

ABSTRACT
The SAT protein (SATp) of porcine parvovirus (PPV) accumulates in the endoplasmic reticulum (ER), and SAT deletion induces the slow-spreading phenotype. The in vitro comparison of the wild-type Kresse strain and its SAT knockout (SAT−) mutant revealed that prolonged cell integrity and late viral release are responsible for the slower spreading of the SAT− virus. During PPV infection, regardless of the presence or absence of SATp, the expression of downstream ER stress response proteins (Xbp1 and CHOP) was induced. However, in the absence of SATp, significant differences in the quantity and the localization of CHOP were detected, suggesting a role of SATp in the induction of irreversible ER stress in infected cells. The involvement of the induction of irreversible ER stress in porcine testis (PT) cell necrosis and viral egress was confirmed by treatment of infected cells by ER stress-inducing chemicals (MG132, dithiothreitol, and thapsigargin), which accelerated the egress and spreading of both the wild-type and the SAT− viruses. UV stress induction had no beneficial effect on PPV infection, underscoring the specificity of ER stress pathways in the process. However, induction of CHOP and its nuclear translocation cannot alone be responsible for the biological effect of SAT, since nuclear CHOP could not complement the lack of SAT in a coexpression experiment.
IMPORTANCE SATp is encoded by an alternative open reading frame of the PPV genome. Earlier we showed that SATp of the attenuated PPV NADL-2 strain accumulates in the ER and accelerates virus release and spreading. Our present work revealed that slow spreading is a general feature of SAT− PPVs and is the consequence of prolonged cell integrity. PPV infection induced ER stress in infected cells regardless of the presence of SATp, as demonstrated by the morphological changes of the ER and expression of the stress response proteins Xbp1 and CHOP. However, the presence of SATp made the ER stress more severe and accelerated cell death during infection, as shown by the higher rate of expression of CHOP and alteration of the localization of CHOP. The beneficial effect of irreversible ER stress on PPV spread was confirmed by treatment of infected cells with ER stress-inducing chemicals.
KEYWORDS: CHOP, ER stress, SAT, Xbp1, alternative ORF, parvovirus, protoparvovirus, viral egress
INTRODUCTION
Porcine parvovirus (PPV) belongs to the species Ungulate protoparvovirus 1 in the genus Protoparvovirus. It is the causative agent of the stillbirth, mummification, embryonic death, infertility (SMEDI) syndrome in swine (1).
A highly pathogenic strain with broad tissue tropism, strain Kresse, was isolated in the 1980s (2, 3). Kresse strain-infected piglets showed atypical signs of PPV infection, including necrotic lesions (4) on the lips, snout, tongue, and foot.
The PPV single-stranded genome contains two major open reading frames (ORFs) and one short, genus-specific ORF. The upstream major ORF encodes two nonstructural proteins (NS1 and NS2). The NS1 protein has helicase and nickase activities, and it can induce apoptosis (5, 6) and cell lysis (7). The NS2 protein is translated from an alternatively spliced RNA (8). The downstream major ORF encodes a nested set of three C-terminally identical structural proteins (VP1 to VP3). A Ca-dependent phospholipase A2 (PLA2) enzyme located in the unique N-terminal region of VP1 (9) is necessary for viral infectivity.
The short genus-specific ORF overlaps the 5′ end of the ORF of the VP2 protein in all Protoparvovirus genomes, and it encodes a small alternatively translated protein (SATp) (10). SATp contains a single membrane-spanning α-helix, and it localizes in the endoplasmic reticulum (ER)-nuclear membrane network. The SAT knockout (SAT−) mutant of the attenuated NADL-2 strain of PPV shows a slow-spreading phenotype in cell culture. On the basis of the results of in vitro complementation studies, it was presumed that the function of SATp is to induce ER stress to facilitate cell lysis (10).
The ER plays a key role in protein folding and maturation. Immature proteins enter the ER lumen, where they are folded by molecular chaperons (e.g., Grp78, Grp94, calnexin) and protein-folding enzymes, like calreticulin and protein disulfide isomerase (11). At the end of the maturation process, only the correctly structured proteins are transported to destination sites with ER vesicles (12). Incorrectly folded proteins accumulate in the ER lumen, causing ER stress. Multiple transmembrane sensors of signaling pathways, including the protein kinase R-like kinase (PERK) (13), the inositol-requiring kinase/endoribonuclease 1 (IRE1), and ATF6 (14), detect the ER stress.
IRE1 has endoribonuclease and serine/threonine protein kinase activities, and its signaling pathway represents the most conserved element of the ER stress response (15, 16). After activation, IRE1 cuts out a 26-bp section from the mRNA of X box-binding protein 1 (Xbp1), which leads to a frameshift and a spliced Xbp1 protein (Xbp1s). Xbp1s is transported to the nucleus and regulates the expression of genes contributing to protein folding, glycosylation, and ER membrane biogenesis (17, 18).
The activated PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2α), which inhibits the translation of most mRNAs (19), thus reducing the protein load and easing ER stress (20, 21). At the same time, the translation rate of some ER stress-related proteins, including activated transcription factor 4 (ATF4), increases. ATF4 induces the translation of genes that regulate amino acid biosynthesis and transport (22). One of the ATF4-stimulated genes is the C/EBP homologous protein (CHOP) (23). Under prolonged or severe ER stress, CHOP irreversibly triggers programmed cell death (24, 25).
In the present study, we show that wild-type (wt) PPV infection induces irreversible ER stress, whereas the loss of SATp through mutations lessens this ER stress and leads to reduced apoptosis and cell lysis in virus-infected cells. ER stress-inducing drugs can compensate for the loss of SAT in vitro, and they accelerate the egress of wt PPV. This process seems to be ER stress specific, since UV stress does not induce a similar effect.
RESULTS
Spread of wild-type and SAT− mutant PPV Kresse strains.
Previously, we reported that the SAT− mutant of the attenuated NADL-2 strain of PPV has a slow-spreading phenotype (10). In the present study, our first step was to investigate whether the loss of SAT causes a similar effect in the virulent Kresse strain.
For this purpose, a SAT− mutant of the Kresse strain was first created by eliminating a potential second initiation codon of SAT (8 codons behind the initiation codon of SAT) and introducing a stop codon into the SAT coding frame in the infectious clone of the Kresse strain without changing the protein sequence of the VP proteins (Fig. 1). Then, porcine testis (PT) cells were infected with the rescued SAT− or wt virus at a low multiplicity of infection (MOI; 0.01). The differences between the spreading of the two virus strains were monitored by immunofluorescence (IF) staining using the 3C9 assembled capsid-sensitive primary antibody.
FIG 1.

DNA and amino acid sequences of the wild-type (A) and the SAT− mutant (B) Kresse strains (based on the U44978.1 sequence). The 2842nd and 2845th nucleotides were changed (T → A and T → C). These modifications did not change the amino acid sequence of VP1; however, they led to a stop codon in the SAT ORF and to the substitution of the nearest methionine.
The first positive cells appeared at 12 h postinfection (p.i.), with both the wt and the SAT− mutant viruses indicating similar kinetics in viral entry, decapsidation, replication, viral protein synthesis, and capsid assembly. The number of positive cells continuously grew until 20 h p.i., when the first secondary infections could be detected in the wt virus-infected cells in the form of fluorescence foci, while the first signs of reinfection were visible only at 24 h p.i. in the case of the SAT− mutant virus (Fig. 2A). Incubation of the cells infected with the virus at a low MOI with the 3C9 neutralizing antibody abolished the appearance of large fluorescent foci (FFs) at 24 h with both the wild-type and the SAT− viruses, proving that large FFs are indeed the result of secondary viral infection. (Fig. 2B; only data for the wild type are shown). The difference in virus spreading became even more obvious as the infection progressed. At 48 h p.i., the wt virus infected almost every cell, in contrast to the findings for the SAT− strain. Changes in the infectious titer and the viral copy number of the supernatant correlated well with the spreading pattern revealed by IF staining (Fig. 3A). Thus, the loss of SATp in the pathogenic Kresse strain and the attenuated NADL-2 strain (10) resulted in similar phenotypes.
FIG 2.

Spread of the wild-type and the SAT− mutant viruses in PT cells at a low multiplicity of infection (MOI, 0.01). Infected cells (red) were visualized with the assembled capsid-specific 3C9 primary antibody and the CF594 secondary antibody. (A) Cells were fixed at the indicated time points, and their nuclei were labeled with Hoechst 33342. (B) 3C9 antibody was added to Kresse-infected cells to monitor the inhibition of the secondary infections. The times of the beginning of the treatments are indicated. The cells were fixed at 24 h p.i.
FIG 3.

Change of the viral copy numbers during infection in the medium of PT cells. (A) Copy numbers and infectious titers (triangles and columns, respectively) at a low multiplicity of infection (MOI, 0.01). (B) Total copy numbers and copy numbers measured after DNase treatment (triangles and columns, respectively) at a high multiplicity of infection (MOI, 3). Error bars indicate standard deviations.
The use of a high multiplicity of infection resulted in impaired egress for the SAT− Kresse strain. The copy number of the wt virus in the supernatant started increasing sharply at 20 h p.i. as a consequence of the mass release of the viruses and viral DNA from a large number of infected cells (Fig. 3B). The increase in the copy number of the SAT− mutant virus started later (between 22 h and 24 h p.i.), and the number of viral genomes in the supernatant remained constantly less than that of the wt virus. The biggest difference between the copy numbers was at about 48 h p.i., and it decreased gradually until the end of the monitoring period.
DNase treatment of the supernatants revealed that the majority of the viral copies detected in the supernatant of the infected cells by quantitative PCR (qPCR) came from DNase-sensitive replicative forms and partially or nonencapsidated genome forms, which were probably released from the dying cells (Fig. 3B). The difference between the DNase-resistant copy numbers in the supernatants of the two strains decreased to a minimal amount by the end of the monitoring period (84 h p.i.), indicating very similar amounts of packaged DNA and infectious virus production. Determination of the titers of the wt and SAT− virus stocks on PT cells (4.85 × 109 and 2.56 × 109, respectively) indeed revealed very similar levels of production of infectious particles. This result was also congruent with our earlier findings with the NADL-2 strain and its SAT− mutant (10).
We theorized that the quicker release of the wt virus must be the consequence of the earlier cell death and lysis induced by SATp in the host cell. To better understand the function of SATp, we first studied the phenotypic effects, including viability, cell lysis, and apoptosis of SAT− and wt viruses on infected cells.
Cytopathogenic effects of viral infection.
First, lactate dehydrogenase (LDH) release, as an indicator of cell lysis, was quantified from cells infected with virus at a high MOI (26) (Fig. 4A). From 18 h p.i. to 48 h p.i., the level of LDH released from wt virus-infected cells started to increase much faster (exponentially; R2 = 0.9946) than that from SAT− virus-infected cells, from which the increase was logarithmic (R2 = 0.9639) during this interval. The measured values reached an approximately 2-fold difference at 48 h p.i. in the supernatant of wt virus-infected cells. At this time point, barely any of the wt virus-infected cells remained alive, while the majority of the SAT− virus-infected cells remained adherent (Fig. 4B). The extracellular LDH activity of SAT− virus-infected cells started to sharply increase after 64 h, and that increase was preceded by a rapid decrease in the attached cell count. Monitoring of the attached cells with propidium iodide (PI) revealed that the membrane integrity of the SAT− virus-infected cells was also sustained for a longer time and in more cells than that of the wt virus-infected cells (Fig. 4C). The number of pyknotic and karyorrhectic nuclei remained relatively low during the course of infection with both viruses (less than 14% and 8% in the case of the wt and SAT− viruses, respectively) (Fig. 4D). Nevertheless, their rates were always higher in wt virus-infected cells than among SAT− virus-infected cells, and the difference became and remained statistically significant (P < 0.046) starting at 28 h p.i. Plasma membrane blebbing could hardly be seen in infected cells, while their nuclei were frequently enlarged (Fig. 4E and 5).
FIG 4.

Different forms of cell death during PPV Kresse infection in porcine testis cells. The error bars represent 1 standard deviation. (A) LDH activity in the supernatant of infected and control cells as an indicator of total cell death. The maximum absorption value (lysed control uninfected cells) was 1.79 at 18 h p.i. (B) Attached cell count as an indicator of viability of cells. The number of uninfected cells at 24 h represents 100%. (C) Proportion of attached propidium iodide-positive (PI+) cells (the values for wt virus-infected cells were calculated until 48 h p.i.). (D) Proportion of attached pyknotic and karyorrhectic cells, calculated by Hoechst staining (values for wt virus-infected cells were calculated until 48 h p.i.). (E) Swelling of nuclei at 22 h p.i.
FIG 5.
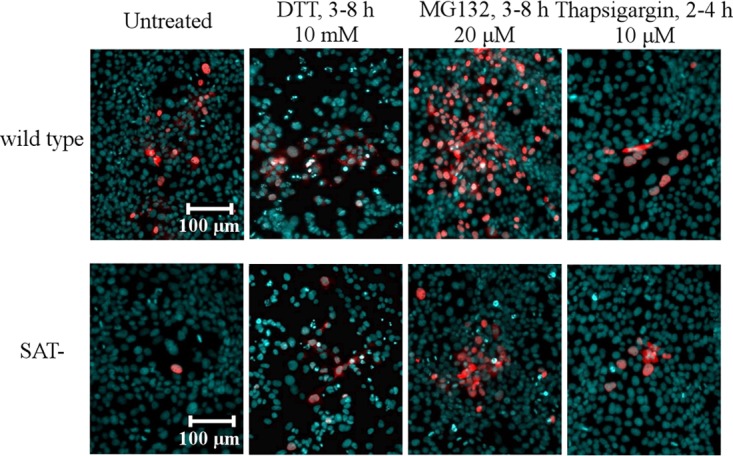
Spreading of the PPV wild-type and SAT− strains after ER stress inducer treatments at a low MOI (MOI, 0.01). Infected PT cells were fixed at 20 h p.i. The 3C9 anticapsid monoclonal antibody was used for the detection of infected cells (red). Cell nuclei (blue) were visualized by Hoechst staining. The concentrations of chemicals and the duration of treatments (for the indicated times [in hours] postinfection) are indicated.
The results of these experiments highlight that lysis rather than apoptosis is the main form of cell death during PPV infection in PT cells. They also reveal that the loss of SAT prolongs cell life throughout PPV infection and it decreases the number of both lysed and necrotic cells during the course of infection.
Effects of ER stress inducers.
Since SATp accumulates in the ER, it seemed plausible to presume that SATp facilitates early cell death and virus release through ER stress induction. To gather evidence on the effect of ER stress on PPV egress, the influence of ER stress-inducing drugs on infected cells was investigated. PT cells were infected with the wt and SAT− Kresse strains at a low MOI (MOI, 0.01), and the cells were treated at different time points with 10 mM dithiothreitol (DTT), 20 μM MG132, or 10 μM thapsigargin. The cells were fixed at 20 h p.i. and monitored for the presence of plaque-like FFs of PPV by IF (Fig. 5). The SAT− virus did not induce FFs in untreated cells at 20 h p.i., and only individual cells were positive for the virus.
Treatments with all ER stress inducers moderately increased the sizes of the FFs in wt virus-infected cells, but most importantly, they also induced FFs in SAT− virus-infected cells, regardless of the starting time of the treatment. MG132 treatment starting at 3 h p.i. gave the biggest and highest number of FFs. Under these conditions, the FFs of the SAT− virus were similar in size or even bigger than the FFs of wt virus in untreated cells (Fig. 5).
The same treatments (10 mM DTT or 20 μM MG132) gave similar results at a high MOI (MOI, 3), where the virus titer in the culture medium at 24 h p.i. was quantified by qPCR (Fig. 6A). Both ER stress inducers substantially increased viral egress into the medium. DTT treatment starting at 7 h p.i. had the strongest effect on wt virus release (4.41 times), while MG132 treatment starting at 3 h p.i. induced the highest increase in titer in the medium of cells infected with the SAT− virus (71.67 times). Unexpectedly, almost all of the tested chemical treatments (with the exception of the DTT treatment starting at 7 h p.i.) increased the SAT− virus titer above not only the titer of basic wt virus but also that of the chemically enhanced wt virus.
FIG 6.
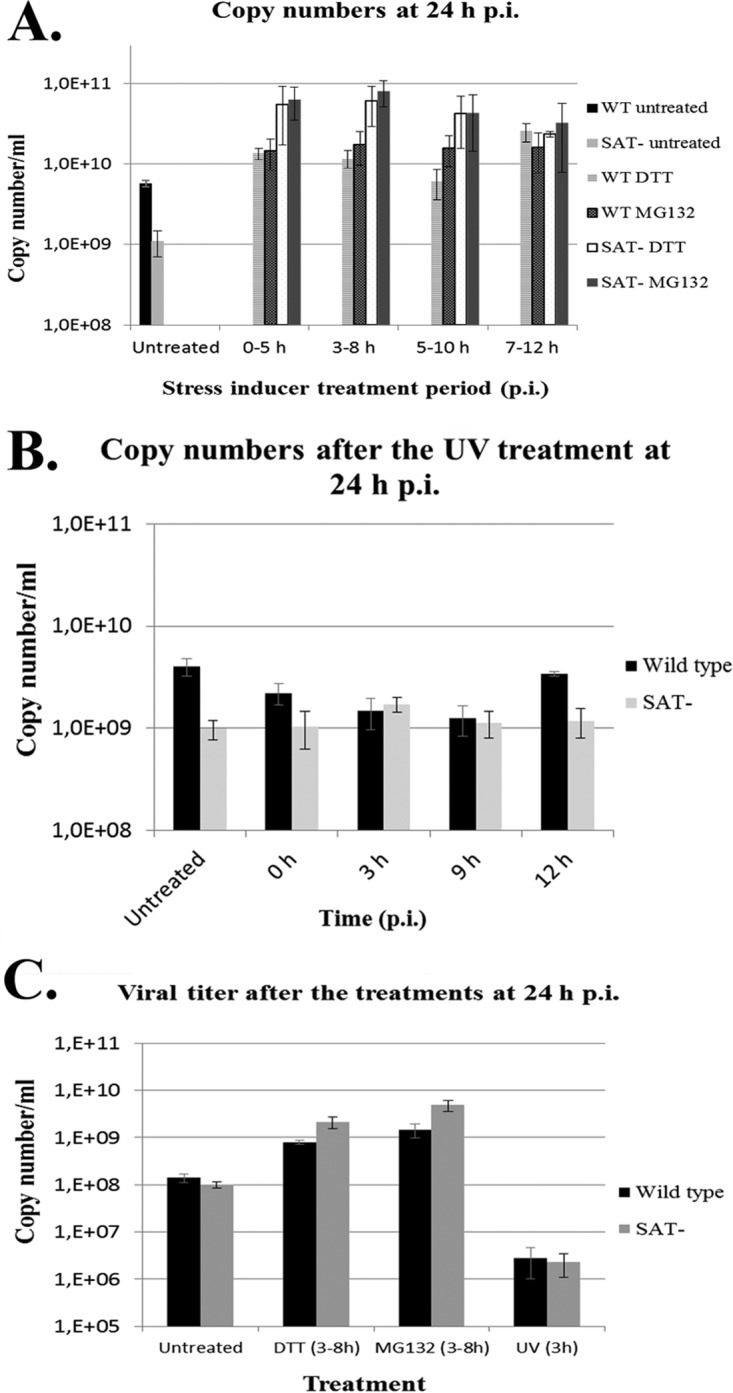
Changes of viral copy numbers in the medium at a high multiplicity of infection (MOI, 3) after stress inducer treatments. All supernatants were harvested at 24 h p.i. (A) Cells were infected with the wild-type and SAT− strains and treated with ER stress inducers (20 μM MG132 and 10 mM DTT) for different times. (B) Cells were infected with the wild-type and SAT− strains and treated with UV-C light (sublethal dose) at different times p.i. for 5 min. (C). Infectious titer of supernatants of differently treated infected cells.
Back titration of the supernatants of ER stress inducer-treated cells verified that the increase in the viral copy number correlates with the increase in the number of infectious particles in the supernatants (Fig. 6C).
To investigate the specificity of the ER stress among other stress factors for acceleration of viral egress, the effect of UV radiation was also examined. Short-term sublethal UV-C light treatment (5 min) did not change the viral copy numbers considerably, while it significantly reduced (to ∼2% of the initial number; P < 0.005) the number of the infectious particles in the supernatant (Fig. 6B and C).
The results of these experiments strongly suggested that ER stress indeed facilitates the release of mature particles from infected cells, and ER stress seems to be specific in this regard, because UV-C radiation was not able to induce a similar effect.
Detection of ER stress.
To further clarify the relation of ER stress and SATp to the acceleration of viral egress, the pattern of ER and ER stress markers in infected cells was studied. PT cells were infected with the SAT− and the wt Kresse strains (MOI, 3), and the levels of calreticulin, as an ER marker, as well as those of Xbp1 and CHOP, as downstream ER stress response markers, were monitored by IF staining. PPV infection—with or without SAT—caused a condensation in the ER membranes of the infected cells. Perinuclear nodes and clots could be detected, implying fragmentation and fusion of the tubular network as obvious signs of ER stress (27–29) (Fig. 7). However, no significant differences between the wt virus- or SAT− virus-infected cells in the starting time or in the pattern of the disintegration in attached cells were found.
FIG 7.

Morphological changes of the ER in PT cells during PPV infection. To visualize the ER and the viral particles, anticalreticulin antibody (red) and anticapsid (green) antibodies, respectively, were used.
There was also no difference between SAT-positive (SAT+) and SAT− viruses until 48 h, when the reversible ER stress marker Xbp1 was monitored during viral infection (Fig. 8A). Xbp1 was mainly detected in the nuclei of infected cells, with the earliest detection time being about 14 h p.i. The presence of the protein was temporary, and the protein level peaked sharply at 16 to 18 h p.i., when about 95% of the infected cells showed positivity. After that it rapidly declined: at 20 h p.i., Xbp1 could be detected in only less than 5% of the cells. Low-level Xbp1 expression was detected again in SAT− virus-infected cells at 60 h p.i. However, that was most probably the consequence of ER stress induced by nutrient starvation (30) because a similar level of Xbp1 expression was also visible in noninfected cells at the same time (Fig. 8C).
FIG 8.
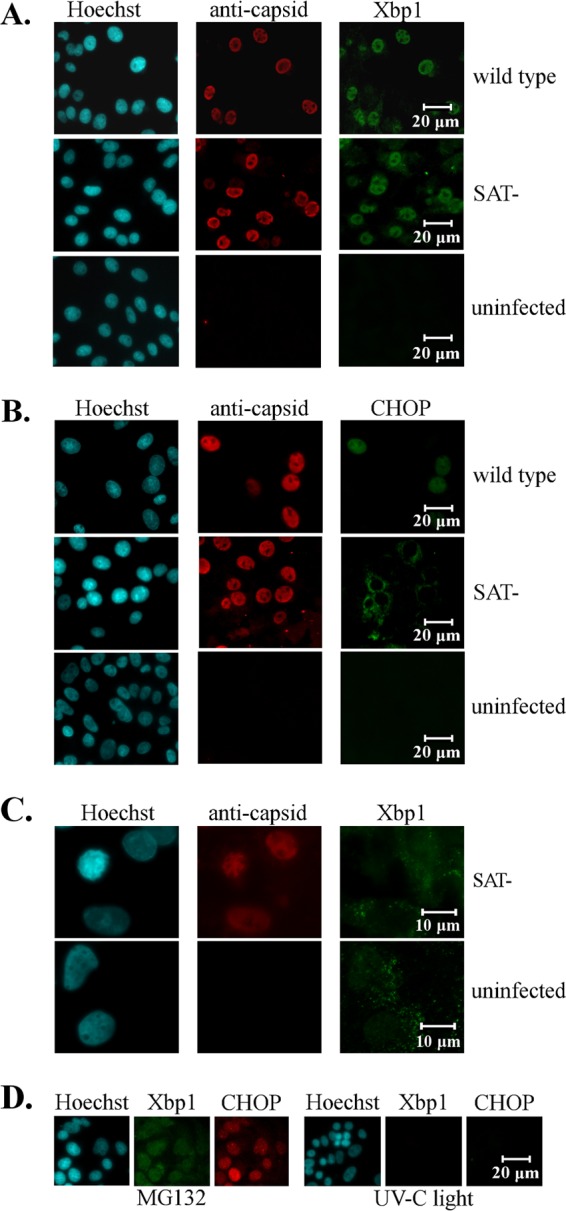
Detection of ER stress protein markers during PPV infection. Cell nuclei (blue) were visualized by Hoechst staining. (A) Activation of the Xbp1 reversible ER stress marker after wt and SAT− strain infection. Xbp1 (green) and viral capsid (red) were labeled with anti-Xbp1 and anticapsid antibodies, respectively, at 18 h p.i. (B) Activation of the CHOP irreversible ER stress marker after wt and SAT− strain infection. CHOP (green) and viral capsid (red) were labeled with anti-CHOP and anticapsid antibodies, respectively, at 24 h p.i. (C) Activation of Xbp1 in SAT− virus-infected and noninfected cells at 60 h p.i. (D) Induction of Xbp1 and CHOP after 20 μM MG132 treatment and sublethal UV treatment at 12 h.
Among the monitored markers, only the irreversible ER stress marker CHOP showed a difference between wt and SAT− virus infection (Fig. 8B). In the wt virus-infected cells, CHOP was first detected at 22 h p.i. in about 20% of the cell nuclei. Its expression plateaued at 24 h p.i., with 75% of the infected cells being positive, and remained relatively unchanged until 36 h p.i. After 36 h, very few attached infected cells showed positivity for CHOP. Although CHOP was also detected at 22 h p.i. in about 20% of the nuclei of SAT− virus-infected cells, instead of nuclear localization, it was detected perinuclearly in the cytoplasm. Its level also plateaued at 24 h p.i., but only 41% of the cells were positive; this value essentially remained unaffected until 36 h for attached cells, and the perinuclear localization of the protein did not change either. CHOP could not be detected in SAT− virus-infected cells after 48 h p.i.
These experiments show that PPV infection induces ER stress regardless of the presence or absence of SATp. However, wt virus expressing SATp was able to activate the expression of CHOP in significantly more cells than the SAT− virus, and SATp also influenced the localization of cell death, triggering CHOP in infected cells.
Since chemical ER stress inducers were able to compensate for the loss of SAT, their effect on the induction of ER stress markers was also monitored. As expected, the chemicals induced Xbp1 and CHOP expression in the treated cells (Fig. 8D). CHOP expression could be detected in 100% of the treated cells at 18 h and 8 h after the beginning of MG132 and DTT treatment, respectively. However, the chemical treatments always triggered the nuclear localization of CHOP similarly to wt virus infection but contrary to SAT− virus infection. Sublethal UV treatment of the PT cells did not induce either Xbp1 or CHOP expression.
These findings give additional support to the possible role of nuclearly localized CHOP in the acceleration of cell lysis.
Effect of the cloned SAT protein in PT cells.
To further explore the role of SATp in the induction of ER stress markers, a SAT-DsRed fusion protein-expressing vector was transfected into PT cells. Transfected cells were fixed every 4 h during the period from 16 to 48 h posttransfection (p.t.) and examined for Xbp1 and CHOP expression by IF. Neither protein was induced in SAT-expressing cells during the monitoring period (data not shown), despite the fact that in their ERs morphological alterations (condensation in the perinuclear region) similar to those seen in infected cells could be detected (Fig. 9). During the monitoring period, SATp always cocompartmentalized with calreticulin (Fig. 9). Furthermore, the SAT-DsRed protein induced apoptosis (Fig. 9) in the late phase of transfection (from 30 h posttransfection). After 48 h p.t., only a very few SAT-DsRed-expressing cells could be detected in the transfected wells, while cells expressing the control DsRed protein remained viable and detectable even after 90 h p.t.
FIG 9.

Morphological changes of the ER in SAT-DsRed fusion protein-expressing PT cells. Cells were transfected with a SAT-DsRed-expressing plasmid and, as a control, with a DsRed (red)-expressing plasmid. The calreticulin was labeled by anticalreticulin monoclonal antibody (green), and the cell nuclei (blue) were visualized by Hoechst staining. The cells were fixed at different times after transfection. Arrows, apoptotic nuclei.
These experiments indicate that SATp alone is toxic and its accumulation has an effect on the morphology of the ER. However, without the other viral proteins, SATp is not able to activate either Xbp1 or CHOP expression.
Effect of nuclear CHOP on viral spread.
Since the presence of the nuclearly localized CHOP in the infected cells showed a strong correlation with accelerated viral egress, we further studied the role of CHOP in the process. We cloned the porcine CHOP and made a construct constitutively expressing the porcine CHOP-DsRed fusion protein. Many studies have reported that transiently expressed mammalian CHOP from a transfected plasmid accumulates in the nucleus. Indeed, the transfection of our construct alone or together with the pSAT-negative (pSAT−) or the pKresse (pUC19 into which the PPV Kresse genome was cloned) infectious clones resulted in the nuclear localization of the CHOP-DsRed protein (Fig. 10A). The CHOP-DsRed protein proved to be functionally active because it could induce apoptosis in the transfected cells as early as 18 h p.t. (Fig. 10B) and it killed almost all transfected cells at 48 h p.t. (data not shown). However, when the CHOP-DsRed-expressing construct was cotransfected with pSAT−, it did not increase the size of FFs, in contrast to the SAT-DsRed-expressing construct, which readily induced large FFs (compared to the size of FFs in cells transfected with the pSAT− plasmid only) (Fig. 10C). This experiment revealed that nuclear CHOP alone is not able to induce in the nucleus such transcriptional changes that would accelerate cell lysis and viral spread.
FIG 10.

Localization of porcine CHOP and its effect on the spreading of the SAT− virus strain. The viral capsid was labeled with anticapsid antibodies (green), while the nuclei (blue) were visualized by Hoechst staining. (A) Nuclear localization of the CHOP-DsRed fusion protein (red) in infected and noninfected cells. CHOP-DsRed-expressing plasmids were transfected with wild-type and pSAT− infectious clones or alone into PT cells and fixed at 24 h posttransfection. (B) Cytopathic effect of CHOP-DsRed at 18 h posttransfection. Arrow, fragmented nuclei. (C) Spreading of the SAT− strain after cotransfection of the pSAT− infectious clone with CHOP-DsRed- and SAT-DsRed-expressing plasmids. As a positive control, the wild-type clone was transfected with DsRed-expressing plasmid. Cells were fixed at 96 h posttransfection, and the blue (nucleus) and green (virus-positive cells) channels were merged.
DISCUSSION
Quantification of virus in the medium of cells infected with PPV at a high and a low MOI by qPCR and titration revealed that SATp induces early viral release but does not increase the final amount of virus produced (Fig. 3).
The dynamics of the increase in the viral titer in the medium of cells infected at high and low MOIs were quite different. At a low MOI, differences in levels of infection of orders of magnitude between the mutant and the wild-type virus could be detected during the 40th to 64th hours of infection (Fig. 3A). The accumulating effect of the 3- to 4-h delays in the first viral release in every consecutive replicative cycle, together with the sharp decrease in the amount of dividing cells that support parvoviral replication after 28 h p.i. (the cell number grew to more than 2.5 times the seeding number in the first 28 h [Fig. 4B], while in the next 60 h the growth rate was drastically reduced), can provide a plausible explanation for the phenomenon.
It may look surprising that at a high MOI, when all of the cells were infected at the same time, the difference was much smaller. However, we could expect to see orders-of-magnitude differences only if either or both of the following premises were fulfilled: (i) one of the viruses produced much more progeny than the other in the cells, or (ii) at a given time point, orders-of-magnitude more cells lyse and release progeny viruses. We showed in an earlier paper as well (10) that SAT− and wild-type viruses produce equal amounts of progeny viruses. In order for the second premise to be true, magnitudes of more cells should be dying among SAT− virus-infected cells than wild-type virus-infected cells at any given time, and that was obviously not the case. The biggest differences in DNase-resistant viral copy numbers (6.5 times) could be seen at about 48 h p.i. (Fig. 3B), after 95% of the wild-type virus-infected cells detached and started to lose their integrity (indicated by the attached cell count at 42 h p.i.) and they lysed en masse (see the jumps in LDH activity at 48 to 64 h p.i.). At the same time, about 15% of the SAT− cells were detached and lysed (Fig. 4). The difference in detached cell numbers of 95% versus 15% is big; however, their ratio was only 6.33-fold, which is a pretty good match to the measured 6.5-fold difference in viral copy number in the medium, given that all the dead cells produced equal amounts of viruses.
Interestingly, wild-type virus-infected cells released more DNase-sensitive viral DNA than SAT− virus-infected cells (Fig. 3B). Since DNA is a potent immunostimulator acting through several DNA-sensing receptors (31), this phenomenon may have potential therapeutic significance for the development of oncolytic parvoviruses (32).
It was reported earlier that minute virus of mice and parvovirus H-1, close relatives of PPV, are actively transported in vesicles from the nucleus to the cell periphery and released into the culture medium (33). However, we were not able to detect any assembled capsid in the cytoplasm of either wt virus- or SAT− virus-infected cells at a low MOI (MOI, 0.01) or even at a high MOI (MOI, 3) (Fig. 2 and 5) until the cells started to lyse en masse (for wt virus, at 40 h p.i. at an MOI of 3; for SAT− virus, at 64 h p.i. at an MOI of 3) and a large amount of virus was released into the culture medium and reinfected the remaining cells. This observation suggests that vesicular transport does not play a significant role in the egress of PPV, at least not in PT cells.
Members of the Protoparvovirus genus can induce either necrosis or apoptosis, depending on the virus and on the cell type (34). Parvovirus infection is also able to activate early apoptotic events that do not go to completion and can lead to necrotic cell death, as demonstrated by H-1 infection in HeLa and P1 cells. In the case of PPV, it was reported that the YL strain induces apoptosis in ST and PK-15 cells and in the late phase of infection (60 h p.i.) the number of apoptotic cells can reach 50% (35, 36). In contrast, during Kresse strain infection in PT cells, the number of apoptotic cells remained low, as indicated by the number of pyknotic and karyorrhectic nuclei, and it never exceeded 14%, regardless of the presence or absence of SATp. Since subtle mutations of the PPV capsid can modify interactions with host factors and change the cytopathic effect of the virus (37, 38), it is hard to pinpoint whether the viral strains or the cell lines used are responsible for the observed differences.
It is worth mentioning that neither pyknosis nor karyorrhexis is an exclusive characteristic of apoptosis, as both of these are frequently detected in nonapoptotic, necrotic cells as well (necrotic pyknosis). Strikingly, we never detected any nuclear fragmentation inside an infected cell with a well-defined nuclear boundary. Apoptotic nuclei were readily produced in SAT- and CHOP-expressing cells (Fig. 9 and 10), and they were also demonstrated in parvoviral NS1-transfected cells (39). Our findings suggest that cell death in PT cells during PPV infection is not characteristically apoptotic even if apoptotic pathways are involved, as indicated by CHOP induction. Swelling of the infected nuclei, a lack of blebbing, early cell membrane failure (as indicated by PI uptake), rapid LDH release, and free viral DNA release all point toward necrosis as the main form of cell death in PT cells during PPV infection.
ER stress inducers could mimic the effect of SATp. To minimize the chance that the observed result is the consequence of unknown side effects, three differently acting chemicals (DTT, MG132, and thapsigargin) that are commonly used for the activation of the unfolded protein response (UPR) (40–42) were tested. DTT reduces the disulfide bonds (43) of the proteins and results in the accumulation of un- or misfolded proteins. MG132 is a proteasome-specific protease inhibitor (44) that blocks the degradation of proteins, while thapsigargin is a sarcoendoplasmic reticulum Ca2+-ATPase inhibitor, and it increases the free Ca2+ concentration in the cytosol (45). Thapsigargin was shown to inhibit parvoviral infection. Boisvert et al. (46) also showed that sustained MG132 treatment starting at the early stage of PPV NADL-2 infection blocks infection (at 0 to 20 h p.i. treatment, ∼100% inhibition; at 8 to 20 h p.i. treatment, ∼50% inhibition). A similar MG132 treatment induced very similar effects on PPV Kresse infection, as we detected it by monitoring the effects of a low MOI (not shown). In fact, all tested inducers had an inhibitory effect on viral infection when they were applied at a higher concentration or over an extended period of time. However, they enhanced the spreading of the SAT− strain at the low multiplicity of infection (MOI, 0.01), and two of them (thapsigargin was not tested) increased the level of viral release at the high MOI under the applied conditions. This happened despite the fact that DTT and thapsigargin were visibly toxic to the cells. Obviously, the concentration and the timing of the treatments fundamentally influenced the outcome of the experiments, indicating a delicate balance between the opposing effects of these chemicals on the infectious process. Many chemicals can be used to demonstrate a negative effect on parvoviral infection. The special feature of this work is that, by using inhibitors, it demonstrates a positive effect on this process.
At the concentration applied, all inducers switched on Xbp1 and CHOP expression, which led to the death of 100% of the cells in 48 h. Interestingly, switching the UPR early on not only beneficially influenced viral egress, as indicated by the titer of the treated cells infected at a high MOI (Fig. 6), but also, at least seemingly, it did not have a large negative effect on parvoviral entry and replication; otherwise, second-cycle replication and the increase in the amount of FFs could not have been detected in cells that were infected at a low MOI and that were already in the stage of UPR (Fig. 5).
UV stress did not produce any enhancement of PPV release, and it did not activate Xbp1 or CHOP expression (Fig. 8C). Earlier it was shown that pretreatment of cells with UV light facilitates the replication of adeno-associated virus (47, 48); however, it does not significantly affect the replication of H-1 parvovirus, though it increases the mutation rate of the virus in a dose-dependent fashion (49). We found that sublethal UV stress applied to the cells after infection did not significantly affect PPV output but, not surprisingly, considerably reduced (∼50 times) the infectious titer of the progeny viral stocks (Fig. 6C). The probing of different stress factors strongly suggested that ER stress indeed facilitates the release of mature particles from infected cells, and ER stress seems to be specific in this regard, because UV-C radiation was not able to induce a similar effect.
Our experiments with wt and SAT− viruses revealed that PPV, similarly to numerous other viruses from different viral families (50–54), induces ER stress and UPR in infected cells, as demonstrated by the morphological changes of the ER and expression of Xbp1 and CHOP (55). It is suspected that the different localization of CHOP depends on the state of the cells and the intensity of the stress effect (56). In wt virus-infected cells, CHOP was detected in the nuclei of the majority of the cells, indicating severe ER stress, while in SAT− virus-infected cells, CHOP was also expressed, but to a lesser extent (41% of SAT− virus-infected cells versus 76% of wt-infected cells), and its localization remained perinuclear (Fig. 8B). It is generally accepted that nuclear CHOP is one of the most important mediators of cell death during ER stress (25, 55). CHOP is a transcriptional activator of several proapoptotic proteins (e.g., GADD34, DR5, Ero1α) and inhibits the transcription of the antiapoptotic Bcl-2 (57, 58). On the other hand, it was also shown in mouse embryo fibroblasts that forced expression of CHOP alone or together with ATF4 does not increase the expression of cell death-related genes and only sensitizes the cell to ER stress-induced cell death (59). Besides CHOP expression, increased protein synthesis and oxidative stress were required to induce cell death. In our case, expressed porcine CHOP was clearly detrimental to PT cells, causing nuclear fragmentation and cell death (Fig. 10B) in the majority of cells at 48 h posttransfection. These data suggest that the outcome of CHOP expression can depend on the cell type and on the actual status of the cell.
The presence of SATp clearly influences the localization of CHOP and the outcome of the ER stress response during PPV infection. It is likely that SATp increases the stress effect in the infected cells, which leads to the nuclear accumulation of CHOP and to the induction of proapoptotic or other pathways that accelerate the death of infected cells. The effect of overexpressed SATp in transfected cells supports this theory. SATp alone is able to change the morphology of the ER and can induce apoptosis (Fig. 9). It is clear that the influence of SATp on the activation and localization of CHOP cannot be direct, because by itself it cannot induce CHOP expression (data not shown). It occurred to us that the DsRed tag can influence the functional interactions of SATp, but we transfected tagless SATp-expressing plasmids into PT cells and were not able to detect Xbp1 or CHOP expression either. The functionality of SAT-DsRed is further supported by the fact that in a cotransfection experiment the protein could complement the missing function of native SATp (Fig. 10C).
However, no matter which way SATp stimulates the expression of CHOP, it seems that the induction of this protein and its nuclear localization cannot alone be responsible for the biological effect of SATp, since nuclear CHOP could not complement the lack of SATp in a coexpression experiment (Fig. 10C). Since severe ER stress emulates the effect of SATp, one of the most likely mechanisms of action of SATp is that it influences protein interactions in the ER, which make the UPR response more severe. This process may lead to the activation of the PERK-eIF2α-ATF4-CHOP pathway, where one or more of the proteins upstream of CHOP also induce alternative pathways. These may supplement the effect of CHOP, leading to early cell death. In an alternative scenario, SATp may induce another ER stress response pathway(s) besides UPR (e.g., the ER overload response), and this effect alone or synergistically with the CHOP pathway could cause early cell death.
MATERIALS AND METHODS
Cells, viruses, and transfection.
The PT cell line was used in all experiments. The cells were grown in a Dulbecco modified Eagle medium (DMEM)-based medium (high glucose [4.5 g/liter]; PAA Laboratories) supplemented with 10% serum (fetal bovine serum gold; PAA Laboratories), 1% penicillin-streptomycin (PAA Laboratories), and 1% sodium pyruvate solution (Lonza) in the presence of 5% CO2 at 37°C.
For infection and transfection, the cells were seeded in 24-well tissue culture plates (1 × 105 cells/well), and they were infected or transfected with either the wt PPV strain Kresse or the SAT− mutant of the PPV Kresse strain at about 50% cell confluence. We used a multiplicity of infection (MOI) of 0.01 to monitor viral spread and an MOI of 3 (the optimal MOI to use a minimal number of viruses to target the maximal number of cells) in the experiments investigating virus release, apoptosis, viability, and ER stress. The infectious titer of the viral stocks was determined as described earlier (10). Data from four independent dilutions were averaged.
The wt and mutant viral stocks were created by transfecting PT cells with pKresse (pUC19 into which the PPV Kresse genome was cloned [37]) and pSAT− plasmids, respectively. By using the method described earlier, two nucleotides of pKresse (T-2842 → A and T-2845 → C) were changed to knock out SAT and to create pSAT− (10) (Fig. 1).
Fusion constructs alone were transfected into adherent cells using the TurboFect transfection reagent (Thermo Scientific) following the supplier's recommendations.
Cotransfection of the infectious clones with fusion constructs was performed in suspension cell cultures as follows. One microgram infectious clone and 1 μg fusion construct DNA were mixed with 3 μl the TurboFect reagent in 100 μl DMEM and incubated for 20 min at room temperature. The transfection mix was then added to 1 ml freshly trypsinized, suspended PT cells (1 × 105 cells/ml in DMEM with 10% FBS) and incubated on a gently rocking platform for 3 h at room temperature. Cells were centrifuged at 1,000 × g for 1 min, resuspended in 1 ml fresh medium, and plated in 24-well plates.
Fusion constructs.
DsRed-labeled SAT and CHOP protein constructs were created in DsRed-monomer-N1 plasmids (Clontech). The SATp and CHOP sequences were amplified by use of the primers PPV-SATf (GC GGTACC ATG TGG AAC AAC ACA ACC CTA), PPV-SATr (CG GGTACC TT GAT GTA TGA GTC TTG ATG CGT), F-CHOP-DsRed (GC AAGCTT ATG GCA GCT GAG TCA TTG CCT), and R-CHOP-DsRed (GC GGATCC CG TGC TTG GTG CAG ATT AAC CAT) and Phusion Hot Start DNA polymerase. To get the template for CHOP, total RNA was purified with the RiboZol RNA extraction reagent (Amresco) and reverse transcribed with SuperScript III reverse transcriptase (Thermo Fisher Scientific) using the R-CHOP-DsRed primer according to the manufacturer's recommendation.
The isolated PCR fragments and the vector were digested with the KpnI or HindIII and BamHI restriction enzymes and were ligated with T4 ligase (Thermo Scientific).
IF staining.
For immunofluorescence (IF) staining, the cells were plated on coverslips. They were fixed at the appropriate time with 3% formaldehyde and incubated for 30 min at room temperature. After two washing steps (1.5 g bovine serum albumin dissolved in 300 ml 1× phosphate-buffered saline [PBS]), they were permeabilized using 1% Triton X-100 (Sigma-Aldrich). After 15 min, the cells were washed twice, 5% inactivated horse serum (diluted in PBS) was loaded into the wells, and the plate was incubated for 30 min at room temperature. After two washings, the cells were exposed to the primary antibodies 3C9 (a mouse anti-PPV capsid-specific monoclonal antibody; CRL-17; ATCC), mouse anti-CHOP (Thermo Scientific), and rabbit anti-Xbp1 (Santa Cruz Biotechnology) and the serum of PPV-infected swine for 60 min at room temperature. After further washings, the cells were incubated with their respective secondary antibodies (anti-mouse immunoglobulin CF594, anti-mouse immunoglobulin CF488, anti-rabbit immunoglobulin CF488a, or anti-swine immunoglobulin CF586 antibody [Biotium]) for 60 min at room temperature in the dark. After the final washings, the cover glasses were removed from the wells and were fixed onto a slide using Fluoroshield histology mounting medium (Sigma-Aldrich) according to the manufacturer's protocol. An Axio Observer D1 inverted fluorescence microscope (Zeiss) was used for visualization.
Fluorescent focus growth inhibition.
To find the neutralizing concentration of the 3C9 monoclonal antibody, the supernatant of the 3C9 hybridoma was diluted 10, 20, 50, or 100 times in complete medium, 50 μl of the diluted solutions was mixed with 50 μl viral stock at an MOI of 0.01, and then the mixture was incubated for 1 h at room temperature. After incubation, 100 μl of the medium containing the antibody-virus complexes was loaded onto 50% confluent PT cells on a 96-well plate, and the cells were incubated at 37°C for viral growth. After 24 h, the cells were fixed and the progression of infection was detected by the standard IF method described above. Supernatant diluted 20 times completely blocked the PPV infection. In the next step, the PT cells were infected with 50 μl low-MOI viral stocks (MOI, 0.01) and incubated at 37°C. After 4 h the supernatant was removed, 50 μl medium with 3C9 diluted 20 times was added, and the cells were incubated at 37°C for an additional 20 h. After the incubation, the cells were fixed and monitored for viral infection by the standard IF method.
Apoptosis and viability experiments.
For the investigation of apoptosis and lysis, the cells were seeded at 50% confluence (2.5 × 105) in 24-well plates, and after 3 h they were infected with viruses. Live, unfixed, infected, and uninfected cells were incubated with 1 μg/ml Hoechst 33342 and with 0.25 μg/ml propidium iodide at different time points (18 h to 88 h p.i.) for 60 min at room temperature in the dark. Then they were washed with PBS and examined under a microscope, and several photographs of the central regions of the wells were taken in the blue and the red channels. All cells, including PI-positive and apoptotic cells, were counted from three to six photographs of each well, and the numbers were averaged. Pyknotic nuclei were identified by strong staining with Hoechst, fragmentation, or shrinkage of the nucleus to 1 to 4 μm (60). More than a thousand cells (on a minimum of four photographs) were counted for every time point (except when the attached cell count went under 20% of the total) by two independent persons, and the data were averaged. The Mann-Whitney U test was applied for statistical analysis of the data, where the null hypothesis was that the two samples came from the same population.
Lysis was quantified on the basis of lactate dehydrogenase (LDH) enzyme activity (61, 62) by use of a cytotoxicity detection kit (Roche) following the manufacturer's instructions. After 1:1 dilution of the supernatants with PBS, the absorbance at 490 nm was measured in the linear range by an EL × 800 enzyme-linked immunosorbent assay plate reader (Dialab GMBH, Austria). The absorbance of a sample was calculated by subtracting the background value for the serum from the measured value.
Real-time quantitative PCR.
The supernatants of the infected cells were sampled at between 0 h p.i. and 84 h p.i. The viral DNAs were purified with a High Pure viral nucleic acid kit (Roche) according to the manufacturer's protocol. The qPCR solution (25 μl) contained 18.25 μl water, 2.5 μl 10× DreamTaq buffer (Thermo Scientific), 0.5 μl a deoxynucleoside triphosphate mix (2 mM each), 1 μl template DNA from the supernatants, 0.5 μl DreamTaq DNA polymerase (Thermo Scientific), 1.25 μl 20× EvaGreen dye (Biotium), 0.5 μl forward primer (CTT TAG CCT TGG AGC CGT GGA), and 0.5 μl reverse primer (AAC TAC CCT TAC CTC TTG CTC TT) (both at a 20-pmol/μl concentration).
The thermal reaction started with a predenaturation step at 95°C for 5 min and was followed by 40 cycles (denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 50 s), and the reaction was finished with a postelongation step at 72°C for 5 min. The specificity of the qPCR was verified by melting curve analysis.
DNase treatment.
A 10× DNase buffer (20 μl) with 25 mM MgCl2 (Thermo Scientific) and 4 μl 50 U/μl bovine pancreatic DNase I (Roche) was added to 200 μl supernatant of the infected cells. After 90 min at room temperature, the viral DNA was purified with the High Pure viral nucleic acid kit (Roche) according to the manufacturer's protocol.
ER stress induction.
The plated and infected cells (MOIs, 0.01 and 3) were incubated with 20 μM MG132 or 10 mM dithiothreitol (DTT) at 0 to 5 h, 3 to 8 h, 5 to 10 h, or 7 to 12 h p.i. or 10 μM thapsigargin at 0 to 2 h, 2 to 4 h, 4 to 6 h, or 6 to 8 h p.i. at 37°C. After the chemical incubation period, the cells were washed twice and 1 ml fresh cell culture medium was added into the wells. The supernatant was harvested, and the cells were fixed at 24 h p.i.
UV stress induction.
The infected cells (MOI, 3) were treated with sublethal UV-C light (30-W light source, 50-cm distance) for 5 min at 0, 3, 9, and 12 h p.i. After the treatment period, the cells were washed twice and 1 ml fresh cell culture medium was added into the wells. The supernatant was harvested at 24 h p.i., and the cells were fixed for further examinations.
ACKNOWLEDGMENTS
The study was supported by the Hungarian Scientific Research Fund (K108607) and the National Research, Development and Innovation Office (K119381).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Johnson RH, Collings DF. 1969. Experimental infection of piglets and pregnant gilts with a parvovirus. Vet Rec 85:446–447. doi: 10.1136/vr.85.16.446. [DOI] [PubMed] [Google Scholar]
- 2.Choi CS, Molitor TW, Joo HS, Gunther R. 1987. Pathogenicity of a skin isolate of porcine parvovirus in swine foetuses. Vet Microbiol 15:19–29. doi: 10.1016/0378-1135(87)90125-8. [DOI] [PubMed] [Google Scholar]
- 3.Oraveerakul K, Choi CS, Molitor TW. 1993. Tissue tropisms of porcine parvovirus in swine. Arch Virol 130:377–389. doi: 10.1007/BF01309668. [DOI] [PubMed] [Google Scholar]
- 4.Kresse JI, Taylor WD, Stewart WW, Eernisse KA. 1985. Parvovirus infection in pigs with necrotic and vesicle-like lesions. Vet Microbiol 10:525–531. doi: 10.1016/0378-1135(85)90061-6. [DOI] [PubMed] [Google Scholar]
- 5.Rayet B, Lopez-Guerrero JA, Rommelaere J, Dinsart C. 1998. Induction of programmed cell death by parvovirus H-1 in U937 cells: connection with the tumor necrosis factor alpha signalling pathway. J Virol 72:8893–8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sol N, Le Junter J, Vassias I, Freyssinier JM, Thomas A, Prigent AF, Rudkin BB, Fichelson S, Morinet F. 1999. Possible interactions between the NS-1 protein and tumor necrosis factor alpha pathways in erythroid cell apoptosis induced by human parvovirus B19. J Virol 73:8762–8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daeffler L, Horlein R, Rommelaere J, Nuesch JP. 2003. Modulation of minute virus of mice cytotoxic activities through site-directed mutagenesis within the NS coding region. J Virol 77:12466–12478. doi: 10.1128/JVI.77.23.12466-12478.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergeron J, Menezes J, Tijssen P. 1993. Genomic organization and mapping of transcription and translation products of the NADL-2 strain of porcine parvovirus. Virology 197:86–98. doi: 10.1006/viro.1993.1569. [DOI] [PubMed] [Google Scholar]
- 9.Zádori Z, Szelei J, Lacoste MC, Gariepy S, Raymond P, Allaire M, Nabi IR, Tijssen P. 2001. A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell 1:291–302. doi: 10.1016/S1534-5807(01)00031-4. [DOI] [PubMed] [Google Scholar]
- 10.Zádori Z, Szelei J, Tijssen P. 2005. SAT: a late NS protein of porcine parvovirus. J Virol 79:13129–13138. doi: 10.1128/JVI.79.20.13129-13138.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozcan L, Tabas I. 2012. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu Rev Med 63:317–328. doi: 10.1146/annurev-med-043010-144749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellgaard L, Helenius A. 2003. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 13.Marciniak SJ, Ron D. 2006. Endoplasmic reticulum stress signaling in disease. Physiol Rev 86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 14.Ron D, Hubbard SR. 2008. How IRE1 reacts to ER stress. Cell 132:24–26. doi: 10.1016/j.cell.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 15.Patil C, Walter P. 2001. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13:349–355. doi: 10.1016/S0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 16.Kohno K. 2010. Stress-sensing mechanisms in the unfolded protein response: similarities and differences between yeast and mammals. J Biochem 147:27–33. doi: 10.1093/jb/mvp196. [DOI] [PubMed] [Google Scholar]
- 17.Lee AH, Iwakoshi NN, Glimcher LH. 2003. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. 2004. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Hinnebusch AG. 2000. Mechanism and regulation of initiator methionyl tRNA binding to ribosomes, p 185–243. In Sonenberg N, Hershey JWB, Mathews MB (ed), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 20.Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. 1998. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18:7499–7509. doi: 10.1128/MCB.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harding HP, Zhang Y, Ron D. 1999. Translation and protein folding are coupled by an endoplasmic reticulum resident kinase. Nature 397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 22.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633. doi: 10.1016/S1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 23.Wang X-Z, Lawson B, Brewer J, Zinszner H, Sanjay A, Mi L, Boorstein R, Kreibich G, Hendershot L, Ron D. 1996. Signals from the stressed endoplasmic reticulum induce C/EBP homologous protein (CHOP/GADD153). Mol Cell Biol 16:4273–4280. doi: 10.1128/MCB.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. 1998. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korzeniewski C, Callewaert DM. 1983. An enzyme-release assay for natural cytotoxicity. J Immunol Methods 64:313–320. doi: 10.1016/0022-1759(83)90438-6. [DOI] [PubMed] [Google Scholar]
- 27.Terrinoni A, Ranalli M, Cadot B, Leta A, Bagetta G, Vousden KH, Melino G. 2004. p73-alpha is capable of inducing scotin and ER stress. Oncogene 23:3721–3725. doi: 10.1038/sj.onc.1207342. [DOI] [PubMed] [Google Scholar]
- 28.Hübener J, Vauti F, Funke C, Wolburg H, Ye Y, Schmidt T, Wolburg-Buchholz K, Schmitt I, Gardyan A, Driessen S, Arnold HH, Nguyen HP, Riess O. 2011. N-terminal ataxin-3 causes neurological symptoms with inclusions, endoplasmic reticulum stress and ribosomal dislocation. Brain 134(Pt 7):1925–1942. doi: 10.1093/brain/awr118. [DOI] [PubMed] [Google Scholar]
- 29.Ngoh GA, Papanicolaou KN, Walsh K. 2012. Loss of mitofusin 2 promotes endoplasmic reticulum stress. J Biol Chem 287:20321–20332. doi: 10.1074/jbc.M112.359174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iurlaro R, Muñoz-Pinedo C. 2016. Cell death induced by endoplasmic reticulum stress. FEBS J 283:2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- 31.Herrada AA, Rojas-Colonelli N, González-Figueroa P, Roco J, Oyarce C, Ligtenberg MA, Lladser A. 2012. Harnessing DNA-induced immune responses for improving cancer vaccines. Hum Vaccin Immunother 8:1682–1693. doi: 10.4161/hv.22345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchini A, Bonifati S, Scott EM, Angelova AL, Rommelaere J. 2015. Oncolytic parvoviruses: from basic virology to clinical applications. Virol J 12:6. doi: 10.1186/s12985-014-0223-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bär S, Daeffler L, Rommelaere J, Nüesch JP. 2008. Vesicular egress of non-enveloped lytic parvoviruses depends on gelsolin functioning. PLoS Pathog 4:e1000126. doi: 10.1371/journal.ppat.1000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen AY, Qiu J. 2010. Parvovirus infection-induced cell death and cell cycle arrest. Future Virol 5:731–743. doi: 10.2217/fvl.10.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Huang Y, Du Q, Luo X, Zhang L, Zhao X, Tong D. 2015. Porcine parvovirus infection induces apoptosis in PK-15 cells through activation of p53 and mitochondria-mediated pathway. Biochem Biophys Res Commun 456:649–655. doi: 10.1016/j.bbrc.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Xiang H, Bai X, Fei N, Huang Y, Song X, Zhang H, Zhang L, Tong D. 2016. Porcine parvovirus infection activates mitochondria-mediated apoptotic signaling pathway by inducing ROS accumulation. Virol J 13:26. doi: 10.1186/s12985-016-0480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergeron J, Hébert B, Tijssen P. 1996. Genome organization of the Kresse strain of porcine parvovirus: identification of the allotropic determinant and comparison with those of NADL-2 and field isolates. J Virol 70:2508–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandes S, Boisvert M, Szelei J, Tijssen P. 2014. Differential replication of two porcine parvovirus strains in bovine cell lines ensues from initial DNA processing and NS1 expression. J Gen Virol 95:910–921. doi: 10.1099/vir.0.059741-0. [DOI] [PubMed] [Google Scholar]
- 39.Gupta SK, Sahoo AP, Rosh N, Gandham RK, Saxena L, Singh AK, Harish DR, Tiwari AK. 2016. Canine parvovirus NS1 induced apoptosis involves mitochondria, accumulation of reactive oxygen species and activation of caspases. Virus Res 213:46–61. doi: 10.1016/j.virusres.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 41.Kondratyev M, Avezov E, Shenkman M, Groisman B, Lederkremer GZ. 2007. PERK-dependent compartmentalization of ERAD and unfolded protein response machineries during ER stress. Exp Cell Res 313:3395–3407. doi: 10.1016/j.yexcr.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Hammadi M, Oulidi A, Gackière F, Katsogiannou M, Slomianny C, Roudbaraki M, Dewailly E, Delcourt P, Lepage G, Lotteau S, Ducreux S, Prevarskaya N, Van Coppenolle F. 2013. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78. FASEB J 27:1600–1609. doi: 10.1096/fj.12-218875. [DOI] [PubMed] [Google Scholar]
- 43.Cleland WW. 1964. Dithiothreitol, a new protective reagent for SH groups. Biochemistry 3:480–482. doi: 10.1021/bi00892a002. [DOI] [PubMed] [Google Scholar]
- 44.Lee DH, Goldberg AL. 1998. Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol Cell Biol 18:30–38. doi: 10.1128/MCB.18.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ali H, Christensen SB, Foreman JC, Pearce FL, Piotrowski W, Thastrup O. 1985. The ability of thapsigargin and thapsigargicin to activate cells involved in the inflammatory response. Br J Pharmacol 85:705–712. doi: 10.1111/j.1476-5381.1985.tb10567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boisvert M, Fernandes S, Tijssen P. 2010. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J Virol 84:7782–7792. doi: 10.1128/JVI.00479-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yalkinoglu AO, Heilbronn R, Burkle A, Schlehofer J, zur Hausen H. 1988. DNA amplification of adeno-associated virus as a response to cellular genotoxic stress. Cancer Res 48:3123–3129. [PubMed] [Google Scholar]
- 48.Yakobson B, Koch T, Winocour E. 1987. Replication of adeno-associated virus in synchronized cells without the addition of a helper virus. J Virol 61:972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cornelis JJ, Su ZZ, Ward DC, Rommelaere J. 1981. Indirect induction of mutagenesis of intact parvovirus H-1 in mammalian cells treated with UV light or with UV-irradiated H-1 or simian virus 40. Proc Natl Acad Sci U S A 78:4480–4484. doi: 10.1073/pnas.78.7.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jordan R, Wang L, Graczyk TM, Block TM, Romano PR. 2002. Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J Virol 76:9588–9599. doi: 10.1128/JVI.76.19.9588-9599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Wang A. 2012. Virus induced ER stress and the unfolded protein response. Front Plant Sci 3:293. doi: 10.3389/fpls.2012.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan SW. 2014. The unfolded protein response in virus infections. Front Microbiol 5:518. doi: 10.3389/fmicb.2014.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dash S, Chava S, Aydin Y, Chandra PK, Ferraris P, Chen W, Balart LA, Wu T, Garry RF. 2016. Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER-stress response. Viruses 8:E150. doi: 10.3390/v8050150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Y, Qi B, Gu Y, Xu F, Du H, Li X, Fang W. 2016. Porcine circovirus 2 deploys PERK pathway and GRP78 for its enhanced replication in PK-15 cells. Viruses 8:E56. doi: 10.3390/v8020056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schönthal AH. 2012. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica (Cairo) 2012:857516. doi: 10.6064/2012/857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu YQ, Liu LC, Wang FC, Liang Y, Cha DQ, Zhang JJ, Shen YJ, Wang HP, Fang S, Shen YX. 2010. Induction profile of MANF/ARMET by cerebral ischemia and its implication for neuron protection. J Cereb Blood Flow Metab 30:79–91. doi: 10.1038/jcbfm.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sano R, Reed JC. 2013. ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. 2013. When ER stress reaches a dead end. Biochim Biophys Acta 1833:3507–3517. doi: 10.1016/j.bbamcr.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 59.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. 2013. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hristov M, Erl W, Linder S, Weber PC. 2004. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 104:2761–2766. doi: 10.1182/blood-2003-10-3614. [DOI] [PubMed] [Google Scholar]
- 61.Legrand C, Bour JM, Jacob C, Capiaumont J, Martial A, Marc A, Wudtke M, Kretzmer G, Demangel C, Duval D, Hache J. 1992. Lactate dehydrogenase (LDH) activity of the cultured eukaryotic cells as marker of the number of dead cells in the medium. J Biotechnol 25:231–243. (Erratum, 31:234, 1993.) [DOI] [PubMed] [Google Scholar]
- 62.Goergen JL, Marc A, Engasser JM. 1993. Determination of cell lysis and death kinetics in continuous hybridoma cultures from the measurement of lactate dehydrogenase release. Cytotechnology 11:189–195. doi: 10.1007/BF00749869. [DOI] [PubMed] [Google Scholar]