

ABSTRACT
During infectious entry, acidification within the endosome triggers uncoating of the human papillomavirus (HPV) capsid, whereupon host cyclophilins facilitate the release of most of the major capsid protein, L1, from the minor capsid protein L2 and the viral genome. The L2/DNA complex traffics to the trans-Golgi network (TGN). After the onset of mitosis, HPV-harboring transport vesicles bud from the TGN, followed by association with mitotic chromosomes. During this time, the HPV genome remains in a vesicular compartment until the nucleus has completely reformed. Recent data suggest that while most of L1 protein dissociates and is degraded in the endosome, some L1 protein remains associated with the viral genome. The L1 protein has DNA binding activity, and the L2 protein has multiple domains capable of interacting with L1 capsomeres. In this study, we report that some L1 protein traffics with L2 and viral genome to the nucleus. The accompanying L1 protein is mostly full length and retains conformation-dependent epitopes, which are recognized by neutralizing antibodies. Since more than one L1 molecule contributes to these epitopes and requires assembly into capsomeres, we propose that L1 protein is present in the form of pentamers. Furthermore, we provide evidence that the L1 protein interacts directly with viral DNA within the capsid. Based on our findings, we propose that the L1 protein, likely arranged as capsomeres, stabilizes the viral genome within the subviral complex during intracellular trafficking.
IMPORTANCE After internalization, the nonenveloped human papillomavirus virion uncoats in the endosome, whereupon conformational changes result in a dissociation of a subset of the major capsid protein L1 from the minor capsid protein L2, which remains in complex with the viral DNA. Recent data suggest that some L1 protein may accompany the viral genome beyond the endosomal compartment. We demonstrate that conformationally intact L1 protein, likely still arranged as capsomeres, remains associated with the incoming viral genome throughout mitosis and transiently resides in the nucleus until after the viral DNA is released from the transport vesicle.
KEYWORDS: HPV entry, HPV16, L1 protein, L2 protein, mitosis, nuclear transport, virus trafficking
INTRODUCTION
Papillomaviruses are a family of nonenveloped DNA viruses that infect a wide range of hosts with a preference for epithelial cells. The papillomaviruses that infect humans (HPVs) are a particular health burden. While most infections with HPVs are cleared by the immune system, a persistent infection with the high-risk types is associated with increased risk of carcinomas. The high-risk type 16 accounts for approximately 50% of the total cervical cancer cases and up to 90% of anogenital and oropharyngeal cancers, causing significant morbidity and mortality in the human population (1, 2). Due to this, HPV16 is the best-studied HPV type. The HPV genome is 8 kb in size and is a chromatinized, double-stranded DNA molecule protected within a 55 -nm icosahedral capsid. The capsid is comprised of two proteins: the major capsid protein, L1, and the minor capsid protein, L2. In each capsid, there are 360 L1 monomers arranged into 72 pentamers, also called capsomeres, which compose the capsid shell. It has been estimated that there can be up to 72 molecules of L2 per capsid arranged as one L2 copy per capsomere (3). However, conservative estimates are likely in the range of 12 to 36 copies per capsid. The majority of the L2 protein is hidden within the mature capsid, whereas only a portion of the N terminus, residues 60 to 120, is exposed on the surface (4). It has been proposed that the N terminus of the L2 protein adopts a loop-like structure where the first 60 or so amino acids on the N terminus are not accessible in the capsid.
During the primary infection, the virion initially attaches to heparan sulfate proteoglycans (HSPGs) located on the extracellular matrix and/or cell surface (5–9). Multiple engagements of the L1 proteins with HSPGs induce conformational changes in both L1 and L2 proteins (10, 11). Host cell cyclophilin B, a chaperone found on the cell surface and in luminal compartments, facilitates the exposure of the previously hidden N terminus and reveals a furin convertase cleavage site (12–14). The cleavage and loss of these 12 amino acids from the very N terminus are essential downstream in the entry process for endosomal escape. In addition, a fraction of L1 protein is cleaved by kallekrein-8 on the cell surface (15). Following these conformational changes, the virion is reported to associate with a number of non-HSPG secondary receptors, including integrins, tetraspanins, growth factor receptors, and annexin A2, that serve as a platform for internalization (16–24). The virion is internalized via the endocytic route and is trafficked to the endosome whereupon low pH triggers disassembly of the capsid. Cyclophilins facilitate release of much of the L1 protein from the L2 protein, which remains in complex with the viral genome (25). During this time, data suggest that the L2 protein undergoes conformational changes resulting in translocation of the majority of the protein across the endosomal membrane through possible solo or concerted efforts of a membrane-destabilizing (residues 445 to 467) and a transmembrane-like (residues 45 to 65) domain (26–29). This translocation event allows for the cytoplasmic region of the L2 protein (likely residues 65 to 473) to directly interact with cytoplasmic factors, most notably, the retromer complex, which facilitates the trafficking of the L2/DNA complex to the trans-Golgi network (TGN) (29–37). Residues 13 to 45 are located on the luminal side of transport vesicles, whereas residues 45 through 65 span the membrane. Incoming viral genomes take advantage of the structural reorganization of the cell during mitosis for trafficking into the nucleus, where nuclear envelope breakdown is a key rate-limiting step (38, 39). After the onset of mitosis, the viral genome buds out of the TGN and/or endoplasmic reticulum (ER) in a transport vesicle and lines up along microtubules to, presumably, migrate to the condensed chromosomes (38, 40). Once the cell has completed division and the nuclear envelope has reformed, the viral DNA is released from this transport vesicle within the nucleus and the viral genome colocalizes at promyelocytic leukemia (PML) nuclear bodies to establish infection (41).
After uncoating, it is almost completely unknown how the viral genome remains associated with the L2 protein within the subviral complex. Since recent evidence suggests that most of the L2 protein is transmembranous during this time, we hypothesized that other proteins may be responsible for stabilizing the viral genome within the subviral complex. It is established that the L1 protein plays dual roles in both inter-L1 interaction between capsomeres and DNA binding (42–45). The C terminus of the L1 protein is quite disordered and flexible, which allows it to invade the neighboring capsomeres to form disulfide bonds at highly conserved cysteine residues to stabilize the interaction between capsomeres (46–48). It also has been shown to interact with DNA and be required for encapsidation of viral DNA (49). Although the L2 protein is essential for infection, its role in DNA packaging is controversial in that some HPV types depend more on it than others (45, 50–52). Curiously, recent data from several groups have observed that not all of the L1 protein is dissociated from the L2 protein during uncoating (34, 40, 53, 54). Based on these data, we hypothesized that the L1 protein accompanies the viral genome, in association with the L2 protein, to the nucleus during infectious entry. Here, we demonstrate that both L1 and L2 protein transiently colocalize with the viral genome in the nucleus of infected cells after the completion of mitosis. The loss of the L1 protein correlated with the release of viral genome from transport vesicles. The L1 protein that accompanies the viral genome throughout the entry process is mostly full length and retains conformational epitopes that are recognized by well-characterized neutralizing antibodies. Although the exact function of the L1 protein after uncoating is still a mystery, we provide biochemical evidence that L1 protein interacts with encapsidated DNA when assembled into virions, suggesting that L1 may help stabilize the L2/DNA complex during trafficking events well beyond what was previously assumed.
RESULTS
TGN-localized L1 protein retains conformational epitopes.
It has been hinted by a few recent publications that some L1 protein remains associated with the L2/DNA complex after uncoating (34, 40, 53, 54). We previously estimated that approximately up to 75% of the L1 protein is dissociated after uncoating (25, 54). Based on these findings, we sought to determine whether the remaining L1 protein would still be arranged as capsomeres after the capsid has uncoated. To address this question, we used well-characterized neutralizing monoclonal antibodies (MAbs) to probe for protein structure. First, we tested the reactivity of H16.V5, which recognizes a conformational epitope to which the FG and HI loops of two adjacent L1 molecules on the apical surface of L1 capsomeres contribute (55–57). HaCaT cells were infected with HPV16 pseudoviruses (PsVs) harboring an 5-ethynyl-2′-deoxyuridine (EdU)-labeled pseudogenome. After 24 hpi, we fixed and stained the L1 protein using MAb H16.V5, followed by staining of the pseudogenome using Click-iT reaction chemistry. We observed that MAb H16.V5 showed reactivity colocalizing with the EdU puncta in the TGN (Fig. 1A). As a control, we treated the cells with Click-iT reaction buffer before incubation with the MAb H16.V5, which has been previously shown to denature the capsids and thus eliminate the conformational epitopes (25). As we expected, we observed a complete loss of MAb H16.V5 reactivity. We also tested a previously characterized neutralizing MAb, H16.56E, which recognizes a conformational epitope that becomes accessible only after the virions bind and undergo conformational changes on the cell surface (5, 8, 11). We observed that the reactivity of MAb H16.56E colocalized with EdU puncta within the TGN (Fig. 1B). This reactivity was also lost after denaturing the capsids with Click-iT reaction buffer prior to staining with the MAbs, as previously demonstrated (25). To test whether most of the L1 protein trafficking with L2 and viral genome to the TGN retains conformational epitopes rather than being denatured, we tested the reactivity of L1 protein with another well-characterized MAb, 33L1-7, which recognizes a linear epitope (residues 303 to 313) that emerges only after the virus has been denatured or uncoats (17, 58). After infectious entry, we did not observe reactivity with MAb 33L1-7 colocalized with EdU puncta in the TGN (Fig. 1C). Following denaturation, we observed robust reactivity with 33L1-7 MAb. Taken together, these data suggest that the subset of L1 protein that associates with viral genome in the TGN is likely still arranged as conformationally intact L1 capsomeres.
FIG 1.
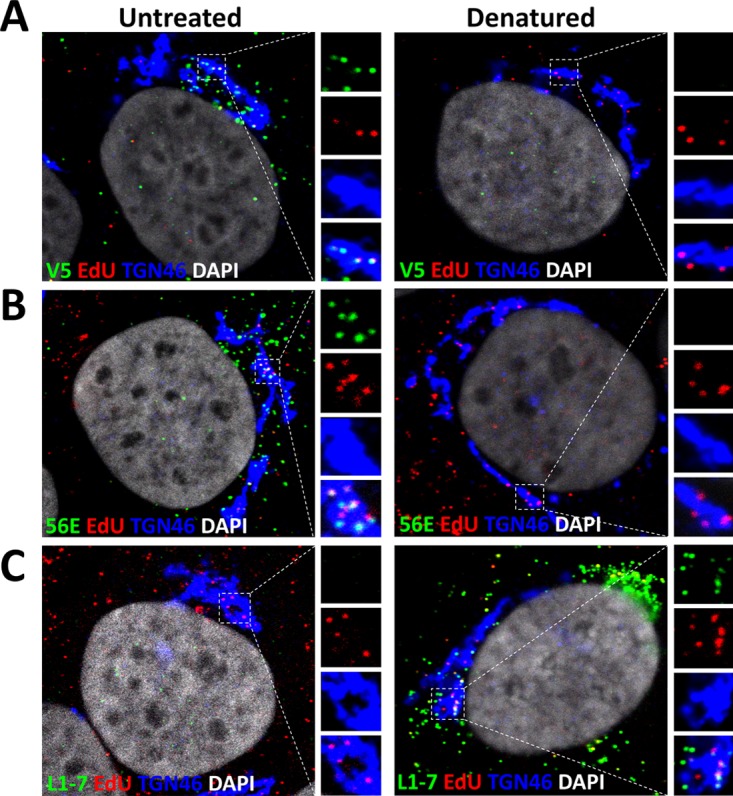
L1 protein localized in the TGN retains conformationally dependent epitopes. (A) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed, permeabilized, and treated with or without Click-iT reaction buffer without dye. The cells were incubated with the L1 conformationally dependent mouse MAb H16.V5 antibody (green), rabbit pAb anti-TGN46 (blue). Next, the cells were treated with Click-iT reaction buffer with AF555 dye (red) to stain EdU-labeled pseudogenome and mounted in DAPI (white). (B) HaCaT cells were infected and stained as described for panel A. Conformationally dependent mouse MAb H16.56E antibody (green) was used for specific detection of the L1 protein. (C) HaCaT cells were infected and stained as described for panel A. Mouse MAb 33L1-7 (green) was used for the specific detection of the L1 protein. Triple colocalization is denoted as a white color.
L1 protein retains conformational epitopes throughout mitosis.
It was previously demonstrated that mitosis is a key rate-limiting step in the HPV infection, and the incoming viral genome associated with condensed chromosomes during mitosis (38, 39). Recent data from our lab suggest that the viral genome buds from the TGN during prophase in a membrane-bound transport vesicle and associates with microtubules for delivery to the nucleus during mitosis (40). We reasoned that if conformationally intact L1 protein does remain associated with the L2 protein and the incoming viral DNA within the TGN, then it may also remain associated throughout mitosis as well. To test this, we infected HaCaT cells with HPV16 PsVs harboring an EdU-labeled pseudogenome for 24 h. We fixed and stained the cells using MAbs H16.V5, H16.56E, and 33L1-7 as described in Fig. 1A to C. We carefully restricted our analysis to cells in different phases of the cell cycle. We observed that EdU puncta associated with condensed chromosomes during mitosis were reactive with MAb H16.V5 (Fig. 2A). Pretreating the cells with Click-iT reaction buffer prior to incubation with the primary antibody eliminated MAb H16.V5 reactivity, as previously observed (Fig. 2B). Next, we tested MAb H16.56E reactivity under the same conditions. Similarly, we observed reactivity with MAb H16.56E colocalized with EdU puncta associated with condensed chromosomes (Fig. 3A) and pretreatment with Click-iT reaction buffer destroyed the H16.56E epitope (Fig. 3B). Lastly, we tested MAb 33L1-7 under the same conditions. Once again, we did not observe reactivity with MAb 33L1-7 colocalized with EdU puncta associated with condensed chromosomes (Fig. 4A). However, once we denatured the capsids using a pretreatment with Click-iT reaction buffer, 33L1-7 reactivity was observed colocalizing with the EdU puncta (Fig. 4B). Taken together, these data support the idea that intact L1 capsomeres remain associated with the viral genome throughout mitosis.
FIG 2.

L1 protein associated with condensed chromosomes retains the reactivity of the H16.V5 epitope. (A and B) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed, permeabilized, and treated with or without Click-iT reaction buffer without dye. Next, the cells were incubated with L1 conformationally dependent mouse MAb H16.V5 antibody (green) and AF488-conjugated anti-α-tubulin (white). Lastly, the cells were treated with Click-iT reaction buffer with AF555 dye (red) to stain the EdU-labeled pseudogenome and mounted in DAPI (blue). Note that colocalization of the EdU puncta and L1 protein is denoted as a yellow color.
FIG 3.

L1 protein associated with condensed chromosomes retains the reactivity of the H16.56E epitope. (A and B) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed, permeabilized, and treated with or without Click-iT reaction buffer without dye. Next, the cells were incubated with L1 conformationally dependent mouse MAb H16.56E antibody (green) and AF488-conjugated anti-α-tubulin (white). Lastly, the cells were treated with Click-iT reaction buffer with AF555 dye (red) to stain the EdU-labeled pseudogenome and mounted in DAPI (blue). Note that colocalization of the EdU puncta and L1 protein is denoted as a yellow color.
FIG 4.

The linear epitope of the 33L1-7 antibody is hidden in the L1 protein associated with condensed chromosomes. (A and B) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed, permeabilized, and treated with or without with Click-iT reaction buffer without dye. Next, the cells were incubated with mouse MAb 33L1-7 antibody (green) and AF488-conjugated anti-α-tubulin (white). Lastly, the cells were treated with Click-iT reaction buffer with AF555 dye (red) to stain the EdU-labeled pseudogenome and mounted in DAPI (blue). Note that colocalization of the EdU puncta and L1 protein is denoted as a yellow color.
The L1 protein is transiently present in newly formed nuclei after the completion of mitosis and is lost after egress of the viral genome from transport vesicles.
Our recently published data suggest that the viral genome resides in a membrane-bound vesicle throughout mitosis and is released, in a time-dependent fashion, into the nucleoplasm after the nuclear envelope has reformed (40). It was previously demonstrated that the L2 protein colocalizes at PML nuclear bodies, but no evidence was reported that L1 protein also localizes in the nucleus during entry (41). However, these experiments were analyzed at 48 h postinfection (hpi), whereupon cells have completed multiple rounds of division. Also, these findings preceded knowledge that mitosis is a key rate-limiting step in the infection (38, 39). We reasoned that if the viral genome is residing in the lumen of a vesicle during mitosis, then the L1 protein would remain with the viral genome until after the completion of mitosis. Indeed, we observed clear colocalization of L1 and viral genome in the nucleus of early interphase cells, giving support for this reasoning (Fig. 5A to D). Quantification of overlapping L1 and EdU puncta revealed that ∼85% of the L1 signal colocalized with EdU puncta associated with condensed chromosomes and was comparable to colocalization within the perinuclear region of interphase cells (Fig. 5E). However, when we analyzed nuclear localized EdU puncta of interphase cells, colocalization between the L1 and EdU signals varied greatly ranging from 0 to 100%, with an average of ∼36% colocalization. These data suggested to us that complete dissociation of the L1 protein from the L2/DNA complex occurs within the nucleus after completion of mitosis. Based on these observations, we hypothesized that the L1 protein would likely be lost after the viral genome becomes accessible in the nucleus. To test whether the L1 and L2 proteins colocalized as a function of genome accessibility, we infected HaCaT cells with HPV16 PsV harboring an EdU-labeled pseudogenome. We first selectively permeabilized the plasma membrane using a low concentration of digitonin. Next, we performed two sequential Click-iT reactions using small-molecular-mass dyes, Alexa Fluor (AF)555 and AF647, as previously described (40). AF555 (denoted in green) was used after digitonin treatment, and AF647 (denoted in red) was used after Triton X-100 (TX-100) treatment. Staining in this order results in differential staining of the viral genome based on limited membrane accessibility. Lastly, we stained for the L1 protein (denoted as cyan) using our MAbs and quantified colocalization (Fig. 6). As a control, we assessed the membrane integrity in our digitonin-treated cells in a parallel experiment by testing the reactivity of an antibody that recognizes a luminal epitope of TGN46 (denoted as cyan) compared to TX-100-treated cells. We observed only reactivity of the luminal epitope of TGN46 in the TX-100-treated cells, but not digitonin-treated cells, suggesting that the plasma membrane but not the internal membranes were permeabilized (Fig. 6A). As a positive control, we treated the cells with TX-100 prior to staining to completely permeabilize the cells. Representative images of infected HaCaT cells in mitosis and interphase are displayed (Fig. 6B and C). We quantified the total number of single red (inaccessible [IN]) or dual green/red (accessible [AC]) EdU puncta and whether L1 colocalized (cyan) (Fig. 6D and E). At 24 hpi, almost 90 and 35% of the EdU puncta were inaccessible in mitotic and interphase cells, respectively (Fig. 6D). In mitotic cells, the vast majority of inaccessible pseudogenome also colocalized with the L1 protein—to nearly comparable levels, as previously observed in Fig. 5E. In contrast, less than 40% of accessible genomes stained positive for the L1 protein (Fig. 6E). During interphase, approximately 35 and 68% of nuclear localized EdU puncta were inaccessible and accessible, respectively (Fig. 6D). Of the inaccessible EdU puncta, 60% colocalized with the L1 protein compared to only 20% of the accessible (Fig. 6E). Colocalization of L1 and genome in interphase was more often observed in newly divided cells compared to “old” interphase cells with fewer numbers of nucleoli. This may explain why we observed various degree of colocalization between nuclear localized EdU and L1 puncta in Fig. 5E. Taken together, our data suggest that, during an infection, the L1 protein does exist in the nucleus, albeit only transiently, and dissociates from the viral genome as it becomes accessible in the nucleus. Next, we wanted to test whether L2 is lost in the same manner (Fig. 7). Using the same experimental design as described in Fig. 6, we differentially stained the viral genome based on limited accessibility to small molecular dyes and quantified colocalization with the L2 protein. Once again, representative images of infected HaCaT cells are displayed (Fig. 7A and B). In mitotic and interphase cells, approximately 45% of inaccessible EdU puncta costained for L2 (Fig. 7C and D), which is due to lower levels of L2 present in viral particles or lower affinity antibody binding. Of the accessible EdU puncta, about 25% had L2 protein present. Taken together, these data suggest that both the L1 and the L2 proteins transiently reside in the nucleus after division is completed, whereupon L1 likely dissociates shortly after viral genome becomes accessible and the L2 protein remains for an undetermined extended period of time.
FIG 5.

L1 protein accompanies the viral genome into the nucleus. (A) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed and permeabilized. Next, the cells were incubated with L1 conformationally dependent mouse MAb H16.V5 antibody (green) and rabbit pAb anti-lamin B receptor (LBR) (white). Lastly, the cells were treated with Click-iT reaction buffer with AF555 dye (red) to stain EdU-labeled pseudogenome and mounted in DAPI (blue). (B) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed, permeabilized, and treated with Click-iT reaction buffer without dye. Next, cells were incubated with mouse MAb 33L1-7 antibody (green) and rabbit pAb anti-LBR (white). Lastly, the cells were treated with Click-iT reaction buffer with AF555 dye (red) to stain EdU-labeled pseudogenome and mounted in DAPI (blue). (C and D) Colocalization of L1 and EdU puncta within the nucleus was assessed by acquiring Z-stack images of infected cells. The signal intensities of single puncta were assessed by analyzing the line profile of individual EdU puncta. Note that the signal intensity is expressed in arbitrary units (AU). (E) Quantifications are from two repeat experiments analyzing single slice images of Z-stacks (n = 25 to 30 cells, and >800 EdU puncta were counted). Data are reported as percentages of the colocalization between L1 and EdU puncta as a function of the EdU localization within the cell. Puncta: perinuclear localized EdU puncta within interphase cells = 87.3% ± 2.2%; condensed chromatin-associated EdU puncta within mitotic cells = 84.0% ± 2.6%; and nuclear localized EdU puncta within interphase cells = 36.3% ± 5.1%.
FIG 6.

L1 proteins that accompany the viral genome into the nucleus dissociate after release of the viral genome. (A) At 24 h, uninfected HaCaT cells were fixed, permeabilized with either digitonin at 0.625 μg/ml or 0.5% TX-100, and treated with AF555 (green) in Click-iT reaction buffer. Next, the cells were incubated with pAb rabbit anti-TGN46 (cyan), which recognizes a luminal epitope of TGN46. Lastly, the cells were permeabilized in 0.5% TX-100, followed by incubation with goat anti-rabbit secondary antibody and subsequent mounting in DAPI (white). Note the lack of reactivity of luminal anti-TGN46 antibody after digitonin treatment. (B and C) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed, permeabilized with digitonin at 0.625 μg/ml (B) or 0.5% TX-100 (C), and treated with AF555 (green) in Click-iT reaction buffer. The cells were permeabilized again with 0.5% TX-100 and treated with AF647 (red) in Click-iT reaction buffer. Lastly, the cells were incubated with mouse MAb 33L1-7 (cyan) for specific detection of the L1 protein and mounted in DAPI (white). (D and E) The percent accessibility of viral genome was determined by counting the number of red-only (inaccessible [IN]) or red/green (accessible (AC) stained EdU puncta associated with condensed chromosomes on mitotic cells or nuclear localized in interphase cells. Colocalization of L1 and EdU puncta was quantified by counting the number of EdU puncta that colocalized with L1 signal. Quantifications are from two repeat experiments analyzing two to three Z-stack images per cell (n = 30 to 40 cells, and >800 EdU puncta were counted per experiment). Mitosis: %IN = 88.58% ± 7.67%, %AC = 11.42% ± 7.67%, %L1 of IN = 82.3% ± 7.30%, %L1 of AC = 42.665% ± 9.335%. Interphase: %IN = 34.53% ± 0.427%, %AC = 68.36% ± 6.463%, %L1 of IN = 58.8% ± 3.805%, %L1 of AC = 20.84% ± 9.159%.
FIG 7.

L2 proteins that accompany the viral genome into the nucleus remain associated with the viral genome. (A and B) At 24 hpi, HaCaT cells infected with EdU-labeled pseudovirus were fixed, permeabilized with digitonin at 0.625 μg/ml of (A) or 0.5% TX-100 (B), and treated with AF555 (green) in Click-iT reaction buffer. The cells were permeabilized again with 0.5% TX-100 and treated with AF647 (red) in Click-iT reaction buffer. Lastly, the cells were incubated with mouse MAb 33L2-1 (cyan) for specific detection of the L2 protein and mounted in DAPI (white). (C and D) The percent accessibility of the viral genome was determined by counting the number of red-only (inaccessible [IN]) or red/green (accessible [AC]) stained EdU puncta associated with condensed chromosomes on mitotic cells or nuclear localized in interphase cells. Colocalization of L2 and EdU puncta was quantified by counting the number of EdU puncta that colocalized with L2 signal. Quantifications are from two repeat experiments analyzing two to three Z-stack images per cell (n = 30 to 40 cells and >800 EdU puncta counter per experiment). Mitosis: %IN = 83.67% ± 8.435%, %AC = 16.34% ± 8.435%, %L2 of IN = 46.15% ± 6.15%, %L2 of AC = 24.45% ± 16.55%. Interphase: %IN = 42.15% ± 1.35%, %AC = 57.85% ± 1.35%, %L2 of IN = 44.65% ± 6.35%, %L2 of AC = 30.65% ± 3.35%.
Full-length L1 protein is present in mitotic transport vesicles.
To analyze this subset of L1 protein accompanying the viral genome to the nucleus further, we needed to enrich for mitotic associated viral genomes. We previously showed that infection in the presence of Eg5 inhibitor III, a potent inhibitor of the Eg5 mitotic kinesin, results in enrichment of viral genomes associated with condensed chromosomes in cells arrested in mitosis. When cells were treated with Eg5i, they become arrested in a monoastral phenotype, which we observed by live-cell tracking of infected HeLa cells after 18 hpi (Fig. 8A). We had previously shown that viral genome is retained in membrane-bound transport vesicles in monoastral cells (Fig. 8B) (40). However, what we did not know is whether or not the L1 protein is still associated with the L2/DNA complex in these mitotically arrested monoastral cells. To test this, we infected HaCaT cells, arrested them in mitosis by treating them with Eg5i, and stained for the L1 protein and viral genome. Analysis of arrested cells infected with pseudovirus depicted robust colocalization between L1 and the EdU puncta associated with the condensed chromosomes (Fig. 8C). It is known that the DNA binding activity of L1 protein was mapped to the carboxy-terminal overlapping with the nuclear localization signal. Recently, it was also shown that L1 protein is partially proteolytically cleaved by kallekrein-8 on the cell surface after conformational changes have occurred (15). This cleavage was suggested to occur at a conserved consensus cleavage site in the carboxy-terminal arm of the L1 protein. To test whether L1 protein trafficking to the nucleus retains the DNA binding domain on the C terminus, we enriched infected HeLa cells by treating them with Eg5i. Monoastral HeLa cells were tapped off the plastic surface at 18 hpi in the presence of Eg5i, treated with trypsin to remove remnant HPV particles attached to the cell surface, and subsequently physically ruptured by passage through a syringe, followed by another trypsin treatment. Even after extended trypsin digestion, a fraction of the L1 protein was running as full-length protein in SDS-PAGE. In addition, two proteolytic fragments were observed, which is consistent with kallekrein-8-cleaved L1 protein (Fig. 8D) (15).
FIG 8.
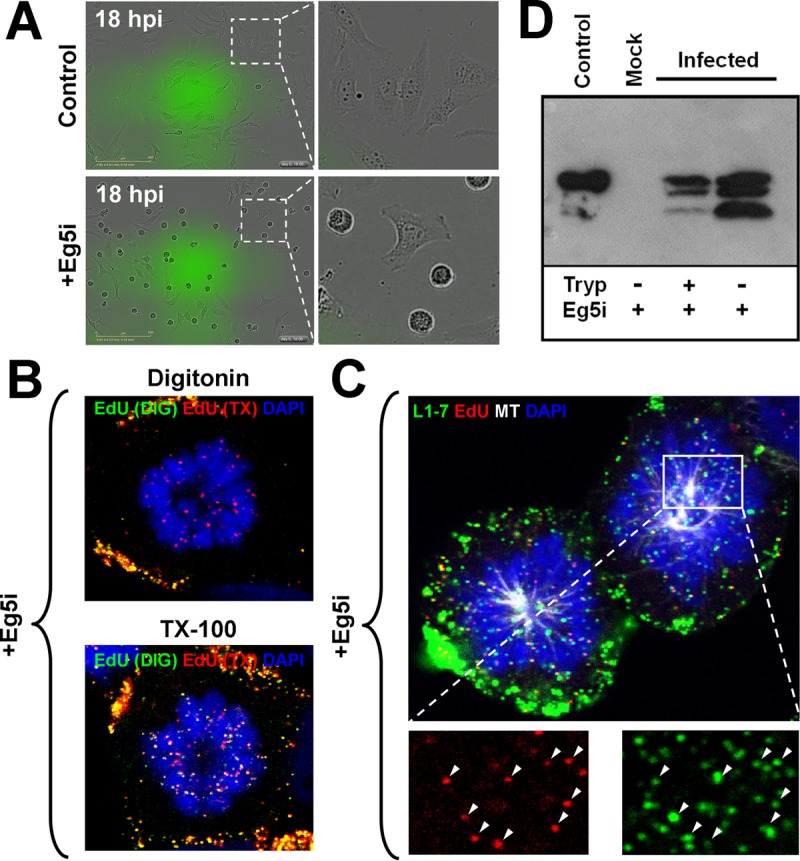
Some full-length L1 protein accompanies the viral genome to the nucleus. (A) HeLa cells were infected with HPV16 pseudovirus with or without 1.5 μM Eg5 inhibitor III (Eg5i) and tracked via live-cell imaging using the IncuCyte Zoom for 48 h. Note that the images are depicted at 18 hpi. (B) HaCaT cells were infected with EdU-labeled HPV16 pseudovirus in the presence of 1.5 μM Eg5i. The cells were fixed at 24 hpi and permeabilized with either digitonin at 0.625 μg/ml or 0.5% TX-100 and then treated with AF555 (green) in Click-iT reaction buffer. The cells were permeabilized again with 0.5% TX-100 and treated with AF647 (red) in Click-iT reaction buffer. Lastly, the cells were mounted in DAPI (blue). (C) HaCaT cells were infected with EdU-labeled pseudovirus and treated with 1.5 μM Eg5i. At 24 hpi, the cells were fixed and permeabilized in 0.5% TX-100. Next, the cells were treated with AF555 (red) in Click-iT reaction buffer, followed by incubation with AF488-conjugated anti-α-tubulin (white) and MAb 33L1-7 (green). Lastly, the cells were mounted in DAPI (blue). Note the colocalization between EdU and L1 signal denoted by white arrows. (D) HeLa cells were infected with HPV16 pseudovirus in the presence of 1.5 μM Eg5i. Cells were trypsinized for 2 min, and monoastral cells were collected. Next, the cells were treated with 15 μl of 0.25% trypsin for 1 h at 37°C. The cells were lysed by passage through a 1-ml syringe with a 25-gauge needle 40 times. Cell lysates were incubated for 1 h at 37°C once more, the trypsin was inactivated, and the samples were analyzed by Western blot analysis with a cocktail of HPV16 L1-specific mouse MAbs (IID5, 33L1-7, and 312F).
The L1 protein interacts with encapsidated DNA in virions.
We hypothesized that L1 protein may be stabilizing the viral DNA by a direct interaction in intracellular transport vesicles. This hypothesis is supported by the notion that the C terminus of the L1 protein has the propensity to bind DNA and was essential for DNA encapsidation (45, 49). However, it was unknown whether L1 protein directly interacts with encapsidated DNA. To test whether this is the case, we generated HPV16 PsVs that encapsidate a reporter plasmid (pseudogenome). Using these purified PsVs, we partially disassembled the capsid structure by treating them with or without dithiothreitol (DTT) to disrupt inter-L1 disulfide bonds between capsomeres (44). Next, we treated the capsids with or without DNase I, performed sedimentation using a linear sucrose gradient fractionation, and analyzed the protein by Western blot analysis (Fig. 9). To determine how disassembly affects sedimentation of L1 and L2 in the absence of packaged DNA, we also generated DNA-free virus-like particles (VLPs). The VLPs were purified by buoyant density gradient centrifugation to remove any DNA-harboring particles. DTT treatment of DNA-free VLPs released a large fraction of L1 protein, which remained close to the top of the gradient (Fig. 9A). Surprisingly, the L2 protein and some L1 protein sedimented far into the gradient in a broad peak. When we analyzed DTT-treated VLPs by electron microscopy, we confirmed disassembly of VLPs but also found that L1 capsomeres were still present in large clusters with a rather regular spacing (Fig. 9B and C). Underlying was a density, which we assume is due to L2 protein. When we analyzed untreated and DTT-treated pseudovirions by sucrose gradient sedimentation, we observed that the L1 and L2 proteins cosedimented in the middle of the gradient (fractions 8 to 12) (Fig. 9D). Slightly slower sedimentation of DTT-treated pseudovirions is consistent with previously observed expansion of HPV virions in the absence of stabilizing disulfide bonds (59). Only additional treatment with DNase I resulted in L1 protein being released and fractionated near the top of the gradient (fractions 1 to 3) (Fig. 9D). Treatment with DTT and DNase I did not result in release of the L2 protein, as observed with L1. Instead, the L2 protein cosedimented with a subset of the L1 protein in the middle fractions similar to what we observed with DNA-free VLPs (fractions 10 to 11). Successful digestion of pseudogenome after DNase treatment was confirmed by PCR (data not shown). Taken together, these data suggest that there is an interaction between the L1 protein and the viral DNA within the capsid.
FIG 9.
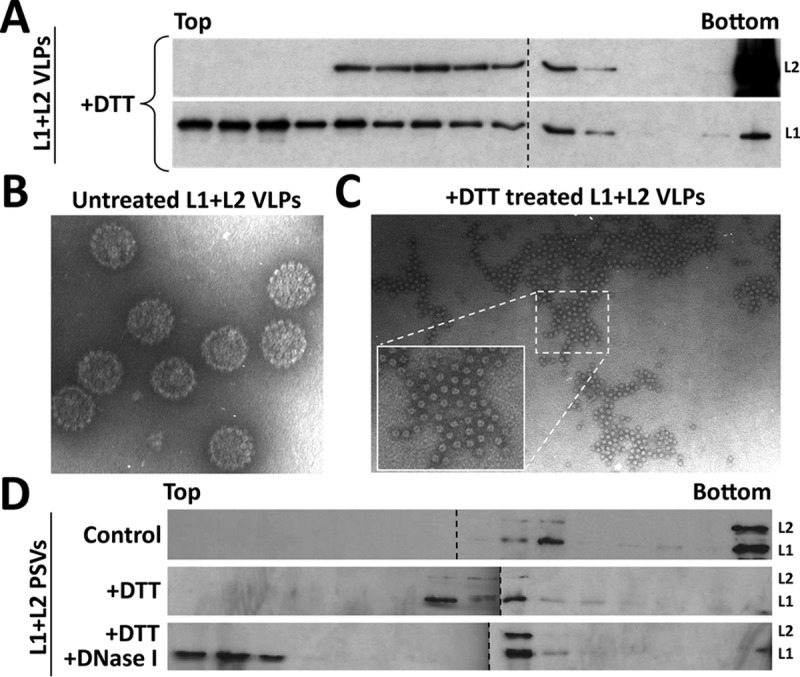
L1 protein interacts with the viral DNA within the capsid. (A) Purified DNA-free HPV16 L1/L2 VLPs were incubated with 20 mM DTT for 15 min at room temperature. Samples were layered on the top of a 20 to 60% sucrose linear gradient and subjected to ultracentrifugation. Collected fractions were analyzed by Western blotting for specific detection of the L1 and L2 proteins using MAbs. (B and C) Cesium chloride-purified DNA-free HPV16 L1/L2 VLPs were analyzed by electron microscopy with or without 20 mM DTT treatment. (D) HPV16 L1/L2 pseudoviruses were incubated with 10 mM MgCl2, with or without 20 mM DTT, and with or without 2 U of DNase I for 30 min at 37°C. Samples were layered on the top of a 20 to 60% sucrose linear gradient and subjected to ultracentrifugation. Collected fractions were analyzed by Western blotting for the specific detection of L1 and L2 proteins using MAbs.
DISCUSSION
The findings presented here suggest that a subset of the L1 protein, likely arranged as capsomeres, accompanies the L2 protein and viral genome to the nucleus. During infectious entry of pseudoviruses, we observed that conformationally intact L1 protein remained associated with viral genome localized within the TGN and associated with condensed chromosomes. Full-length L1 protein was present in HPV-harboring vesicles throughout mitosis, suggesting that the known DNA binding domain at the C terminus of the L1 protein is retained. Furthermore, we provide biochemical evidence that DNase I digestion of DTT-treated capsids releases some L1 protein from the capsid, suggesting that the L1 protein interacts with encapsidated DNA. However, we cannot exclude that structural changes of the capsid proteins after the removal of encapsidated DNA rather than loss of DNA binding are responsible for the release of L1 protein. Quantification revealed that the L1 protein dissociates after the viral genome is released from transport vesicles and becomes accessible in the nucleus, whereas the L2 protein likely remains longer.
We envision a scenario where, after internalization and cleavage of the N terminus of L2 protein and partial cleavage of L1 protein by kallekrein-8, acidification of the endosome triggers disassembly of the viral capsid. Host cell cyclophilins dissociate the subset of L1 capsomeres that interact with the C terminus of the L2 protein via the central hydrophobic core at the base of the capsomere. This release of the L1 capsomeres, which could be facilitated by cleavage of the carboxyl-terminal DNA binding domain through the activity of kallekrein-8, frees up the C-terminal membrane-destabilization domain of the L2 protein, which would allow it to penetrate the limiting endocytic membrane for subsequent interactions with cytosolic factors. The N-terminal putative transmembrane domain may serve as a stop-translocation signal and anchor L2 protein in the membrane resembling type I transmembrane protein topology.
During preparation of the manuscript, two studies were published supporting the hypothesis that L2 protein partially translocates across intracellular membranes to complete trafficking to the nucleus (60, 61). Although the exact mechanism of how the L2 protein translocates across intracellular membranes is unknown, the Schelhaas and Campos groups provide two alternative hypotheses. (i) The L2 protein may homodimerize through its GxxxG motif and form a pore that would be used for translocation. (ii) The L2 protein may even span the TGN membrane twice, whereas both the N and C termini and the viral genome reside in the lumen during trafficking. Theoretically, this would allow for the L2 protein to interact with its cytosolic binding partners needed for intracellular trafficking preceding true translocation of the L2/DNA complex (29–31, 33–37), while at the same time allowing the carboxyl-terminal putative DNA binding domain on the luminal side to interact with viral genome. However, at the onset of mitosis, the carboxyl terminus has been shown by the Campos group to be accessible in the cytosol, suggesting that at this time the L2 protein assumes the possible type I transmembrane configuration previously suggested by us. If the L2 protein does translocate as recent data suggest, we wondered how the viral genome would remain associated with the subviral protein complex.
Our previous work suggested that the majority of the L2 protein is accessible on the cytosolic side, but the viral genome does not become fully accessible until after the completion of mitosis (40). The new data presented here suggest that a subset of intact L1 capsomeres, which interact with the encapsidated viral DNA, remains associated throughout the entry process. Based on these findings, we speculate that these L1 capsomeres interact with both the L2 protein and the viral DNA and may act as a linker that stabilizes the entire subviral complex within the lumen of intracellular vesicles. Our previously published trypsin sensitivity assays suggest that essentially all the L2 protein becomes sensitive to trypsin during infectious entry (27). These results were also recapitulated in a recent publication (61). Here, we used a similar digestion assay, which suggests that some of the L1 protein associated with mitotic cells is still full length. We infected HeLa cells instead of HaCaT cells to limit the amount of background signal from the viral particles bound to the extracellular matrix. The reduction of total L1 protein we observed after trypsin digestion is likely due to digestion of uninternalized particles located on the cell surface since the entry process is highly asynchronous. Since we observed that some full-length L1 remains associated during mitosis, we think these interactions between the L1 capsomeres and the viral DNA within the membranous compartment would likely be facilitated by the C-terminal arm of the L1 protein. However, further studies would need to address whether the C-terminal arm is indeed involved in the entry process.
At this time, we also cannot completely rule out that L1 trafficking is coincidental and that interaction between the L2 protein and viral genome is mediated by L2 protein that has not yet penetrated the endocytic membrane or, as recently suggested, may pass the limiting membrane twice, resulting in the carboxyl terminus facing the lumen of transport vesicles. In addition, we cannot rule out accidental cotrafficking; however, we deem this unlikely because essentially every HPV-harboring mitotic transport vesicle also contains easily detectable L1 protein, all of which retains conformational epitopes. Unequivocal proof for our hypothesis, however, would require identification of L1 protein mutants that allow attachment, entry, uncoating, and trafficking of L2 protein to condensed mitotic chromosomes but fail to deliver viral genome into the nucleus. Due to the current lack of structural information detailing the interaction of L1 and L2 on the capsid surface and the manifold roles the L1 protein plays in cell surface events, such mutants will be difficult to identify.
Even though we do not have a very good understanding of the interactions of the major and minor capsid proteins within the capsid, it has become clear that the C terminus of the L2 protein engages with the central base of capsomeres. Some published evidence suggests that L2 protein harbors sites capable of interacting with L1 capsomeres (42–44). However, these have not been clearly mapped, and structural data are not yet available. Recent high-resolution reconstruction of cryoelectron microscopic images of HPV16 pseudovirions provided evidence for L2 density present on the outer capsid surface is suggestive of interaction between the L1 and L2 proteins (62). This supports previous findings based on the accessibility of L2 epitopes, which have suggested that L2 protein penetrates the capsid barrier allowing binding of L2 specific antibodies to N-terminal segments (residues 60 to 120) (4). The inaccessibility of the very N terminus to antibody binding also suggested that the very N terminus folds back into the capsid or engages with the surface of capsomeres. Passage through the capsid base and inaccessibility imply that N-terminal portions of L2 protein directly interact with L1 capsomeres providing a rationale for our model of how we envision interactions within the subviral complex. We have unsuccessfully tried to demonstrate interaction of L2 residues 13 through 44 with L1 capsomeres by pulldown assays or in sandwich enzyme-linked immunosorbent assays (ELISAs). Our failure to measure this interaction may be partially due to the fact that interactions of L2 protein with L1 capsomeres outside this region (residues 1 through 12 or further C terminal) may be required to initiate and stabilize the additional binding via L2 residues 13 to 44.
Our data suggested that the L1 and L2 proteins and viral genome traffic as a subviral complex to mitotic chromosomes residing in transport vesicles and are retained in the nuclei of interphase cells. Although colocalization with the L1 protein and EdU-labeled pseudogenome was very prominent and seen in ∼85 to 90% of all inaccessible EdU puncta associated with mitotic chromosomes, colocalization with the L2 protein was observed to a lesser extent. However, we attribute this to lower sensitivity of L2 antibodies used, either due to the lower abundance of the minor capsid protein or to lower affinity binding. At some point after the completion of mitosis, both the L1 and the L2 proteins dissociate from the viral genome. Loss of L1 protein coincides with egress of the viral genome from transport vesicles. At this time, we cannot rule out whether the L1 protein is being degraded or simply dispersing within the nucleus below our detection limits. While the reason for this loss is unknown, we speculate that the L1 protein may only be required during trafficking to stabilize and hold the subviral complex together. Once the viral genome is safely released in the nucleus, the L1 protein may no longer be required. In contrast, L2 protein seems to reside inside the nucleus and stay associated with the viral genome for an extended period of time. The longer residence of L2 protein after delivery of viral genome suggests that it may serve functions important for subsequent events such as establishment of infection. It is well established that incoming viral genome targets PML nuclear bodies. The Schiller group had previously reported that the L2 protein but not L1 protein colocalizes with viral genome within the nucleus of infected cells (41). These cells were analyzed at 48 hpi. This late in infection, we also see a complete loss of L1 protein. It will be interesting to investigate the exact timing of PML nuclear body association in relation to the egress of viral genome from transport vesicles and the loss of capsid protein. This may help us to gain a better understanding of the role both capsid proteins play late in the entry process.
In summary, we report that some full-length L1 protein, likely arranged as capsomeres, accompanies the viral genome to the nucleus following infectious entry. Further analysis determined that the L1 protein remains associated with the viral genome until after the completion of mitosis. Once the genome becomes accessible within the newly formed nuclei, the L1 protein dissociates. Lastly, we provide biochemical evidence that the L1 protein interacts with the encapsidated viral genome and the L2 protein. Taken together, these data suggest that the L1 protein may be involved in trafficking events well beyond the endosome as numerous studies have previously hinted at (34, 53, 54). Our new findings open up the possibility of future studies to define the role the L1 protein in the less understood later trafficking events of HPV entry. It is also worth noting that future studies should also include analyses of mitotic cells and early interphase cells since it has become apparent in the recent years that cell division is a key rate-limiting step for the nuclear entry of HPVs (38, 39).
MATERIALS AND METHODS
Cell lines.
The 293TT cells used in the generation of pseudovirions were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), l-glutamine, and antibiotics. HaCaT cells used in the infection studies were grown in low-glucose DMEM supplemented with 5% FBS and antibiotics. The HeLa cells used in this study were grown in DMEM supplemented with 10% FBS, l-glutamine, and antibiotics.
Generation of HPV16 pseudovirus.
HPV16 pseudoviruses encapsidating a green fluorescent protein (GFP) expression plasmid, pfwB, were generated in the 293TT cell line as described previously using expression plasmid pShell16L1L2HA-3′ (50, 51, 59, 63). The pfwB plasmid was kindly provided by John Schiller, National Cancer Institute, Bethesda, MD. The L1 and L2 expression plasmids harbor codon-optimized genes, previously described, and were provided graciously by Martin Müller, Deutsches Krebsfoschungszentrum, Heidelberg, Germany (64). Viral DNA within the virions was isolated using NucleoSpin Blood QuickPure (catalog no. 740569.250; Macherey-Nagel) supplemented with 20 mM EDTA and DTT. The genome copy number was quantified by quantitative real-time PCR using pfwB-specific primers. Pseudovirus preparations in our hands have a particle/infectivity ratio of 1:100 to 1:300. For pseudogenome detection by immunofluorescence microscopy, the growth medium during the generation of pseudoviruses was supplemented with 100 μM EdU at 6 h posttransfection, as previously described (65). DNA-free VLPs were generated using recombinant vaccinia virus and were subjected to buoyant density cesium chloride gradients as previously described (49, 66). DNA-free VLPs were collected from 1.29- to 1.30-g/cm3 light density fractions.
Antibodies and other reagents.
The L1 protein was detected using mouse MAbs H16.V5, H16.56E, and 33L1-7 for immunofluorescence microscopy. MAb H16.V5 is a type-specific neutralizing antibody that recognizes an epitope that maps to several exposed loops on the apical surface of capsomeres simultaneously and was generously provided by Neil Christensen (55, 67, 68). H16.56E is a conformational HPV16 L1-specific neutralizing antibody. The binding site includes—but is not restricted to—the N-terminal portion of the FG loop (HPV16 L1 residues 260 to 270) (58, 69, 70). The 33L1-7 MAb has been previously characterized and recognizes a linear epitope at amino acids 303 to 313 that is buried inside the capsid structure (11, 25, 58, 71). The L2 protein was detected with mouse MAb 33L2-1, which recognizes an epitope at amino acids 163 to 170 (25, 27, 72). The TGN was detected using rabbit polyclonal antibody (pAb) anti-TGN46, which recognizes a luminal epitope within amino acids residues 200 to 350 (PA5-23068; Thermo Fisher Scientific). The microtubule network was detected using mouse MAb anti-α-tubulin conjugated to Alexa Fluor 488 (AF488; catalog no. 8058S; Cell Signaling). Primary antibodies were detected using pAb goat anti-rabbit or anti-mouse AF488 or AF647 conjugated secondary antibodies (A-11034, A-11029, A-21244, and A-21236; Thermo Fisher Scientific). The L1 and L2 proteins were detected by Western blot analysis using a cocktail of mouse MAbs 16L1-312F, IID5, and 33L1-7 and 33L2-1 in combination with the secondary peroxidase-conjugated AffiniPure pAb goat anti-mouse (catalog no. 115-035-003; Jackson ImmunoResearch). Eg5 inhibitor III (dimethylenastron) was purchased from Calbiochem (catalog no. 324622).
Immunofluorescence microscopy.
HaCaT cells were grown on coverslips for 24 h to approximately 50% confluence and infected with EdU-labeled HPV16 pseudovirus at approximately 106 viral genome equivalents per coverslip. At 24 hpi, the cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, washed with phosphate-buffered saline (PBS; pH 7.5), and permeabilized with 0.5% TX-100 in PBS for 10 min, washed, and blocked with 5% normal goat serum (NGS) for 30 min; the Click-iT reaction mixture containing AF555 was used for specific detection of the EdU-labeled pseudogenome (C10338; Invitrogen) for 30 min at room temperature protected from light (65). After cells were washed, they were incubated with primary antibodies in 2.5% NGS for 1 h at 37°C in a humidified chamber. After extensive washing, the cells were incubated with AF-tagged secondary antibodies for 1 h. After another round of extensively washing the cells in PBS, the cells were mounted in ProLong Gold and SlowFade Antifade containing DAPI (4′,6′-diamidino-2-phenylindole; catalog no. P36931; Invitrogen). Single-slice images and Z-stacks were acquired with the Leica TCS SP5 spectral confocal microscope. All images from each individual experiment were acquired under the same laser power settings and enhanced uniformly in Adobe Photoshop. Quantifications are from two repeat experiments analyzing single-slice images of Z-stacks (n = 25 to 30 cells, and >800 EdU puncta were counted).
Immunofluorescence microscopy using Click-iT reaction chemistry after selective permeabilization.
HaCaT cells were grown on coverslips for 24 h to ∼50% confluence and infected with HPV16 pseudovirus encapsidating the EdU-labeled pseudogenomes. After 24 hpi, the cells were fixed with 4% PFA for 10 min at room temperature, washed with PBS, and selectively permeabilized with 0.625 μg/ml of digitonin in PBS for 10 min at room temperature. The cells were washed in PBS and blocked in 5% NGS for 20 min. The cells were treated with the first round of AF555 in Click-iT reaction buffer for 30 min at room temperature protected from light. (In a parallel experiment, the cells were incubated for 1 h with primary rabbit pAb anti-TGN46 in 2.5% NGS at 37°C, followed by extensive washing and incubation with a secondary pAb, goat anti-rabbit conjugated with AF488 in 2.5% NGS for 1 h at 37°C. This parallel experiment acts as a permeabilization control, as previously described [27].) The cells were washed again in PBS and permeabilized using 0.5% TX-100 for 10 min at room temperature. After another wash with PBS, the cells were treated with a second round with AF647 in Click-iT reaction buffer for 30 min at room temperature protected from light. The cells were washed extensively again in PBS and incubated with mouse MAb 33L1-7 or 33L2-1 in 2.5% NGS for 1 h at 37°C. After a washing step, the cells were incubated with a secondary pAb goat anti-mouse conjugated with AF488 in 2.5% NGS for 1 h at 37°C. The cells were extensively washed in PBS one last time and mounted using DAPI. Differential staining of EdU-labeled pseudogenome of selectively permeabilized HaCaT cells using two sequential Click-iT reactions was previously described in greater detail (40). Single-slice images and Z-stacks were acquired with a Leica TCS SP5 spectral confocal microscope. All images from each individual experiment were acquired under the same laser power settings and enhanced uniformly in Adobe Photoshop. Colocalization of L1, L2, and EdU puncta was quantified by selecting two to three Z-stack images per cell and counting the number of nuclear puncta as a function of single or double EdU staining and colocalization with L1 or L2 signal. The quantification data depicts the average of two independent experiments (n = 30 to 40 cells, and >800 EdU puncta were counted for each experiment).
DNase I digestion and linear gradient fractionation.
Purified HPV16 pseudoviruses were incubated 10 mM MgCl2, with or without 20 mM DTT, and with or without 2 U of DNase I for 30 min (min) at 37°C. Samples were transferred to ice, followed by incubation with 20 mM EDTA to stop the reaction. A linear gradient of 20% to 60% sucrose in 1× PBS plus BSA at 10 μg/ml was prepared in advance using a gradient mixer for a total of 12 ml. Each sample was layered on top of the gradient. The samples were centrifuged at 36,000 rpm for 3.5 h in a SW40 rotor (prechilled) at 4°C; fractions of 750 μl were collected from the top. Samples were concentrated using trichloroacetic acid (TCA) precipitation. Then, 750-μl portions of 20% TCA were added to each fraction, followed by vigorous mixing and incubation on ice for 10 min. Proteins within samples were isolated via centrifugation at 13,000 rpm for 10 min, and the supernatant was discarded. Pelleted proteins were then washed twice, first with 500 μl of 5% TCA and then with chilled (−20°C) acetone. Proteins were isolated between washes via centrifugation at 13,000 rpm (5 and 10 min, respectively). Isolated proteins were finally dried out and resuspended in 20 μl of 1× Laemmli buffer plus 10% 2-mercaptoethanol. Proteins from fractions were loaded onto two Novex WedgeWell 4 to 20% Tris-Glycine Mini Gels (XP04200BOX; Thermo Fisher Scientific) and analyzed by Western blot analysis.
Live-cell tracking.
HeLa cells were grown overnight at 37°C in a 24-well plate to 30 to 50% confluence and infected with HPV16 pseudovirus in the presence or absence of 1.5 μM Eg5 inhibitor III. Plates were incubated in the IncuCyte ZOOM system (Essen Bioscience) at 37°C. Images were obtained every hour for up to 72 h for both phase-contrast and GFP excitation. Still composite images were taken at 18 hpi.
Electron microscopy.
Purified DNA-free HPV16 VLPs were spotted onto carbon grids and negatively stained with 5% ammonium molybdate (pH 7.0) containing 1% trehalose. Samples were examined under a Zeiss EM900 transmission electron microscope at an instrumental magnification of ×30,000.
Trypsin digestion of HPV16-infected monoastral cells.
HeLa cells growing in a 6-well dish were infected with HPV16 pseudovirus in the presence of 1.5 μM Eg5 inhibitor III. At 18 hpi, the cells were washed with PBS and trypsinized for 2 min at room temperature. Monoastral cells were gently tapped off the dish and collected. Trypsin was inactivated with DMEM containing FBS and spun down. The cells were washed in PBS and resuspended in 85 μl of 10 mM EDTA in PBS. Next, 15 μl of 0.25% trypsin was added to the cells or an equivalent buffer with no trypsin as a control, followed by incubation for 1 h at 37°C. The cells were lysed by passing cells through a 1-ml syringe with a 25-gauge needle 40 times. Next, the lysates were incubated for 1 h more at 37°C. The trypsin was inactivated with trypsin inhibitor at 10 mg/ml, followed by incubation at room temperature for 15 min. Lastly, 4× Laemmli buffer containing 2-mercaptoethanol was added to the samples; the samples were then boiled for 10 min at 98°C and analyzed by Western blotting.
ACKNOWLEDGMENTS
This study was supported by R01AI081809 from the National Institute of Allergy and Infectious Diseases to M.S. and in part by grants from the National Institute of General Medical Sciences (P20GM103433). M.S. received additional support from the Feist Weiller Cancer Center. S.D. was supported by a Carroll Feist Predoctoral Fellowship.
We are grateful to John Schiller, Martin Müller, and Neil Christensen for generously providing reagents.
REFERENCES
- 1.Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, Franceschi S. 2012. Global burden of human papillomavirus and related diseases. Vaccine 30(Suppl 5):F12–F23. doi: 10.1016/j.vaccine.2012.07.055. [DOI] [PubMed] [Google Scholar]
- 2.Crow JM. 2012. HPV: the global burden. Nature 488:S2–S3. doi: 10.1038/488S2a. [DOI] [PubMed] [Google Scholar]
- 3.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. 2008. Arrangement of L2 within the papillomavirus capsid. J Virol 82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu WJ, Gissmann L, Sun XY, Kanjanahaluethai A, Muller M, Doorbar J, Zhou J. 1997. Sequence close to the N terminus of L2 protein is displayed on the surface of bovine papillomavirus type 1 virions. Virology 227:474–483. doi: 10.1006/viro.1996.8348. [DOI] [PubMed] [Google Scholar]
- 5.Knappe M, Bodevin S, Selinka HC, Spillmann D, Streeck RE, Chen XS, Lindahl U, Sapp M. 2007. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. J Biol Chem 282:27913–27922. doi: 10.1074/jbc.M705127200. [DOI] [PubMed] [Google Scholar]
- 6.Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J Virol 75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell surface glycosaminoglycans on human keratinocytes. J Biol Chem 274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- 8.Selinka HC, Florin L, Patel HD, Freitag K, Schmidtke M, Makarov VA, Sapp M. 2007. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J Virol 81:10970–10980. doi: 10.1128/JVI.00998-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Culp TD, Budgeon LR, Christensen ND. 2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 347:147–159. doi: 10.1016/j.virol.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 10.Dasgupta J, Bienkowska-Haba M, Ortega ME, Patel HD, Bodevin S, Spillmann D, Bishop B, Sapp M, Chen XS. 2011. Structural basis of oligosaccharide receptor recognition by human papillomavirus. J Biol Chem 286:2617–2624. doi: 10.1074/jbc.M110.160184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards KF, Bienkowska-Haba M, Dasgupta J, Chen XS, Sapp M. 2013. Multiple heparan sulfate binding site engagements are required for the infectious entry of human papillomavirus type 16. J Virol 87:11426–11437. doi: 10.1128/JVI.01721-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bienkowska-Haba M, Patel HD, Sapp M. 2009. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards RM, Lowy DR, Schiller JT, Day PM. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A 103:1522–1527. doi: 10.1073/pnas.0508815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Day PM, Schiller JT. 2009. The role of furin in papillomavirus infection. Future Microbiol 4:1255–1262. doi: 10.2217/fmb.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerqueira C, Samperio Ventayol P, Vogeley C, Schelhaas M. 2015. Kallikrein-8 proteolytically processes human papillomaviruses in the extracellular space to facilitate entry into host cells. J Virol 89:7038–7052. doi: 10.1128/JVI.00234-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16: involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheffer KD, Gawlitza A, Spoden GA, Zhang XA, Lambert C, Berditchevski F, Florin L. 2013. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J Virol 87:3435–3446. doi: 10.1128/JVI.02906-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abban CY, Meneses PI. 2010. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 403:1–16. doi: 10.1016/j.virol.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon CS, Kim KD, Park SN, Cheong SW. 2001. α6 Integrin is the main receptor of human papillomavirus type 16 VLP. Biochem Biophys Res Commun 283:668–673. doi: 10.1006/bbrc.2001.4838. [DOI] [PubMed] [Google Scholar]
- 21.Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NA. 1997. Identification of the α6 integrin as a candidate receptor for papillomaviruses. J Virol 71:2449–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Surviladze Z, Dziduszko A, Ozbun MA. 2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog 8:e1002519. doi: 10.1371/journal.ppat.1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Surviladze Z, Sterk RT, DeHaro SA, Ozbun MA. 2013. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J Virol 87:2508–2517. doi: 10.1128/JVI.02319-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grassel L, Fast LA, Scheffer KD, Boukhallouk F, Spoden GA, Tenzer S, Boller K, Bago R, Rajesh S, Overduin M, Berditchevski F, Florin L. 2016. The CD63-Syntenin-1 complex controls post-endocytic trafficking of oncogenic human papillomaviruses. Sci Rep 6:32337. doi: 10.1038/srep32337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol 86:9875–9887. doi: 10.1128/JVI.00980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronnimann MP, Chapman JA, Park CK, Campos SK. 2013. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J Virol 87:464–473. doi: 10.1128/JVI.01539-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiGiuseppe S, Keiffer TR, Bienkowska-Haba M, Luszczek W, Guion LG, Muller M, Sapp M. 2015. Topography of the human papillomavirus minor capsid protein L2 during vesicular trafficking of infectious entry. J Virol 89:10442–10452. doi: 10.1128/JVI.01588-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamper N, Day PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. 2006. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol 80:759–768. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popa A, Zhang W, Harrison MS, Goodner K, Kazakov T, Goodwin EC, Lipovsky A, Burd CG, DiMaio D. 2015. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog 11:e1004699. doi: 10.1371/journal.ppat.1004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergant M, Peternel S, Pim D, Broniarczyk J, Banks L. 2017. Characterizing the spatio-temporal role of sorting nexin 17 in human papillomavirus trafficking. J Gen Virol 98:715–725. doi: 10.1099/jgv.0.000734. [DOI] [PubMed] [Google Scholar]
- 31.Bergant-Marusic M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467. doi: 10.1111/j.1600-0854.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Day PM, Thompson CD, Schowalter RM, Lowy DR, Schiller JT. 2013. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol 87:3862–3870. doi: 10.1128/JVI.03222-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Florin L, Becker KA, Lambert C, Nowak T, Sapp C, Strand D, Streeck RE, Sapp M. 2006. Identification of a dynein interacting domain in the papillomavirus minor capsid protein l2. J Virol 80:6691–6696. doi: 10.1128/JVI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, DiMaio D. 2013. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A 110:7452–7457. doi: 10.1073/pnas.1302164110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pim D, Broniarczyk J, Bergant M, Playford MP, Banks L. 2015. A novel PDZ domain interaction mediates the binding between human papillomavirus 16 L2 and sorting nexin 27 and modulates virion trafficking. J Virol 89:10145–10155. doi: 10.1128/JVI.01499-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider MA, Spoden GA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol 13:32–46. doi: 10.1111/j.1462-5822.2010.01515.x. [DOI] [PubMed] [Google Scholar]
- 37.Wustenhagen E, Hampe L, Boukhallouk F, Schneider MA, Spoden GA, Negwer I, Koynov K, Kast WM, Florin L. 2016. The cytoskeletal adaptor obscurin-like 1 interacts with the human papillomavirus 16 (HPV16) capsid protein L2 and is required for HPV16 endocytosis. J Virol 90:10629–10641. doi: 10.1128/JVI.01222-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aydin I, Weber S, Snijder B, Samperio Ventayol P, Kuhbacher A, Becker M, Day PM, Schiller JT, Kann M, Pelkmans L, Helenius A, Schelhaas M. 2014. Large-scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog 10:e1004162. doi: 10.1371/journal.ppat.1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog 5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiGiuseppe S, Luszczek W, Keiffer TR, Bienkowska-Haba M, Guion LG, Sapp MJ. 2016. Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proc Natl Acad Sci U S A 113:6289–6294. doi: 10.1073/pnas.1600638113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A 101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finnen RL, Erickson KD, Chen XS, Garcea RL. 2003. Interactions between papillomavirus L1 and L2 capsid proteins. J Virol 77:4818–4826. doi: 10.1128/JVI.77.8.4818-4826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okun MM, Day PM, Greenstone HL, Booy FP, Lowy DR, Schiller JT, Roden RB. 2001. L1 interaction domains of papillomavirus l2 necessary for viral genome encapsidation. J Virol 75:4332–4342. doi: 10.1128/JVI.75.9.4332-4342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sapp M, Volpers C, Muller M, Streeck RE. 1995. Organization of the major and minor capsid proteins in human papillomavirus type-33 virus-like particles. J Gen Virol 76:2407–2412. doi: 10.1099/0022-1317-76-9-2407. [DOI] [PubMed] [Google Scholar]
- 45.Schafer F, Florin L, Sapp M. 2002. DNA binding of L1 is required for human papillomavirus morphogenesis in vivo. Virology 295:172–181. doi: 10.1006/viro.2002.1361. [DOI] [PubMed] [Google Scholar]
- 46.Li M, Cripe TP, Estes PA, Lyon MK, Rose RC, Garcea RL. 1997. Expression of the human papillomavirus type 11 L1 capsid protein in Escherichia coli: characterization of protein domains involved in DNA binding and capsid assembly. J Virol 71:2988–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sapp M, Fligge C, Petzak I, Harris JR, Streeck RE. 1998. Papillomavirus assembly requires trimerization of the major capsid protein by disulfides between two highly conserved cysteines. J Virol 72:6186–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolf M, Garcea RL, Grigorieff N, Harrison SC. 2010. Subunit interactions in bovine papillomavirus. Proc Natl Acad Sci U S A 107:6298–6303. doi: 10.1073/pnas.0914604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Unckell F, Streeck RE, Sapp M. 1997. Generation and neutralization of pseudovirions of human papillomavirus type 33. J Virol 71:2934–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2004. Efficient intracellular assembly of papillomaviral vectors. J Virol 78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol Med 119:445–462. [DOI] [PubMed] [Google Scholar]
- 52.Zhou J, Sun XY, Louis K, Frazer IH. 1994. Interaction of human papillomavirus (HPV) type 16 capsid proteins with HPV DNA requires an intact L2 N-terminal sequence. J Virol 68:619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aksoy P, Meneses PI. 2017. The Role of DCT in HPV16 Infection of HaCaTs. PLoS One 12:e0170158. doi: 10.1371/journal.pone.0170158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DiGiuseppe S, Bienkowska-Haba M, Hilbig L, Sapp M. 2014. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 458–459:93–105. doi: 10.1016/j.virol.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee H, Brendle SA, Bywaters SM, Guan J, Ashley RE, Yoder JD, Makhov AM, Conway JF, Christensen ND, Hafenstein S. 2015. A cryo-electron microscopy study identifies the complete H16.V5 epitope and reveals global conformational changes initiated by binding of the neutralizing antibody fragment. J Virol 89:1428–1438. doi: 10.1128/JVI.02898-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carter JJ, Wipf GC, Madeleine MM, Schwartz SM, Koutsky LA, Galloway DA. 2006. Identification of human papillomavirus type 16 L1 surface loops required for neutralization by human sera. J Virol 80:4664–4672. doi: 10.1128/JVI.80.10.4664-4672.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guan J, Bywaters SM, Brendle SA, Lee H, Ashley RE, Makhov AM, Conway JF, Christensen ND, Hafenstein S. 2015. Structural comparison of four different antibodies interacting with human papillomavirus 16 and mechanisms of neutralization. Virology 483:253–263. doi: 10.1016/j.virol.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sapp M, Kraus U, Volpers C, Snijders PJ, Walboomers JM, Streeck RE. 1994. Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J Gen Virol 75(Pt 12):3375–3383. doi: 10.1099/0022-1317-75-12-3375. [DOI] [PubMed] [Google Scholar]
- 59.Buck CB, Thompson CD, Pang YY, Lowy DR, Schiller JT. 2005. Maturation of papillomavirus capsids. J Virol 79:2839–2846. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aydin I, Villalonga-Planells R, Greune L, Bronnimann MP, Calton CM, Becker M, Lai K-Y, Campos SK, Schmidt MA, Schelhaas M. 2017. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog 13:e1006308. doi: 10.1371/journal.ppat.1006308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calton CM, Bronnimann MP, Manson AR, Li S, Chapman JA, Suarez-Berumen M, Williamson TR, Molugu SK, Bernal RA, Campos SK. 2017. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog 13:e1006200. doi: 10.1371/journal.ppat.1006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guan J, Bywaters SM, Brendle SA, Ashley RE, Makhov AM, Conway JF, Christensen ND, Hafenstein S. 2017. Cryoelectron microscopy maps of human papillomavirus 16 reveal L2 densities and heparin binding site. Structure 25:253–263. doi: 10.1016/j.str.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 63.Buck CB, Thompson CD. 2007. Production of papillomavirus-based gene transfer vectors. Curr Protoc Cell Biol Chapter 26:Unit 26.1. doi: 10.1002/0471143030.cb2601s37. [DOI] [PubMed] [Google Scholar]
- 64.Leder C, Kleinschmidt JA, Wiethe C, Muller M. 2001. Enhancement of capsid gene expression: preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J Virol 75:9201–9209. doi: 10.1128/JVI.75.19.9201-9209.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishii Y, Tanaka K, Kondo K, Takeuchi T, Mori S, Kanda T. 2010. Inhibition of nuclear entry of HPV16 pseudovirus-packaged DNA by an anti-HPV16 L2 neutralizing antibody. Virology 406:181–188. doi: 10.1016/j.virol.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 66.Fligge C, Schafer F, Selinka HC, Sapp C, Sapp M. 2001. DNA-induced structural changes in the papillomavirus capsid. J Virol 75:7727–7731. doi: 10.1128/JVI.75.16.7727-7731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Christensen ND, Dillner J, Eklund C, Carter JJ, Wipf GC, Reed CA, Cladel NM, Galloway DA. 1996. Surface conformational and linear epitopes on HPV-16 and HPV-18 L1 virus-like particles as defined by monoclonal antibodies. Virology 223:174–184. doi: 10.1006/viro.1996.0466. [DOI] [PubMed] [Google Scholar]
- 68.Christensen ND, Cladel NM, Reed CA, Budgeon LR, Embers ME, Skulsky DM, McClements WL, Ludmerer SW, Jansen KU. 2001. Hybrid papillomavirus L1 molecules assemble into virus-like particles that reconstitute conformational epitopes and induce neutralizing antibodies to distinct HPV types. Virology 291:324–334. doi: 10.1006/viro.2001.1220. [DOI] [PubMed] [Google Scholar]
- 69.Roth SD, Sapp M, Streeck RE, Selinka HC. 2006. Characterization of neutralizing epitopes within the major capsid protein of human papillomavirus type 33. Virol J 3:83. doi: 10.1186/1743-422X-3-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bergsdorf C, Beyer C, Umansky V, Werr M, Sapp M. 2003. Highly efficient transport of carboxyfluorescein diacetate succinimidyl ester into COS7 cells using human papillomavirus-like particles. FEBS Lett 536:120–124. doi: 10.1016/S0014-5793(03)00039-5. [DOI] [PubMed] [Google Scholar]
- 71.Rommel O, Dillner J, Fligge C, Bergsdorf C, Wang X, Selinka HC, Sapp M. 2005. Heparan sulfate proteoglycans interact exclusively with conformationally intact HPV L1 assemblies: basis for a virus-like particle ELISA. J Med Virol 75:114–121. doi: 10.1002/jmv.20245. [DOI] [PubMed] [Google Scholar]
- 72.Volpers C, Sapp M, Snijders PJF, Walboomers JMM, Streeck RE. 1995. Conformational and linear epitopes on virus-like particles of human papillomavirus type-33 identified by monoclonal antibodies to the minor capsid protein L2. J Gen Virol 76:2661–2667. doi: 10.1099/0022-1317-76-11-2661. [DOI] [PubMed] [Google Scholar]