

ABSTRACT
The invention of next-generation sequencing (NGS) techniques marked the coming of a new era in the detection of the genetic diversity of intrahost viral populations. A good understanding of the genetic structure of these populations requires, first, the ability to identify the different isolates or variants and, second, the ability to accurately quantify them. However, the initial amplification step of NGS studies can impose potential quantitative biases, modifying the variant relative frequencies. In particular, the number of target molecules (NTM) used during the amplification step is vastly overlooked although of primary importance, as it sets the limit of the accuracy and sensitivity of the sequencing procedure. In the present article, we investigated quantitative biases in an NGS study of populations of a multipartite single-stranded DNA (ssDNA) virus at different steps of the procedure. We studied 20 independent populations of the ssDNA virus faba bean necrotic stunt virus (FBNSV) in two host plants, Vicia faba and Medicago truncatula. FBNSV is a multipartite virus composed of eight genomic segments, whose specific and host-dependent relative frequencies are defined as the “genome formula.” Our results show a significant distortion of the FBNSV genome formula after the amplification and sequencing steps. We also quantified the genetic bottleneck occurring at the amplification step by documenting the NTM of two genomic segments of FBNSV. We argue that the NTM must be documented and carefully considered when determining the sensitivity and accuracy of data from NGS studies.
IMPORTANCE The advent of next-generation sequencing (NGS) techniques now enables study of the genetic diversity of viral populations. A good understanding of the genetic structure of these populations first requires the ability to identify the different isolates or variants and second requires the ability to accurately quantify them. Prior to sequencing, viral genomes need to be amplified, a step that potentially imposes quantitative biases and modifies the viral population structure. In particular, the number of target molecules (NTM) used during the amplification step is of primary importance, as it sets the limit of the accuracy and sensitivity of the sequencing procedure. In this work, we used 20 replicated populations of the multipartite faba bean necrotic stunt virus (FBNSV) to estimate the various limitations of ultradeep-sequencing studies performed on intrahost viral populations. We report quantitative biases during rolling-circle amplification and the NTM of two genomic segments of FBNSV.
KEYWORDS: DNA sequencing, FBNSV, faba bean, Medicago truncatula, next-generation sequencing, number of target molecules, rolling-circle amplification, sequencing accuracy, sequencing sensitivity
INTRODUCTION
Until recently, the study of natural viral populations was technically limited. The coming of next-generation sequencing (NGS) technologies has opened this field of research by vastly improving the detection and quantification of genetic variability in viral populations. Many metagenomic studies have already been dedicated to describing viral communities in many different environments, such as ocean coastal seawater (1), freshwater lakes (2), stromatolites (3), and soil (4, 5; see reference 6 for a more exhaustive review). NGS technologies and, more specifically, ultradeep sequencing (UDS) (7) also made it possible to investigate the intraspecific and intrahost variations of viral populations. The high sequencing coverage provided by UDS enables the detection of mutants at low frequencies and the quantification of them with precision. Those studies highlighted major features of intrahost viral population dynamics, such as the existence of strong bottlenecks during transmission (8, 9), the appearance and dynamics of drug-resistant mutants (10, 11), or the existence of unexpected multiple infections (12, 13).
Studying viral populations with NGS can be achieved with different approaches. While some NGS techniques do not require the amplification of the target nucleic acid (14), for most methods, the first step in the NGS pipeline is the amplification of viral sequences from the collected samples through either PCR, reverse transcription-PCR (RT-PCR), or rolling-circle amplification (RCA). When the template DNA is ready, the two remaining steps of the NGS pipeline consist of the preparation of DNA libraries and sequencing (15). Various errors and biases may occur at each of these steps.
The amplification step is particularly susceptible to the introduction of (i) sequence errors (artifactual mutations) during replication and (ii) biases in sequence relative frequencies. At the viral community level, the linker amplification shotgun library (LASL) method (1) amplifies only double-stranded DNA (dsDNA) viruses (therefore missing single-stranded DNA [ssDNA] and RNA viruses) and suffers from differential amplification rates related to the size of amplicons (16) and GC content (17). The multiple-displacement amplification method (MDA), based on RCA (18), tends to overrepresent small circular genomes (e.g., ssDNA viruses) (19, 20) and, depending on the kit, tends to overrepresent sequences rich in GC content or frequent sequences in the population (21). Thus, at the interspecific level, the sequence divergence (single or double stranded, DNA or RNA, genome length, and GC content, etc.) challenges the accuracy of NGS technologies. However, given that the sequence divergence is much lower at the intraspecific level than at the interspecific level, it remains unclear if amplification steps can introduce biases in the estimation of relative frequencies of sequences of the same species.
Some biases may also occur during library preparation. For instance, with the Nextera Illumina method, library preparation requires a step of fragmentation and transposition of adapter sequences. As the activity of the transposases generally results in nonrandom integrations (22, 23), some biases in sequence coverage may arise if the distribution of the fragmentation and tagging is not homogeneous throughout the genome or between genotypes. Finally, sequencing errors occur whatever the method used, and appropriate data analyses are required to distinguish them from actual polymorphisms (24–26).
While these various biases and errors have been fairly well studied and described, a new kind of limitation has been identified recently. In their study, McCrone and Lauring (27) showed that the sensitivity of single-nucleotide variant detection was limited at low nucleic acid concentrations, probably because of the small number of template molecules, analogous to population bottlenecks occurring at the amplification and/or library preparation step. Bottlenecks can seriously reduce the detection of this variability (at the intraspecific and interspecific levels), depending on how severe they are. Suppose that we extract viral genomes from an infected host in order to detect and quantify the relative frequency of viral variants or viral species. We first amplify these viral genomes, then perform NGS analysis, and end up with 10,000-fold coverage. If the number of target viral molecules actually amplified is 1,000, the 10,000-fold coverage will show the genetic diversity contained in those 1,000 molecules. If the number of target molecules is much smaller (e.g., 100 or fewer), ultradeep sequencing may potentially reveal only a very small and quantitatively biased fraction of the genetic variability contained in the infected host. The sensitivity and accuracy of the NGS procedure depend on the actual number of target molecules (NTM) that were amplified, which may be much smaller than the coverage. Importantly, the NTM as defined here is not the total amount of viral genomes present in a sample but the fraction that is actually amplified.
The ssDNA virus faba bean necrotic stunt virus (FBNSV) is a nanovirus composed of eight circular genomic segments, with each segment being encapsidated independently. All segments have similar lengths (∼1 kb) and carry different genes but also have some homologous noncoding sequences. A recent study showed that these different genomic segments consistently accumulate at different relative frequencies in the host plant, defining the genome formula (28). It has been hypothesized that the genome formula could correspond to a gene expression regulation system through the modification of the gene copy number (29). The multipartite structure of the FBNSV genome, the fact that its segments are of similar sizes and share common sequences, and the obvious importance of the genome formula make FBNSV a great model to investigate potential quantitative NGS biases at the intraspecific level. In the present article, we investigated such putative biases during an NGS study of 40 replicated FBNSV populations. We performed an experiment in which this multipartite virus was inoculated into 20 faba bean plants and later transmitted to 20 Medicago plants via aphids. We used these 20 independent lines to evaluate the impact of RCA and NGS Illumina Nextera techniques on the estimation of the relative frequencies of FBNSV segments. In an additional experiment, we were also able to infer for two segments (one frequent and one rare) the NTM actually amplified during RCA and thus quantify the bottleneck occurring in our ultradeep-sequencing method.
RESULTS
Rolling-circle amplification modifies the FBNSV genome formula.
To investigate whether RCA could bias the genome formula, we performed quantitative PCRs (qPCRs) before and after RCA (Fig. 1) and compared the relative frequencies of each FBNSV segment. Figure 2 (and Table 1) illustrates how the FBNSV genome formula was altered by RCA. Our statistical analysis (Table 2) confirms that the segment relative frequencies (i.e., the genome formula) differed in faba bean and Medicago (segment*plant factor F value = 377.5, where the asterisk indicates an interaction between the segment and plant factors; P < 2.2e−16), as previously described (28). More surprisingly, it also shows that the genome formula was distorted during RCA amplification (segment*RCA factor F value = 67.6; P < 2.2e−16) and that the effect of RCA was different in faba bean and Medicago (segment*plant*RCA factor F value = 11.2; P = 2.04e−13). While segment U4 experienced the highest relative proportion increase (26.5% before and 47% after RCA, i.e., a 1.77-fold increase, in faba bean, and 12.4% before and 17.8% after RCA, i.e., a 1.43-fold increase, in Medicago), segment U2 suffered the highest relative proportion decrease (18.4% before and 4.6% after RCA, i.e., a 4-fold decrease, in faba bean, and 21.8% before and 12.3% after RCA, i.e., a 1.77-fold decrease, in Medicago). In order to understand how RCA affected the genome formula, we calculated the amplification rates for each segment. Figure 3 and the associated Tukey honestly significant difference (HSD) test confirm that (i) FBNSV segments were not all amplified at the same rates and (ii) these amplification rates were different in faba bean (Fig. 3A) and Medicago (Fig. 3B). In faba beans, C, N, and U4 were the most amplified segments, while R and U2 were the least amplified. In Medicago, the most amplified segments were C, U1, and U4, while the least amplified were R and U2 (like in faba bean). Finally, in order to tentatively explain why the FBNSV genome formula varied during RCA, we tested whether the initial segment relative frequencies could bias amplification, with the most frequent segments being potentially the most amplified ones (21). We tested this hypothesis by plotting the relative segment frequencies (i.e., the genome formula) versus the RCA rates and found no correlation (Fig. 4).
FIG 1.

Overview of the protocol for experiment 1. (A) Faba bean plants (V. faba) were agroinoculated with a mix of A. tumefaciens cultures carrying plasmids containing the FBNSV segments. Ten A. pisum aphids were used as vectors to transmit FBNSV from symptomatic faba bean plants to Medicago truncatula plants. Total DNA was collected from faba bean and Medicago plants. This procedure was performed on 20 independent lines. Vf1, Vicia faba; Mt, Medicago truncatula. (B) Rolling-circle amplifications were performed on each of the 40 DNA samples collected from faba bean and Medicago plants (from the 20 lines described above for panel A). DNA purification was performed after RCA in order to remove the RCA buffer from the samples. Cleaned DNA samples were sent for sequencing. Genome formulae were measured by qPCR on pre- and post-RCA samples and estimated by counting the number of reads per segment after NGS analysis.
FIG 2.

FBNSV genome formulas after DNA extraction (white), after RCA (light gray), and after NGS (dark gray) in faba bean (A) and Medicago (B) plants. Each box plot represents the distribution of the relative frequencies of one segment in 20 replicated plants.
TABLE 1.
Evolution of the average FBNSV genome formula after RCA and NGS proceduresa
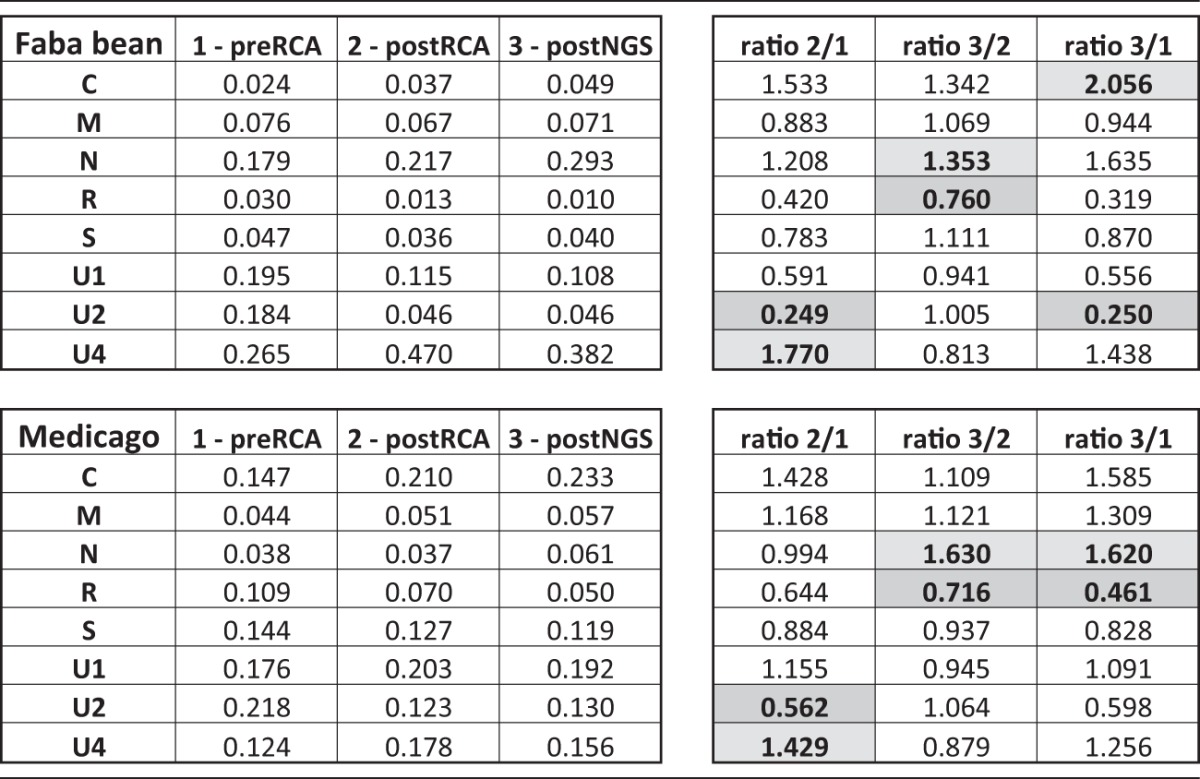
Relative frequency ratios were calculated between steps 1 and 2, 2 and 3, and 1 and 3. The highest of the 8 ratios calculated for each segment is shown in light-gray cells and the lowest in gray cells.
TABLE 2.
Statistical analysis of the impact of RCA on FBNSV segment relative frequenciesa
| Factor | df | Sum sq | Mean sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Segment | 7 | 44.57 | 6.3671 | 339.649 | <2.20e−16*** |
| Plant | 1 | 2.33 | 2.3305 | 124.319 | <2.20e−16*** |
| RCA | 1 | 0.514 | 0.5136 | 27.399 | 2.28e−07*** |
| Segment*plant | 7 | 49.53 | 7.0757 | 377.451 | <2.20e−16*** |
| Segment*RCA | 7 | 8.864 | 1.2663 | 67.55 | <2.20e−16*** |
| Plant*RCA | 1 | 0.402 | 0.4017 | 21.429 | 4.49e−06*** |
| Segment*plant*RCA | 7 | 1.474 | 0.2106 | 11.232 | 2.04e−13*** |
| Residuals | 608 | 11.398 | 0.0187 |
Sum sq, sum of the squares; Mean sq, mean of the squares; Pr(>F), F-test P value (***, P < 0.001).
FIG 3.

Rolling-circle amplification rates for the different segments of FBNSV with DNA samples from faba bean (A) or Medicago (B) plants. Box plots show the distribution of RCA rates observed for 20 independent plants. Letters on top of the box plots determine groups of values not statistically different after a Tukey HSD test was performed on these data.
FIG 4.

Correlation between the relative proportions of segments (genome formula) and rolling-circle amplification rates in faba bean (A) and Medicago (B) samples. The regression formulae are y = 0.0001x + 0.087 (R2 = 0.056) for panel A and y = −5.105x + 0.165 (R2 = 0.042) for panel B. Pearson's product-moment correlations were not significant for faba bean (t = 0.60, df = 6, P value = 0.57, and correlation coefficient = 0.24) or Medicago (t = −0.51, df = 6, P value = 0.63, and correlation coefficient = −0.21).
NGS estimates of the FBNSV genome formula are biased.
We were interested to know whether the FBNSV genome formula could be reliably estimated by the NGS procedure used in this study. For this, we compared qPCR estimates of the genome formula in RCA products to the relative proportion of reads matching on each segment obtained after the HiSeq procedure was performed on the same RCA products (Fig. 2). Our statistical analysis (Table 3) shows that the segment relative frequencies estimated by both qPCR and HiSeq are different in faba bean and Medicago (segment*plant factor F value = 440.3; P < 2.2e−16). It also shows that the genome formulae estimated by qPCR and by HiSeq are significantly different (segment*NGS factor F value = 14.4; P < 2.2e−16) but that the distortions induced by the NGS estimates are not statistically different in samples from faba beans and Medicago (segment*plant*RCA factor F value = 1.1; P = 0.37). Comparison of the average genome formulae in RCA products estimated by qPCR and by HiSeq (Fig. 2 and Table 1) confirmed a quantitative difference with the HiSeq method. While the N segment showed the highest relative proportion increase (21.7% estimated by qPCR and 29.3% estimated by HiSeq, i.e., a 1.35-fold increase, in faba bean, and 4% by qPCR and 6% by HiSeq, i.e., a 1.63-fold increase, in Medicago), and the R segment suffered the highest relative proportion decrease (1.3% by qPCR and 1% by HiSeq, i.e., a 1.3-fold, decrease in faba bean, and 7% by qPCR and 5% by HiSeq, i.e., a 1.4-fold decrease, in Medicago).
TABLE 3.
Statistical analysis of the impact of NGS on FBNSV segment relative frequenciesa
| Factor | df | Sum sq | Mean sq | F value | Pr(>F) |
|---|---|---|---|---|---|
| Segment | 7 | 65.18 | 9.3115 | 530.8806 | <2e−16*** |
| Plant | 1 | 4.699 | 4.6988 | 267.8934 | <2e−16*** |
| NGS | 1 | 0.033 | 0.0328 | 1.8701 | 0.172 |
| Segment*plant | 7 | 54.06 | 7.7229 | 440.3093 | <2e−16*** |
| Segment*NGS | 7 | 1.766 | 0.2523 | 14.3823 | <2e−16*** |
| Plant*NGS | 1 | 0 | 0.0001 | 0.003 | 0.9563 |
| Segment*plant*NGS | 7 | 0.134 | 0.0191 | 1.0904 | 0.3676 |
| Residuals | 608 | 10.664 | 0.0175 |
Sum sq, sum of the squares; Mean sq, mean of the squares; Pr(>F), F-test P value (***, P < 0.001).
Estimation of the number of target molecules amplified during RCA.
As mentioned in the introduction, the initial NTM amplified by RCA could limit the sensitivity of our ultradeep-sequencing effort. Because it is an important issue for the final interpretation of NGS results, we performed an additional experiment to evaluate this putative limitation. First, we estimated the number of molecules present in 1 μl of DNA extract used for the RCA reaction by qPCR. We came up with estimations of 6.5 × 106 and 1 × 106 molecules of the N and S segments, respectively. Second, we estimated the size of the bottleneck undergone by the viral segments N and S during RCA using a statistical method based on variations of the relative frequencies of two genetic markers inserted into the same segment (N or S), before and after RCA. This method encompasses that the various segments (bearing different markers) can be amplified at different rates. A model comparison revealed that the model assuming differential replication best explained the results over the model assuming equal replication (posterior model probability of >0.99). The model that best explains our experimental results corresponds to NTM of 3,250 (confidence interval [CI], 1,065 to 8,974) and 617 (CI, 255 to 1,478) for the N and S segments, respectively (see Table 4 for detailed parameter estimation results). Therefore, only 0.05% and 0.06% of the N and S segment molecules were used as targets during RCA, respectively.
TABLE 4.
Parameter estimation (medians, means, and 95% credibility intervals) of the best model explaining experiment 2a
| Segment | Parameter | Value |
|||
|---|---|---|---|---|---|
| Quantile (2.5%) | Median | Mean | Quantile (97.5%) | ||
| N | s | −0.36 | −0.34 | −0.34 | −0.33 |
| N | λN | 1,065 | 3,286 | 3,250 | 8,974 |
| S | s | −0.54 | −0.51 | −0.51 | −0.48 |
| S | λN | 255 | 610 | 617 | 1,478 |
Two parameters are estimated for each viral segment: (i) λN corresponds to the number of target molecules (NTM), and (ii) s corresponds to a differential replication coefficient of the marker of interest during RCA.
DISCUSSION
RCA affects the FBNSV genome formula.
We observed significant genome formula variations during RCA (Fig. 2 and Table 1). This bias could be explained by the fact that either (i) the NTM of each segment was not proportional to the genome formula, (ii) the different segments were amplified at different rates in RCA, or (iii) both (i) and (ii) occurred concomitantly. Our estimation of the NTM (experiment 2) showed that more N molecules (3,250) than S molecules (617) were used as targets during RCA. Interestingly, the ratios of N/S molecules present initially in the DNA sample (genomic formula estimated by qPCR) and those of the corresponding NTM in RCA were very similar (initial N/S ratio = 6.12, NTMN/NTMS ratio = 3,250/617 = 5.27). Comparison of these ratios indicates that the NTM of these segments were proportional to their relative frequencies in the genome formula. Thus, the distortion between the genome formulae before and after RCA cannot be explained by the first hypothesis.
The distortion of the genome formula during RCA was due mostly to the fact that the different FBNSV segments were not amplified at the same rate (Fig. 3). Interestingly, the relative frequencies of five of the eight FBNSV segments consistently showed similar RCA-associated variation patterns between replicate populations and even across host plants (faba bean and Medicago, with an increase in the relative frequencies of C and U4 and a decrease in the relative frequencies of R, S, and U2). Such results suggest the existence of some sort of mechanism underlying the relative frequency modifications induced by RCA. Contrary to results reported previously by Yilmaz et al. (21), we did not detect the existence of a frequency dependence that would result in the overamplification of frequent segments compared to rare ones (Fig. 4). As RCA is initiated by the annealing of random hexamer primers on the target DNA, sequence variation or various secondary folding structures may differentially affect priming on distinct segments and explain why certain segments were more replicated than others. Clearly, more investigations are necessary to mechanistically understand the effect of rolling-circle amplification on the FBNSV genome formula.
In our experiment, most of the bias introduced by RCA was due to differential rates of amplification between segments and not the effects of the bottleneck at the initial step of RCA. Given our estimates of a large NTM amplified by RCA (>600, even for the rarest segment), this is not surprising. It is unclear, however, how general this may be. More severe bottlenecks in other systems could mediate the relative importance of these processes.
The NGS Illumina Nextera procedure affects the FBNSV genome formula.
After RCA, the FBNSV genome formula was estimated by qPCR and by measuring the relative proportions of Illumina-generated reads that mapped onto each genomic segment. These two independent estimations were slightly but significantly different (Fig. 2 and Tables 1 and 3). These variations were quite consistent between replicates as well as between faba bean and Medicago. There are a few additional amplification steps during the Illumina Nextera procedure, dedicated to the introduction of tag sequences at the 5′ and 3′ ends of the reads. Because these PCR amplifications are performed on adapter sequences, we cannot see how a bias in the FBNSV genome formula could be introduced at these steps. One possibility is that this occurred during library preparation when transposons were inserted into the target DNA sequence. It is well known that transposases promote nonrandom integration (22, 23). Thus, at least partially, we may explain the biases of the FBNSV genome formula occurring during NGS by the fact that Nextera fragmentation and tagging are not homogeneously distributed among the distinct FBNSV segments. Other library preparation kits (not tested here) are now available, and whether they alleviate such biases is an open question (30, 31).
Bottlenecks at the amplification step can impose a limitation on the sensitivity and accuracy of NGS.
As mentioned above, the NTM used as the templates during the amplification steps (e.g., RCA, PCR, and RT-PCR) in NGS pipelines is of crucial importance. Given that the S segment represents around 2% of the within-plant segment population in this experiment and that more than 600 molecules of this segment were used as the templates during RCA, a simple rule of thumb leads to a rough approximation of the size of the target population of 30,000 molecules in total (30,000 amplified genome segments of FBNSV). This figure could appear large enough to prevent the sensitivity and accuracy limitations discussed above. However, according to the proportion of molecules used as targets during RCA (0.05% and 0.06% of the N and S segments, respectively), 30,000 molecules correspond to only a small fraction of the total number of viral molecules present in the DNA extract before the amplification step.
Natural samples do not contain marked sequences allowing easy estimation of the NTM by qPCR. In order to estimate the NTM in these samples, we recommend the use of a nucleic acid solution containing marked sequences as a calibrator. For instance, in studies of double-stranded DNA viruses, a solution containing two dsDNA viruses carrying genetic markers could be introduced into the sample, and their relative proportions could be quantified before and after amplification. Whether various viral species are amplified at different rates should also be estimated in order to take these biases into account in the NTM estimation.
Whether the NTM may or may not be a limitation in precisely detecting or measuring relative variant frequencies depends on a question addressed by deep sequencing. In metagenomic studies, for instance, the NTM can potentially limit the detection of viral species present at low frequencies or the detection of rare intraspecific polymorphisms. Whatever the question asked, we argue that the NTM should be systematically estimated and accounted for, to evaluate how this number limits the sensitivity of detection and therefore allow the accurate interpretation of NGS results. More generally, we also suggest that the NTM should contribute to a more strict definition of the limit of detection in NGS studies dedicated to the detection of polymorphisms (at the intra- and interspecific levels).
Conclusion.
The aim of the present study was to investigate to what extent an NGS study may be quantitatively reliable at the intraspecific level, using the example of experimental populations of a multipartite nanovirus. More specifically, we looked for potential biases that could occur at different steps of such an experiment, i.e., during the RCA and the NGS Illumina Nextera procedures. We observed significant deviations (4-fold maximal variation) of the FBNSV genome formula during both steps. The importance of the FBNSV genome formula in the functioning of the virus is currently under study, and therefore, it remains unclear how such biases could be misleading, but it is certainly important to account for them. Finally, we also quantified the bottleneck occurring at the amplification step. This quantification allowed a rough approximation of a total of 30,000 viral molecules amplified by RCA. We believe that such estimations should be performed by NGS studies aiming to investigate the diversity of viral populations in order to estimate the actual sensitivity of these procedures and help rationalize sequencing efforts. For example, in the setting of our experiment, whatever the level of ultradeep sequencing, the sensitivity of the technique for the detection of polymorphisms was limited to around 0.05% of the available molecules. This limitation uncovered here with RCA potentially also applies to PCR amplification: the NTM sets the limit.
MATERIALS AND METHODS
Experiment overview.
Two independent experiments were performed in this study. Experiment 1 (see Fig. 1 for a schematic description) was dedicated to monitoring the FBNSV genome formula during different steps for the preparation of NGS DNA samples. Nine-day-old faba bean plants (Vicia faba “Seville”) were infected by FBNSV via agroinoculation. At 3 weeks postinfection, aphid transmission of the virus from these faba bean plants to Medicago plants (Medicago truncatula) was performed by using Acyrthosiphon pisum as the vector species. Twenty infected faba bean plants and their associated 20 infected Medicago plants (constituting 20 independent lines) were picked for subsequent analyses. Total DNA extraction was performed on systemically infected faba bean (21 days postinfection) and Medicago (26 days postinfection) plants. qPCR was performed on all 40 DNA extracts in order to measure the FBNSV genome formulae in the two host species. Rolling-circle amplification (amplifying FBNSV single-stranded circular DNA genome segments) was performed on all DNA samples in order to enrich the samples in viral DNA sequences. Another qPCR was performed on these RCA products to investigate the potential effect of RCA on the FBNSV genome formula. Finally, the samples were purified with a NucleoSpin gel and PCR cleanup purification kit (Macherey-Nagel GmbH & Co. KG) for subsequent deep-sequencing analyses.
Experiment 2 was dedicated to estimating the number of viral target molecules (NTM) used in the RCA reaction. To do so, a faba bean plant was infected with FBNSV in which two segments (one rare and one frequent) carried genetic markers. DNA extraction was performed on this plant, and 19 independent RCAs were run on this DNA sample. The relative frequency of each marker was measured by qPCR before and after RCA, and a statistical method based on the variation of these frequencies was used to estimate the number of target molecules actually used in RCA reactions (see the section on statistics, below, for a full description).
Viral strain and agroinoculations. (i) Experiment 1: agroinoculation and aphid transmission.
We used FBNSV isolate JKI-2000, provided by the Gronenborn laboratory and described previously (32). Faba bean plants were agroinoculated with cultures of Agrobacterium tumefaciens COR308 strains, each carrying a pbin19 plasmid containing a tandem repeat of 1 of the 8 FBNSV segments. All 8 A. tumefaciens cultures were mixed together at equal proportions and inoculated into plants as described previously (28). Aphid transmission of the virus was performed by caging 10 A. pisum aphids on infected faba bean plants for 3 days and then transferring them to 15-day-old Medicago plants (Medicago truncatula) for three more days.
(ii) Experiment 2: determination of the number of amplified molecules during RCA.
In experiment 2, we used six wild-type genome segments of FBNSV (C, M, R, U1, U2, and U4) and two engineered alleles of each of the N and S segments. The N and S segments contained 22-base-long genetic markers inserted between the ends of the coding regions and the poly(A) signal sequences (available upon request). The markers mys2 and mys7 were introduced into the N segment, while the markers mys1 and mys8 were introduced into the S segment.
Faba bean plants were agroinoculated with cultures of Agrobacterium tumefaciens COR308 strains, each carrying a pbin19 plasmid containing one of the FBNSV segments mentioned above. All 10 A. tumefaciens cultures were mixed together (1, 1, 1, 1, 1, 1, 0.5, 0.5, 0.5, and 0.5 volumes of C, M, R, U1, U2, U4, Nmys2, Nmys7, Smys1, and Smys8, respectively, in order to inoculate equal proportions of each of the eight segments composing the FBNSV genome) and inoculated into plants.
Estimation of the frequencies of segments and genetic markers by qPCR.
All qPCRs (40 cycles of 95°C for 10 s, 60°C for 10 s [when estimating genome formulae] or 63°C for 10 s [when estimating relative frequencies of marked segments], and 72°C for 10 s) were carried out immediately after DNA collection (same day) by using a LightCycler 480 thermocycler (Roche) and the LightCycler FastStart DNA Master Plus SYBR green I kit (Roche), according to the manufacturer's instructions. Sample DNA (1.2 μl of a 10-fold dilution) was added to the qPCR mix (5 μl of Roche 2× qPCR Mastermix, 3.5 μl of H2O, and 0.3 μl of primer mix, for 8.8 μl total) after distribution into 384-well microtiter plates. Primers (available upon request) were used at a final concentration of 0.3 μM.
Along with samples containing viral DNA, serial dilutions of plitmus28 plasmids, each carrying one of the eight FBNSV segments (32), were placed onto each qPCR plate (8 serial dilutions per PCR plate in total, one for each FBNSV segment). These dilutions were used as an internal control in order to draw a standard curve for each segment and in each PCR plate, thereby taking potential “between-qPCR-plate” variations into account. Thus, with this method, no calibrator was necessary to homogenize between-qPCR plate results. Fluorescence data were first analyzed with the LinRegPCR program (33) and later converted into nanograms of DNA by using standard curves (data are available upon request). Genome formulas could then be estimated by calculating the relative proportions of each segment as described previously (34). Genome formulas were estimated before and after RCA. The rate of rolling-circle amplification could be calculated for each segment as the ratio of the quantity of one given segment (in nanograms) before and the quantity after RCA. All qPCRs were duplicated (two wells on the same PCR plate).
DNA sample preparation and deep sequencing.
Four 6-mm-diameter leaf disks were collected from infected plants, frozen in liquid nitrogen, and ground with steal beads in an MM 301 mixer mill (Retsch). Total DNA was extracted from these samples by using the DNeasy plant minikit (Qiagen). Circular DNA molecules were amplified by rolling-circle amplification in order to obtain a majority of viral sequences in the samples. RCAs were performed with the Illustra TempliPhi amplification kit (GE Health Care Life Sciences, USA) according to the manufacturer's recommendations.
RCA products were purified with a NucleoSpin gel and PCR cleanup purification kit (Macherey-Nagel GmbH & Co. KG) and sequenced on two replicated lanes on the Illumina HiSeq-High Output (HO) system (Fasteris SA, Switzerland). Forty independent libraries were created, one per initial plant extract. For a single plant, around 18 million 150-base-long reads were obtained on average. These reads were analyzed with the following pipeline by using Toggle (35). First, we trimmed the remaining adapter sequences using Cutadapt (1.8.1) and additionally filtered the reads by quality, with a cutoff value of 28, and by read length, with a cutoff value of 70. The efficiency of this procedure was checked with Fastqc (0.11.3). Filtered reads were then mapped to the FBNSV JKI-2000 reference genome (GenBank accession numbers GQ150780.1 for segment C, GQ150781.1 for M, GQ150782.1 for N, GQ150778.1 for R, GQ150779.1 for S, GQ150783.1 for U1, GQ150784.1 for U2, and GQ150785.1 for U4) (32) by using the Burrows-Wheeler aligner and the Samse algorithm (0.7.15). As aligner tools consider only linear sequences, the circular sequences of the FBNSV segments were linearized, and the first 149 bases of the sequence at the 5′ extremity were duplicated at the 3′ extremity of the sequence. We did so to allow the mapping of reads that would fall onto the region where the sequence was opened. Duplication of 149 bases in reference sequences allowed the mapping of 18.8% more reads. Only best-match reads were kept for analysis. Reads were sorted by using Picard Tools Sort Sam (2.7.0) to enable read counting by using the idxstat tool (1.3).
Statistics. (i) Experiment 1: estimation of the effect of RCA and NGS procedures on the FBNSV genome formula.
Segment relative frequencies were logit transformed (log fi/1 − fi, where fi is the relative frequency of the ith segment) in order to normalize the data. These transformed data were analyzed with linear models, including segments, plant species (faba bean or Medicago), and treatments (RCA or NGS) as the main effects and their interactions.
(ii) Experiment 2: estimation of the number of target molecules of viral DNA amplified during RCA.
The general idea of the method for experiment 2 is based on analogy to population genetics. Consider that several replicate populations were founded from a single source population, and the frequency of genetic variants in the replicate populations was monitored through time. The variance in the frequency of the variants across replicate populations can inform us on the average number of individuals that founded the replicate populations. This is called the founder effective size in population genetics, and in the context of RCA, we call it the NTM. Any systematic bias in the mean frequency of genetic variants across replicate populations and their frequency in the source population would indicate that the markers replicated at different rates, analogous to the effect of selection in population genetics. The statistical methods that we detail below consider both processes to yield the NTM for each segment and an estimation of their amplification rate.
More specifically, let us denote by and the observed frequencies of the marker of interest in plant p before and after RCA, respectively. We modeled RCA as a binomial sampling process. The model is as follows:
The size parameter of the binomial process, np, corresponding to the NTM for RCA in each plant, varies from plant to plant as a result of a zero-truncated Poisson process (ZTPois) of unknown parameter λN. We use a zero-truncated Poisson distribution because it ensures that the value of np cannot be zero. Its mean, λN/[1 − exp(−λN)], is nearly equal to λN as soon as λN is ≥10 and corresponds to the NTM. The overall replication probability, prp, of the binomial process depends on the initial frequency of this marker and on s, an unknown differential replication coefficient of the marker of interest during RCA. Thus, if s is equal to 0, the replication probability of the marker of interest will be equal to its initial frequency . If s is >0, the replication probability of the marker of interest is more than proportional to its initial frequency, . We do not have any a priori reason to believe that the different markers for a given segment will replicate differentially, but we wanted to use a general method that could account for such potential biases. The variable mp corresponds to the effective number of copies of the marker of interest generated in plant p given the initial number of target molecules in this plant, np, and the replication probability, prp. Finally, we also assumed that the uncertainty for the measures of and is negligible.
The parameter θ = (λN, s) was estimated by approximate Bayesian computation (ABC) with two summary statistics. The statistic averaged over the plants the difference between the frequencies of the marker of interest before and after RCA. Similarly to the effect of selection in population genetics, S1 measures the tendency of the marker of interest to increase (or decrease) in average over a set of independent samples, the different plants, during RCA. The second statistic, , where , corresponds to an unbiased estimator of genetic drift averaged over the plants (36). Similarly to the effect of genetic drift, the higher the effective number of molecules amplified during RCA, the lower the mean increase of the variance of marker frequencies during the reaction.
Estimations were performed with the adaptive ABC algorithm (37) implemented in the EasyABC package (R software [https://www.r-project.org/]) with the tuning parameters nb_simul = 5,000, p_acc_min = 0.04, and alpha = 0.5. Noninformative uniform priors were used: s ∼ Unif[−0.9, 5] and λN ∼ log − unif[1, 104]. Additionally, we conducted model selection to test if differential amplification occurs during RCA. We compared the above-described model (model 1 with 2 parameters, λN and s) and a model assuming equal amplification (model 2 with only the parameter λN, assuming that s is null) using the multinomial logistic regression method implemented in the ABC package. We also verified the identifiability of the model through numerical simulations to check if data sets with 19 samples, as in our experiment, are informative enough to efficiently estimate the parameter θ = (λN,s). We proceeded in 3 steps. First, the parameter value θtrue was independently drawn from dedicated distributions (s ∼ Unif[−0.9, 3] and λN ∼ log − unif[10, 5,000]) that encompass a large diversity of possible scenarios. Second, data sets with 19 samples (1 ≤ p ≤ 19), as in our real experimental design, were simulated given θtrue and by setting , an intermediate frequency (initial frequency of the Nmys2 marker of 0.27 and initial frequency of the Smys1 marker of 0.16) of the N segment population in our experiment. Steps 1 and 2 were iterated until the acceptance of 350 simulated data sets. Data sets were accepted if, as in the real data set analyzed in experiment 2, the two markers were detected in all plants . Third, for each accepted data set, θ was reestimated by using the approximate Bayesian computation method detailed above. Practical identifiability is assessed by fitting a linear regression between true and estimated parameter values and assessing the relative bias. Overall, the practical identifiability was very satisfactory, with mean relative biases of 0.02 for s and 0.06 for λN. Specifically, for parameter s (differential replication coefficient), the R2 value of the best-fit line was 0.99, the slope was 1, and the intercept was 0.01. For parameter λN (NTM), logarithm-transformed values were used. The R2 value of the best-fit line was 0.91, the intercept was 0.3, and the slope was 0.92. Finally, the credibility intervals were also satisfactory, with 91% of the true values of λN (82% of the true values of s) included in 90% credibility intervals. All statistical analyses were performed with R software (R 3.1.3 GUI 1.65).
ACKNOWLEDGMENTS
We thank Sebastien Ravel for his help with bioinformatics and Mikail Pooggin, Denis Filloux, and Philippe Roumagnac for helpful comments and discussions.
This project was funded by the French National Research Funding Agency (ANR) (ANR-Nano grant number ANR-14-CE02-0014-01). Y.M. acknowledges support from the CNRS and IRD.
We declare no conflict of interest.
REFERENCES
- 1.Breitbart M, Salomon P, Andresen B, Mahaffy JM, Segall AM, Mead D, Azam F, Rohwer F. 2002. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci U S A 99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.López-Bueno A, Tamames J, Velázquez D, Moya A, Quesada A, Alcamí A. 2009. High diversity of the viral community from an Antarctic lake. Science 326:858–861. doi: 10.1126/science.1179287. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida M, Takaki Y, Eitoku M, Nunoura T, Takai K. 2013. Metagenomic analysis of viral communities in (hado)pelagic sediments. PLoS One 8:e57271. doi: 10.1371/journal.pone.0057271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R, Robeson M, Edwards RA, Felts B, Rayhawk S, Knight R, Rohwer F, Jackson RB. 2007. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol 73:7059–7066. doi: 10.1128/AEM.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim K-H, Chang H-W, Nam Y-D, Roh SW, Kim M-S, Sung Y, Jeon CO, Oh H-M, Bae J-W. 2008. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl Environ Microbiol 74:5975–5985. doi: 10.1128/AEM.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosario K, Breitbart M. 2011. Exploring the viral world through metagenomics. Curr Opin Virol 1:289–297. doi: 10.1016/j.coviro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Beerenwinkel N, Zagordi O. 2011. Ultra-deep sequencing for the analysis of viral populations. Curr Opin Virol 1:413–418. doi: 10.1016/j.coviro.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Wang GP, Sherrill-Mix SA, Chang K-M, Quince C, Bushman FD. 2010. Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J Virol 84:6218–6228. doi: 10.1128/JVI.02271-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, Cameron B, Maher L, Dore GJ, White PA, Lloyd AR. 2011. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog 7:e1002243. doi: 10.1371/journal.ppat.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solmone M, Vincenti D, Prosperi MCF, Bruselles A, Ippolito G, Capobianchi MR. 2009. Use of massively parallel ultradeep pyrosequencing to characterize the genetic diversity of hepatitis B virus in drug-resistant and drug-naive patients and to detect minor variants in reverse transcriptase and hepatitis B S antigen. J Virol 83:1718–1726. doi: 10.1128/JVI.02011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margeridon-Thermet S, Shulman NS, Ahmed A, Shahriar R, Liu T, Wang C, Holmes SP, Babrzadeh F, Gharizadeh B, Hanczaruk B, Simen BB, Egholm M, Shafer RW. 2009. Ultra-deep pyrosequencing of hepatitis B virus quasispecies from nucleoside and nucleotide reverse-transcriptase inhibitor (NRTI)-treated patients and NRTI-naive patients. J Infect Dis 199:1275–1285. doi: 10.1086/597808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghedin E, Fitch A, Boyne A, Griesemer S, DePasse J, Bera J, Zhang X, Halpin RA, Smit M, Jennings L, St George K, Holmes EC, Spiro DJ. 2009. Mixed infection and the genesis of influenza virus diversity. J Virol 83:8832–8841. doi: 10.1128/JVI.00773-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramakrishnan MA, Tu ZJ, Singh S, Chockalingam AK, Gramer MR, Wang P, Goyal SM, Yang M, Halvorson DA, Sreevatsan S. 2009. The feasibility of using high resolution genome sequencing of influenza A viruses to detect mixed infections and quasispecies. PLoS One 4:e7105. doi: 10.1371/journal.pone.0007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pham TT, Yin J, Eid JS, Adams E, Lam R, Turner SW, Loomis EW, Wang JY, Hagerman PJ, Hanes JW. 2016. Single-locus enrichment without amplification for sequencing and direct detection of epigenetic modifications. Mol Genet Genomics 291:1491–1504. doi: 10.1007/s00438-016-1167-2. [DOI] [PubMed] [Google Scholar]
- 15.Mardis ER. 2008. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet 9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 16.Henn MR, Sullivan MB, Stange-Thomann N, Osburne MS, Berlin AM, Kelly L, Yandava C, Kodira C, Zeng Q, Weiand M, Sparrow T, Saif S, Giannoukos G, Young SK, Nusbaum C, Birren BW, Chisholm SW. 2010. Analysis of high-throughput sequencing and annotation strategies for phage genomes. PLoS One 5:e9083. doi: 10.1371/journal.pone.0009083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duhaime MB, Deng L, Poulos BT, Sullivan MB. 2012. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: a rigorous assessment and optimization of the linker amplification method. Environ Microbiol 14:2526–2537. doi: 10.1111/j.1462-2920.2012.02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, Chan AM, Haynes M, Kelley S, Liu H, Mahaffy JM, Mueller JE, Nulton J, Olson R, Parsons R, Rayhawk S, Suttle CA, Rohwer F. 2006. The marine viromes of four oceanic regions. PLoS Biol 4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polson SW, Wilhelm SW, Wommack KE. 2011. Unraveling the viral tapestry (from inside the capsid out). ISME J 5:165–168. doi: 10.1038/ismej.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim KH, Bae JW. 2011. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl Environ Microbiol 77:7663–7668. doi: 10.1128/AEM.00289-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yilmaz S, Allgaier M, Hugenholtz P. 2010. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat Methods 7:943–944. doi: 10.1038/nmeth1210-943. [DOI] [PubMed] [Google Scholar]
- 22.Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon JM, Westerman RP, SanMiguel PJ, Bennetzen JL. 2009. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet 5:e1000732. doi: 10.1371/journal.pgen.1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao GC, Rehm EJ, Rubin GM. 2000. Insertion site preferences of the P transposable element in Drosophila melanogaster. Proc Natl Acad Sci U S A 97:3347–3351. doi: 10.1073/pnas.97.7.3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flaherty P, Natsoulis G, Muralidharan O, Winters M, Buenrostro J, Bell J, Brown S, Holodniy M, Zhang N, Ji HP. 2012. Ultrasensitive detection of rare mutations using next-generation targeted resequencing. Nucleic Acids Res 40:e2. doi: 10.1093/nar/gkr861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson SJ, Welkers MRA, Depledge DP, Coulter E, Breuer JM, de Jong MD, Kellam P. 2013. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos Trans R Soc Lond B Biol Sci 368:20120205. doi: 10.1098/rstb.2012.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCrone JT, Lauring AS. 2016. Measurements of intrahost viral diversity are extremely sensitive to systematic errors in variant calling. J Virol 90:6884–6895. doi: 10.1128/JVI.00667-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sicard A, Yvon M, Timchenko T, Gronenborn B, Michalakis Y, Gutierrez S, Blanc S. 2013. Gene copy number is differentially regulated in a multipartite virus. Nat Commun 4:2248. doi: 10.1038/ncomms3248. [DOI] [PubMed] [Google Scholar]
- 29.Sicard A, Michalakis Y, Gutiérrez S, Blanc S. 2016. The strange lifestyle of multipartite viruses. PLoS Pathog 12:e1005819. doi: 10.1371/journal.ppat.1005819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roux S, Solonenko NE, Dang VT, Poulos BT, Schwenck SM, Goldsmith DB, Coleman ML, Breitbart M, Sullivan MB. 2016. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 4:e2777. doi: 10.7717/peerj.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurihara L, Banks L, Chupreta S, Couture C, Laliberte J, Sandhu S, Schumacher C, Spurbeck R, Makarov V. 2014. A new method for low-input, PCR-free NGS libraries with exceptional evenness of coverage. Swift Biosciences, Ann Arbor, MI. [Google Scholar]
- 32.Grigoras I, Timchenko T, Katul L, Grande-Pérez A, Vetten H-J, Gronenborn B. 2009. Reconstitution of authentic nanovirus from multiple cloned DNAs. J Virol 83:10778–10787. doi: 10.1128/JVI.01212-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, Van Den Hoff MJB, Moorman AF. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sicard A, Zeddam J-L, Yvon M, Michalakis Y, Gutiérrez S, Blanc S. 2015. Circulative nonpropagative aphid transmission of nanoviruses: an oversimplified view. J Virol 89:9719–9726. doi: 10.1128/JVI.00780-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monat C, Tranchant-Dubreuil C, Kougbeadjo A, Farcy C, Ortega-Abboud E, Amanzougarene S, Ravel S, Agbessi M, Orjuela-Bouniol J, Summo M, Sabot F. 2015. TOGGLE: toolbox for generic NGS analyses. BMC Bioinformatics 16:374. doi: 10.1186/s12859-015-0795-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jorde PE, Ryman N. 2007. Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935. doi: 10.1534/genetics.107.075481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lenormand M, Jabot F, Deffuant G. 2013. Adaptive approximate Bayesian computation for complex models. Comput Stat 28:2777–2796. doi: 10.1007/s00180-013-0428-3. [DOI] [Google Scholar]