions of negative-sense single-stranded RNA virus nucleocapsid proteins. Protein Cell 3:893–902. doi: 10.1007/s13238-012-2087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou H, Sun Y, Guo Y, Lou Z. 2013. Structural perspective on the formation of ribonucleoprotein complex in negative-sense single-stranded RNA viruses. Trends Microbiol 21:475–484. doi: 10.1016/j.tim.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Muhlberger E. 2007. Filovirus replication and transcription. Future Virol 2:205–215. doi: 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruigrok RW, Crepin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol 14:504–510. doi: 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Hastie KM, Liu T, Li S, King LB, Ngo N, Zandonatti MA, Woods VL, de la Torre JC Jr, Saphire EO. 2011. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc Natl Acad Sci U S A 108:19365–19370. doi: 10.1073/pnas.1108515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. 2011. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A 108:2396–2401. doi: 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Y, Wang W, Ji W, Deng M, Sun Y, Zhou H, Yang C, Deng F, Wang H, Hu Z, Lou Z, Rao Z. 2012. Crimean-Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc Natl Acad Sci U S A 109:5046–5051. doi: 10.1073/pnas.1200808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Li B, Guo Y, Shen S, Zhao L, Zhang P, Sun Y, Sui SF, Deng F, Lou Z. 2016. Molecular basis for the formation of ribonucleoprotein complex of Crimean-Congo hemorrhagic fever virus. J Struct Biol 196:455–465. doi: 10.1016/j.jsb.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Ng AK, Zhang H, Tan K, Li Z, Liu JH, Chan PK, Li SM, Chan WY, Au SW, Joachimiak A, Walz T, Wang JH, Shaw PC. 2008. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J 22:3638–3647. doi: 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Y, Wang W, Sun Y, Ma C, Wang X, Wang X, Liu P, Shen S, Li B, Lin J, Deng F, Wang H, Lou Z. 2015. Crystal structure of the core region of hantavirus nucleocapsid protein reveals the mechanism for ribonucleoprotein complex formation. J Virol 90:1048–1061. doi: 10.1128/JVI.02523-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou H, Sun Y, Wang Y, Liu M, Liu C, Wang W, Liu X, Li L, Deng F, Wang H, Guo Y, Lou Z. 2013. The nucleoprotein of severe fever with thrombocytopenia syndrome virus processes a stable hexameric ring to facilitate RNA encapsidation. Protein Cell 4:445–455. doi: 10.1007/s13238-013-3901-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudolph MG, Kraus I, Dickmanns A, Eickmann M, Garten W, Ficner R. 2003. Crystal structure of the Borna disease virus nucleoprotein. Structure 11:1219–1226. doi: 10.1016/j.str.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA Jr, Jensen MR, Blackledge M, Ruigrok RW, Jamin M. 2011. Structure of the vesicular stomatitis virus N(0)-P complex. PLoS Pathog 7:e1002248. doi: 10.1371/journal.ppat.1002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green TJ, Luo M. 2009. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc Natl Acad Sci U S A 106:11713–11718. doi: 10.1073/pnas.0903228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 21.Alayyoubi M, Leser GP, Kors CA, Lamb RA. 2015. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein-RNA complex. Proc Natl Acad Sci U S A 112:E1792–E1799. doi: 10.1073/pnas.1503941112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yabukarski F, Lawrence P, Tarbouriech N, Bourhis JM, Delaforge E, Jensen MR, Ruigrok RW, Blackledge M, Volchkov V, Jamin M. 2014. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat Struct Mol Biol 21:754–759. doi: 10.1038/nsmb.2868. [DOI] [PubMed] [Google Scholar]
- 23.Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. 2010. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol 28:600–605. doi: 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, Zhai W, Pendri A, Li G, Poss MA, Edavettal S, McDonnell PA, Lewis HA, Maskos K, Mortl M, Kiefersauer R, Steinbacher S, Baldwin ET, Metzler W, Bryson J, Healy MD, Philip T, Zoeckler M, Schartman R, Sinz M, Leyva-Grado VH, Hoffmann HH, Langley DR, Meanwell NA, Krystal M. 2011. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc Natl Acad Sci U S A 108:15366–15371. doi: 10.1073/pnas.1107906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muhlberger E, Lotfering B, Klenk HD, Becker S. 1998. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J Virol 72:8756–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bharat TA, Riches JD, Kolesnikova L, Welsch S, Krahling V, Davey N, Parsy ML, Becker S, Briggs JA. 2011. Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLoS Biol 9:e1001196. doi: 10.1371/journal.pbio.1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bharat TA, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, Kawaoka Y, Briggs JA. 2012. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc Natl Acad Sci U S A 109:4275–4280. doi: 10.1073/pnas.1120453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leung DW, Prins KC, Basler CF, Amarasinghe GK. 2010. Ebolavirus VP35 is a multifunctional virulence factor. Virulence 1:526. doi: 10.4161/viru.1.6.12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramanan P, Edwards MR, Shabman RS, Leung DW, Endlich-Frazier AC, Borek DM, Otwinowski Z, Liu G, Huh J, Basler CF, Amarasinghe GK. 2012. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci U S A 109:20661–20666. doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker S, Rinne C, Hofsass U, Klenk HD, Muhlberger E. 1998. Interactions of Marburg virus nucleocapsid proteins. Virology 249:406–417. doi: 10.1006/viro.1998.9328. [DOI] [PubMed] [Google Scholar]
- 31.Moller P, Pariente N, Klenk HD, Becker S. 2005. Homo-oligomerization of Marburgvirus VP35 is essential for its function in replication and transcription. J Virol 79:14876–14886. doi: 10.1128/JVI.79.23.14876-14886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curran J, Marq J-B, Kolakofsky D. 1995. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J Virol 69:849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gubbay O, Curran J, Kolakofsky D. 2001. Sendai virus genome synthesis and assembly are coupled: a possible mechanism to promote viral RNA polymerase processivity. J Gen Virol 82:2895–2903. doi: 10.1099/0022-1317-82-12-2895. [DOI] [PubMed] [Google Scholar]
- 34.Kirchdoerfer RN, Abelson DM, Li S, Wood MR, Saphire EO. 2015. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep 12:140–149. doi: 10.1016/j.celrep.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leung DW, Borek D, Luthra P, Binning JM, Anantpadma M, Liu G, Harvey IB, Su Z, Endlich-Frazier A, Pan J, Shabman RS, Chiu W, Davey RA, Otwinowski Z, Basler CF, Amarasinghe GK. 2015. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein-RNA interactions. Cell Rep 11:376–389. doi: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong S, Yang P, Li G, Liu B, Wang W, Liu X, Xia B, Yang C, Lou Z, Guo Y, Rao Z. 2015. Insight into the Ebola virus nucleocapsid assembly mechanism: crystal structure of Ebola virus nucleoprotein core domain at 1.8 A resolution. Protein Cell 6:351–362. doi: 10.1007/s13238-015-0163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y, Xu L, Sun Y, Nabel GJ. 2002. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol Cell 10:307–316. doi: 10.1016/S1097-2765(02)00588-9. [DOI] [PubMed] [Google Scholar]
- 38.Noda T, Hagiwara K, Sagara H, Kawaoka Y. 2010. Characterization of the Ebola virus nucleoprotein-RNA complex. J Gen Virol 91:1478–1483. doi: 10.1099/vir.0.019794-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noda T, Kolesnikova L, Becker S, Kawaoka Y. 2011. The importance of the NP:VP35 ratio in Ebola virus nucleocapsid formation. J Infect Dis 204(Suppl 3):S878–S883. doi: 10.1093/infdis/jir310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe S, Noda T, Kawaoka Y. 2006. Functional mapping of the nucleoprotein of Ebola virus. J Virol 80:3743–3751. doi: 10.1128/JVI.80.8.3743-3751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mavrakis M, Mehouas S, Real E, Iseni F, Blondel D, Tordo N, Ruigrok RW. 2006. Rabies virus chaperone: identification of the phosphoprotein peptide that keeps nucleoprotein soluble and free from non-specific RNA. Virology 349:422–429. doi: 10.1016/j.virol.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 42.Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360. doi: 10.1126/science.1126953. [DOI] [PubMed] [Google Scholar]
- 43.Albertini AA, Wernimont AK, Muziol T, Ravelli RB, Clapier CR, Schoehn G, Weissenhorn W, Ruigrok RW. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360–363. doi: 10.1126/science.1125280. [DOI] [PubMed] [Google Scholar]
- 44.Das K, Aramini JM, Ma LC, Krug RM, Arnold E. 2010. Structures of influenza A proteins and insights into antiviral drug targets. Nat Struct Mol Biol 17:530–538. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raymond DD, Piper ME, Gerrard SR, Skiniotis G, Smith JL. 2012. Phleboviruses encapsidate their genomes by sequestering RNA bases. Proc Natl Acad Sci U S A 109:19208–19213. doi: 10.1073/pnas.1213553109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B, Wang Q, Pan X, Fernandez de Castro I, Sun Y, Guo Y, Tao X, Risco C, Sui SF, Lou Z. 2013. Bunyamwera virus possesses a distinct nucleocapsid protein to facilitate genome encapsidation. Proc Natl Acad Sci U S A 110:9048–9053. doi: 10.1073/pnas.1222552110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Li L, Liu X, Dong S, Wang W, Huo T, Guo Y, Rao Z, Yang C. 2013. Crystal structure of Junin virus nucleoprotein. J Gen Virol 94:2175–2183. doi: 10.1099/vir.0.055053-0. [DOI] [PubMed] [Google Scholar]
- 48.Wang W, Liu X, Wang X, Dong H, Ma C, Wang J, Liu B, Mao Y, Wang Y, Li T, Yang C, Guo Y. 2015. Structural and functional diversity of Nairovirus-encoded nucleoproteins. J Virol 89:11740–11749. doi: 10.1128/JVI.01680-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Surtees R, Ariza A, Punch EK, Trinh CH, Dowall SD, Hewson R, Hiscox JA, Barr JN, Edwards TA. 2015. The crystal structure of the Hazara virus nucleocapsid protein. BMC Struct Biol 15:24. doi: 10.1186/s12900-015-0051-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guryanov SG, Liljeroos L, Kasaragod P, Kajander T, Butcher SJ. 2015. Crystal structure of the measles virus nucleoprotein core in complex with an N-terminal region of phosphoprotein. J Virol 90:2849–2857. doi: 10.1128/JVI.02865-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu X, Wang Z, Li L, Dong S, Li Z, Jiang Z, Wang Y, Shui W. 2016. Novel chemical ligands to Ebola virus and Marburg virus nucleoproteins identified by combining affinity mass spectrometry and metabolomics approaches. Sci Rep 6:29680. doi: 10.1038/srep29680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu G, Nash PJ, Johnson B, Pietzsch C, Ilagan MX, Bukreyev A, Basler CF, Bowlin TL, Moir DT, Leung DW, Amarasinghe GK. 2017. A sensitive in vitro high-throughput screen to identify pan-filoviral replication inhibitors targeting the VP35-NP interface. ACS Infect Dis 3:190–198. doi: 10.1021/acsinfecdis.6b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. 2006. HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 54.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 55.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 57.Lovell SC, Davis IW, Arendall WB III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. 2003. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 58.DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA: http://www.pymol.org. [Google Scholar]
- 59.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 61.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]