

ABSTRACT
HIV-1 Nef clones isolated from a rare subset of HIV-1-infected elite controllers (EC), with the ability to suppress viral load to undetectable levels in the absence of antiretroviral therapy, are unable to fully downregulate CD4 from the plasma membrane of CD4+ T cells. Residual CD4 left at the plasma membrane allows Env-CD4 interaction, which leads to increased exposure of Env CD4-induced epitopes and increases susceptibility of infected cells to antibody-dependent cellular cytotoxicity (ADCC). ADCC is mediated largely by natural killer (NK) cells, which control their activation status through the cumulative signals received through activating and inhibitory receptors. Recently, the activating NKG2D receptor was demonstrated to positively influence ADCC responses. Since HIV-1 Nef has been reported to reduce the expression of NKG2D ligands, we evaluated the relative abilities of Nef from EC and progressors to downmodulate NKG2D ligands. Furthermore, we assessed the impact of EC and progressor Nef on the ADCC susceptibility of HIV-1-infected cells. We observed a significantly increased expression of NKG2D ligands on cells infected with viruses coding for Nef from EC. Importantly, NKG2D ligand expression levels correlated with enhanced susceptibility of HIV-1-infected cells to ADCC. The biological significance of this correlation was corroborated by the demonstration that antibody-mediated blockade of NKG2D significantly reduced ADCC of cells infected with viruses carrying Nef from EC. These results suggest the involvement of NKG2D-NKG2D ligand interactions in the enhanced susceptibility of EC HIV-1-infected cells to ADCC responses.
IMPORTANCE Attenuated Nef functions have been reported in HIV-1 isolated from EC. The inability of elite controller Nef to fully remove CD4 from the surface of infected cells enhanced their susceptibility to elimination by ADCC. We now show that downregulation of NKG2D ligands by HIV-1 Nef from EC is inefficient and leaves infected cells susceptible to ADCC. These data suggest a critical role for NKG2D ligands in anti-HIV-1 ADCC responses.
KEYWORDS: HIV-1, Nef, elite controllers, ADCC, NKG2D, gp120, CD4
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) has developed multiple mechanisms to evade the immune system and establish a chronic infection. Several of these strategies are mediated by viral accessory proteins, including Vif, Vpr, Vpu, and Nef, which together ensure viral persistence and survival. The negative regulatory factor (Nef) is a 27- to 35-kDa accessory protein that is highly expressed in the early phase of HIV-1 infection (1, 2). It has been shown that infection with HIV-1 or simian immunodeficiency virus (SIV) strains unable to code for functional Nef proteins leads to a slow or nonprogressive disease (3, 4). The positive effects of Nef on viral pathogenicity and persistence are based largely on its ability to decrease the surface expression levels of important cellular molecules present on the surface of infected cells, including CD4, CD28, NKG2D ligands, and HLA-I molecules (5–8). The ability of Nef to decrease surface expression of certain cell surface proteins, such as CD4 and HLA-I, is impaired in Nef clones isolated from elite controllers (EC) (9, 10), a rare group of infected individuals with the ability to suppress plasma viremia to <50 RNA copies/ml in the absence of antiretroviral therapy. The impaired ability of Nef proteins isolated from EC to decrease CD4 expression is associated with increased susceptibility of HIV-1-infected cells to antibody-dependent cellular cytotoxicity (ADCC) (11).
ADCC efficacy relies on several parameters imposed by the characteristics of the target and effector cells involved, as well as the features of the antibodies. Natural killer (NK) cells, monocytes, and neutrophils serve as effector cells for anti-HIV-1 ADCC (12). The ADCC function of NK cells is a topic of much research interest due to the extensiveness of NK cell regulation. Indeed, NK cell regulation occurs on two levels (reviewed in reference 13). Initially, NK cells undergo a process termed “NK cell education,” where NK cells that carry inhibitory receptors capable of interacting with self-major histocompatibility complex class I (MHC-I or HLA-I) ligands are conferred functional potential. Alternatively, NK cells that do not carry self-HLA-I-binding inhibitory NK cell receptors remain hypofunctional. The second level of NK cell regulation occurs when NK cells encounter a putative target cell. At this point the outcome of the effector-target cell interaction is determined by cumulative signal received through the plethora of inhibitory and activating receptors on the NK cell surface. It is through these cosignaling molecules that the characteristics of the target cell influence the capacity of the effector cell to mediate ADCC. If the target cell expresses sufficient HLA-I that can bind to inhibitory NK cell receptors, such as killer immunoglobulin receptors of the NKG2A receptor, to create a cumulative inhibitory signal, the target will be spared from cytolysis. Alternatively, if the target cell expresses a sufficiently high enough density of antigen targeted by ADCC antibodies that are recognized by the activating receptor FcγRIIIa/CD16, the target will likely induce a cumulative activating signal and be lysed. HIV-1 utilizes this level of NK cell regulation to evade ADCC. Indeed, HIV-1 Nef downregulates HLA-I molecules important for cytotoxic T-lymphocyte recognition of infected cells, including HLA-A and HLA-B, but spares HLA-C and HLA-E, which interact with inhibitory NK cell receptors and can inhibit antiviral ADCC (14, 15). Besides characteristics of effector and target cells, the features of the antibodies involved are important determinants of ADCC. Indeed, glycosylation features of the antibody constant region (Fc) influence the interaction of the antibody with Fc receptors and the degree of ADCC observed (16). Additionally, access of the antibody paratope to the antigenic epitope is essential for efficient ADCC. In the case of HIV-1, it was recently shown that ADCC-mediating antibodies present in HIV sera preferentially recognize Env in its CD4-bound conformation (17). However, to limit the exposure of CD4-bound Env on the surface of infected cells, HIV-1 evolved sophisticated mechanisms to efficiently internalize Env (18), to counteract the host restriction factor BST-2 with the viral Vpu protein (19–21), and to downregulate CD4 by Nef and Vpu (17, 21).
In addition to FcγRIIIa/CD16, there are several other activating and coactivating receptors capable of activating NK cell effector functions. Ligands for these receptors are generally induced as a result of activation, cellular stress, transformation, or viral infection. Accordingly, previous studies demonstrated that ligands for the activating NK cell receptors, NKG2D, are induced during HIV-1 infection (22–25). Importantly, cell surface expression of these ligands on HIV-1-infected cells was found to trigger autologous NK cells to lyse infected cells (22–26). While NK cell-mediated killing of HIV-1-infected cells occurs mainly through NKG2D, this response appears to be optimal when the activating receptor DNAM-1 and the coactivating receptor NTB-A are also simultaneously triggered (27–29).
In addition to its capacity to influence NK cell functionality through decreasing surface expression of select HLA-I and decreasing the availability of cell surface CD4 required for CD4-induced (CD4i) ADCC-mediating epitopes (11), Nef has been demonstrated to interfere with NK cell activation by limiting the expression of NKG2D ligands on the surface of infected cells (8). This observation is interesting in the context of additional research suggesting that NKG2D contributes to HIV-1 disease status. Indeed, a previous study linked low viral set point and low viral loads in long-term nonprogression patients (LTNPs) with the expression level of NKG2D (30). The role of Nef in decreasing NKG2D ligand expression on infected cells is also interesting in the context of recent research demonstrating that NKG2D acts as a coreceptor for NK-mediated ADCC against HIV-1-infected cells (31). Since Nef is known to diminish the expression of NKG2D ligands, including MICA, ULBP1, and ULBP2, at the cell surface (8), here we investigated whether enhanced susceptibility of HIV-1-infected cells expressing EC Nef to ADCC was linked to modulation of NKG2D ligands.
RESULTS
CD4 and NKG2D ligands are inefficiently downregulated by Nef proteins from elite controllers.
The interaction of Env and CD4 induces conformational changes in Env that result in the exposure of CD4i epitopes, including those recognized by anti-cluster A antibodies, known to mediate potent ADCC responses (17, 21, 32–34). As previously reported, when primary CD4+ T cells were infected with HIV-1NL4.3 infectious viral particles coding for Nef clones from ECs, we observed an impaired CD4 downregulation compared to cells infected with viruses coding for Nef clones isolated from chronic progressors (CP) (Fig. 1A and B) (9, 11). Of note, no mutations in motif L166-L168 involved in CD4 downregulation (7) were found in Nef clones from ECs (not shown). Suboptimal CD4 downregulation translated into a significant exposure of anti-cluster A epitopes, such as the one recognized by the ADCC-mediating A32 antibody (Fig. 1C and D) and those recognized by antibodies within HIV+ sera (Fig. 1E and F) despite similar levels of Env being expressed at the cell surface (Fig. 1G and H). Enhanced recognition of infected cells by A32 and HIV+ serum antibodies was previously reported to result in enhanced susceptibility of infected cells to ADCC (11). To mediate ADCC, however, effector cells, such as NK cells, must be activated. It is well known that NK cell effector functions are modulated by a tight balance between signals delivered through inhibitory (KIR and CD94/NKG2A), activating (CD16, NKG2D, and DNAM-1), or coactivating (NTB-A and 2B4) receptors that either suppress or enhance NK cell activity (35). Interestingly, Nef decreases the expression of NKG2D ligands (MICA, ULBP1, and ULBP2) when expressed alone in Jurkat cells or in the context of HIV-1-infected Jurkat or primary CD4+ T cells (8, 36), thus preventing their interaction with the NK cell-activating NKG2D receptor. Since previous work indicated that Nef proteins from EC are impaired in several functions (9), we evaluated if the enhanced susceptibility of HIV-1-infected cells, infected with viruses coding for Nef proteins from EC, to ADCC could be linked to an incomplete downregulation of NKG2D ligands. To evaluate this, primary CD4+ T cells were isolated from HIV-uninfected individuals and infected with pNL4.3 viruses encoding Nef from EC or CP. Primary CD4+ T cells were also infected with wild-type NefSF2 (wt) or Nef-deleted (N−) viruses as positive and negative controls for NKG2D ligand downregulation. Cultures containing infected cells were then stained with a recombinant human NKG2D-Fc chimera which recognizes several NKG2D ligands (23–25, 31) or a matched isotype control, and infected cells within these cultures were identified by intracellular p24 staining. As previously reported (22), HIV-1 infection enhanced NKG2D-Fc detection compared to mock-infected cells (Fig. 2A and B). This enhancement, however, was significantly higher when cells were infected with the nef-defective virus compared to its wt counterpart. Interestingly, we observed that cells infected with viruses coding for Nef from EC expressed higher levels of NKG2D ligands, as demonstrated by a significant increase in NKG2D-Fc binding compared to cells infected with viruses coding for Nef isolated from CP (Fig. 2C). Despite the differential expression of NKG2D ligands at the surface of cells expressing Nef from CP versus EC, we failed to identify a particular motif linked to Nef-mediated NKG2D ligand downregulation (not shown).
FIG 1.

Attenuated CD4 downregulation by Nef alleles from elite controllers enhances exposure of ADCC-mediating epitopes. Primary CD4+ T cells from healthy donors were infected with a panel of pNL4.3-based viruses: wild type (wt; coding for NefSF2), lacking Nef (N−), or encoding Nef from 18 and 19 randomly selected clones from ECs or CPs, respectively. Infected primary CD4 T cells were stained at 48 h postinfection with an anti-CD4 (OKT4) (A and B) or A32 (C and D) antibody, with HIV+ sera from 7 different donors (shown in different colors) (E and F), or with the conformation-independent 2G12 antibody (G and H). On the left are histograms depicting representative staining obtained with wt versus Nef-infected cells. On the right, the median fluorescence intensities (MFI) obtained for multiple staining using cells infected with viruses coding for Nef from ECs (red) or CPs (blue) are shown. Error bars indicate means ± standard errors of the means (SEM). Statistical significance was evaluated using unpaired t test. *, P < 0.05; **, P < 0.01; ns, not significant.
FIG 2.

Attenuated downregulation of NKG2D ligands by Nef alleles from elite controllers. Primary CD4+ T cells from healthy donors were infected with a panel of pNL4.3-based viruses: wild type (wt; coding for NefSF2), lacking Nef (N−), or encoding Nef from 18 and 19 randomly selected clones from ECs or CPs, respectively. Infected primary CD4 T cells were stained at 48 h postinfection with 5 μg/ml of a recombinant NKG2D-Fc chimera or matched IgG Fc molecules and then fluorescently labeled with an Alexa Fluor 647-conjugated anti-human IgG secondary Ab. (A and B) Histogram (A) and graph (B) depicting representative staining of infected (p24+) cells with mock (gray), wt (blue), and N− (in red) virus. (C) Recognition of infected (p24+) cells by viruses coding for Nef from ECs (red) or CPs (blue) with NKG2D-Fc. Data shown are the results of four different experiments, and error bars depict the SEM. Statistical significance was tested using paired one-way analyses of variance (ANOVAs) and unpaired t test (*, P < 0.05; **, P < 0.01; ****, P < 0.0001).
Attenuated downregulation of NKG2D ligands by Nef proteins from elite controllers enhances susceptibility of infected cells to ADCC.
Using a previously described fluorescence-activated cell sorter (FACS)-based ADCC assay (21, 37), we next addressed whether the increased expression of NKG2D ligands on cells infected with viruses coding for Nef alleles from EC enhanced susceptibility to ADCC. Primary CD4+ T cells infected with wild-type and Nef-defective viruses were used as positive and negative controls, respectively, for NKG2D ligand downregulation. As previously reported (11, 17, 21, 33), wild-type-infected cells were not sensitive to ADCC mediated by A32 (Fig. 3A) or antibodies within HIV+ sera (Fig. 3B) using autologous peripheral blood mononuclear cells (PBMCs) as effector cells. Supportive of the role for Nef in viral evasion of ADCC (11, 17, 21, 33), cells infected with Nef-deleted virus were more susceptible to ADCC mediated by either A32 (Fig. 3A) or HIV+ sera from seven different HIV-1-infected individuals (Fig. 3B). Intriguingly, this enhanced susceptibility to ADCC was observed despite similar levels of Env expression, as determined by detection of binding of the conformationally independent 2G12 antibody, on cells infected with viruses encoding wild-type Nef or having Nef deleted (Fig. 1G and H). Corroborating a role for NKG2D ligands in ADCC responses against HIV-1-infected cells (31), addition of a blocking anti-NKG2D antibody significantly decreased, but did not abrogate, the susceptibility of Nef-deleted infected cells to ADCC mediated by A32 and antibodies within HIV-1+ sera (Fig. 3A and B).
FIG 3.

Attenuated downregulation of NKG2D ligands by Nef alleles from elite controllers enhances the susceptibility of infected cells to ADCC mediated by A32 and HIV+ sera. Primary CD4+ T cells from 5 healthy individuals infected with a panel of pNL4.3-based viruses, wild type (wt; coding for NefSF2), lacking Nef (N−), or encoding Nef from at least 18 randomly selected clones from ECs or CPs, were used at 48 h postinfection as target cells using a previously reported FACS-based ADCC assay (11, 21) to determine their susceptibility to ADCC by autologous PBMCs. ADCC mediated by A32 (A and C) or HIV+ sera from two to seven different donors (B and D) is shown. Data shown are the results from at least six different experiments, with means ± SEM. Statistical significance was tested using a paired or unpaired t test (*, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001; ns, not significant).
Given the capacity of Nef to decrease surface expression of NKG2D ligands and our data suggesting Nef from EC are inefficient at decreasing NKG2D ligand expression, we next evaluated the role played by NKG2D ligands in ADCC responses against primary CD4+ T cells infected with viruses coding for Nef proteins from EC or CP. As previously reported (11), we observed enhanced susceptibility of cells infected with viruses coding for Nef from EC versus CP to ADCC mediated by A32 or antibodies within HIV-positive sera (Fig. 3C and D). Suggestive of a role for NKG2D in this enhanced susceptibility to ADCC, addition of an anti-NKG2D blocking antibody, but not of an isotype control, significantly decreased ADCC of cells infected with viruses encoding Nef from EC but not from CP (Fig. 3C and D). In further agreement with a role for NKG2D in anti-HIV-1 ADCC, ADCC mediated by A32 or antibodies within HIV-1-infected sera correlated with not only cell surface levels of CD4 (Fig. 4A) but also NKG2D-Fc binding to infected cells (Fig. 4B). Cumulatively, this suggests that in addition to the exposure of ADCC-mediating epitopes induced by the presence of CD4 at the cell surface, the accumulation of NKG2D activating ligands might promote NK cell cytotoxicity.
FIG 4.
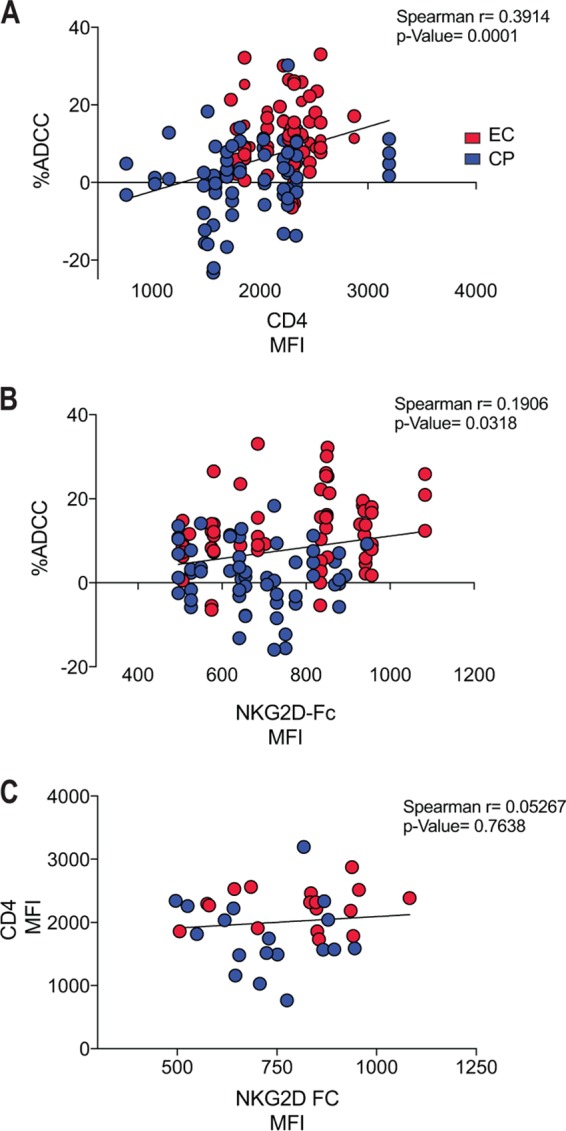
Enhanced levels of CD4 and NKG2D ligands at the surface of HIV-1-infected cells correlate with enhanced ADCC. The presence of CD4 and NKG2D ligands at the surface of HIV-1-infected cells enhances the sensitivity of infected cells to ADCC mediated by HIV+ sera and A32 monoclonal antibody. CD4 expression (A) and the presence of NKG2D ligands (B) on the surface of cells infected with viruses encoding Nef from at least 18 randomly selected clones from ECs (in red) or CPs (in blue) correlated positively with ADCC killing mediated by HIV+ sera and A32 antibody. (C) No statistically significant correlation was observed between cell surface levels of CD4 and NKG2D ligand expression. Statistical analysis was tested utilizing a Spearman rank correlation.
DISCUSSION
HIV-1 has evolved several mechanisms to prevent exposing the CD4-bound conformation of Env at the cell surface. These mechanisms are potentially important, as ADCC-mediating antibodies present in HIV+ sera preferentially target the CD4-bound conformation of Env (11, 17, 33). Accordingly, the HIV-1 Nef accessory protein protects HIV-1-infected cells from ADCC by decreasing cell surface levels of CD4 (11, 17, 21, 33), which otherwise engages with Env and induces the CD4-bound conformation to expose CD4i epitopes. These epitopes are recognized by well-established CD4i ADCC-mediating antibodies, such as A32 (21), or antibodies within sera from HIV-1-infected individuals (11, 17, 21, 33). Here, we confirmed that infection of cells with viruses encoding Nef from EC inefficiently decreased cell surface CD4 expression and resulted in the exposure of viral epitopes recognized by ADCC-mediating antibodies, such as the A32 antibody and antibodies within HIV+ sera. We now show that in addition to an impaired capacity to decrease cell surface CD4 expression, Nef from EC also inefficiently decreases the expression of NKG2D ligands on HIV-1-infected cells. This observation is highly important in the context of recently published data demonstrating NKG2D acts as a coreceptor for anti-HIV-1 ADCC (31).
While we and others (8, 31, 36) observed Nef-mediated interference with NKG2D ligand expression, another report failed to see any effect of Nef on NKG2D ligands (23). The reasons for this are unclear but could be due to the different viral constructs used in the different studies. Indeed, we used fully replicative viruses, while in the Ward et al. study, Env-defective and therefore nonreplicative viruses were used.
It has been reported that Nef-mediated NKG2D ligand downregulation occurs through the utilization of domains that differ from those required for CD4 downregulation (8). Accordingly, we found that the capacity of Nef to decrease cell surface CD4 expression did not correlate with its capacity to decrease cell surface NKG2D ligand expression (Fig. 4C). This could represent an overall impaired function of Nef clones from EC, as illustrated by their decreased ability to downregulate CD4, HLA-I, and CCR5 and their reduced capacity to enhance HIV-1 infectivity and replication (9, 10).
While the presented data are highly relevant for understanding viral mechanisms of susceptibility and evasion of ADCC, they also provide clues to how NK cell-mediated anti-HIV-1 ADCC is regulated on the level of effector-target cell interactions. The mechanism through which NKG2D is contributing to anti-HIV-1 ADCC remains open to debate. Assuming, however, that NKG2D contributes to anti-HIV-1 ADCC through the propagation of activating signals, this observation provides further confirmation of the importance of cumulative activating/inhibitory signals in the functionality of NK cells (35). Indeed, this observation might direct us toward a better understanding of why cells infected with viruses coding for wild-type Nef are poorly susceptible to ADCC. While it is evident that cells infected with wild-type virus inadequately bind antibodies directed to CD4-induced epitopes, these cells do bind low levels of antibodies within the sera of HIV-1-infected donors. This low level of antibody binding to cells infected with viruses encoding wild-type Nef might be sufficient to trigger signals through FcγRIIIa/CD16, but little to no ADCC is observed against these target cells. This raises the possibility that cells infected with viruses encoding wild-type Nef are protected from NK cell-mediated ADCC by their incapacity to ligate sufficient activating receptors and capacity to ligate sufficient inhibitory NK cell receptors to generate a cumulative inhibitory NK cell signal. Indeed, previous research has demonstrated that HIV-1-infected targets express sufficient HLA-C and HLA-E to inhibit anti-HIV-1 ADCC (14, 15) despite the ability of primary Vpu to downregulate HLA-C to various extents (38). Given that cumulative signals are important determinants of ADCC susceptibility, future research should assess if target cells infected with viruses coding for wild-type Nef become susceptible to ADCC after blockade of inhibitory NK cell receptors. Such studies could generate potential therapeutic avenues for eliminating HIV-1-infected cells important for curative efforts to eliminate reactivated HIV-1 (39). Finally, whether ligands for other activating or coactivating receptors expressed on HIV-1-infected cells also participate in this cumulative activating signal and modulate ADCC susceptibility remain to be determined.
Altogether, our results suggest a model (Fig. 5) where the activities of Nef from CP, including efficient downregulation of CD4 and NKG2D ligands, protect infected cells from ADCC by diminishing the propensity of Env to sample the CD4-bound conformation and also by decreasing the amount of NK cell-activating ligands. On the other hand, infected cells expressing Nef from EC become more susceptible to ADCC responses by exposing Env in its CD4-bound conformation, as well as their inability to remove NK cell-activating ligands from the cell surface. The accumulation of NKG2D ligands at the surface of infected cells synergizes with CD16 to elevate ADCC killing. Among different mechanisms of control, including the presence of protective HLA alleles (40), we believe that enhanced susceptibility of cells infected with HIV-1 from EC to anti-HIV-1 ADCC might contribute to the durable suppression of viral replication and low plasma viremia in this rare subset of infected individuals.
FIG 5.

Nef-mediated CD4 and NKG2D ligand downregulation modulates susceptibility of HIV-1-infected cells to ADCC. Nef clones isolated from CPs provide protection to infected cells from ADCC by downregulating CD4 and NKG2D ligands from the surface of infected cells (left panel). Impaired abilities of Nef clones from ECs to reduce surface levels of CD4 and NKG2D ligands expose Env CD4i epitopes targeted by ADCC-mediating Abs and contribute to strengthening the activation of NK cells (right panel).
MATERIALS AND METHODS
Cell lines and isolation of primary cells.
The 293T human embryonic kidney cell line was obtained from ATCC. PBMCs from uninfected healthy donors (n = 5) were obtained from leukapheresis under research regulations approved by CRCHUM; written informed consent was obtained from each individual. Cells were grown as previously described (11, 21). CD4 T lymphocytes were purified from rested PBMCs by negative selection and activated as previously described (32).
Study participants and nef cloning.
Nef clones were obtained from plasma of 47 untreated EC (plasma viral load [pVL] of <50 RNA copies/ml plasma) and 48 untreated CP (median pVL of 80,500; interquartile range [IQR], 25,121 to 221,250) and were previously described (9, 10, 40–43). HIV RNA was extracted and amplified using nested reverse transcription-PCR (RT-PCR) as described previously (43, 44). At least three Nef clones were sequenced per patient, and a single clone having an intact Nef reading frame that closely resembled the sequence of the original bulk plasma RNA was chosen (9). Nef clones were transferred into pNL4.3 lacking Nef (N−) plasmid and confirmed by DNA sequencing, as described previously (45). Recombinant viruses harboring nef from HIV-1SF2 (wt NefSF2) and lacking nef (N−) were used as positive and negative controls, respectively. All EC and CP were HIV-1 subtype B infected, were from the Boston area, and were comparable with respect to ethnicity and diagnosis date of HIV (EC, 1985 to 2006; CP, 1981 to 2003). The study was approved by the institutional review board of Massachusetts General Hospital, Boston, MA; all participants provided written informed consent.
Viral production and infections.
Vesicular stomatitis virus G (VSVG)-pseudotyped pNL4.3 encoding NefSF2, Nef-deleted clones, and Nef clones from EC (18 clones) or CP (19 clones) viruses were produced in 293T cells and titrated as previously described (21). A random number generator (GraphPad QuickCalcs) was used to randomly select EC and CP nef proviruses. Viruses were then used to infect approximately 20% to 30% of primary CD4 T cells from healthy donors by spin infection at 800 × g for 1 h in 96-well plates at 25°C. The same viral stock was used to perform all of the experiments reported in the manuscript.
Antibodies and sera.
The A32 anti-gp120 cluster A antibody was previously reported (21, 34, 46). The monoclonal antibody anti-CD4 OKT4 (BioLegend) binds to the D3 domain of CD4 and was used to measure cell surface levels of CD4, as described previously (21). The soluble NKG2D-IgG Fc fusion proteins (R&D Systems) were used to detect NKG2D ligands (ULBPs, MICA, and MICB), while matched IgG Fc fusion molecules (R&D systems) were used as a control, as previously reported (22). Secondary goat anti-mouse and anti-human antibodies coupled to Alexa Fluor 647 (Invitrogen) were used in flow cytometry experiments.
HIV+ sera were obtained from the Montreal Primary HIV Infection Cohort (47, 48) and the Canadian Cohort of HIV Infected Slow Progressors (40, 49, 50). Research adhered to the ethical guidelines of CRCHUM, and informed consent was obtained from each volunteer. Sera were collected during Ficoll isolation of PBMCs and stored at −80°C. Serum aliquots were heat inactivated for 30 min at 56°C and stored at 4°C until they were used in subsequent experiments, as previously reported (17, 21). A random number generator (GraphPad QuickCalcs) was used to randomly select a number of sera from each cohort.
Flow cytometry: cell surface staining and ADCC responses.
Cell surface staining was performed as previously described (17, 21). Briefly, binding of HIV-1-infected cells by plasma antibodies (1:1,000 dilution) or relevant monoclonal antibodies (1 μg/ml) was performed 48 h after infection. The soluble NKG2D-IgG Fc fusion proteins were used at 5 μg/ml, and binding was detected with an Alexa Fluor 647-conjugated goat anti-human IgG antibody. After surface staining, infected cells were permeabilized using the Cytofix/Cytoperm fixation/permeabilization kit (BD Biosciences, Mississauga, ON, Canada) to detect infected cells (p24+ cells) with the fluorescent anti-p24 monoclonal antibody (phycoerythrin-anti-p24, clone KC57; Beckman Coulter/Immunotech, Hialeah, FL) (1:100 final concentration), as previously described (11, 32). The percentage of infected cells was determined by gating on the living cell population based on the AquaVivid viability dye (Invitrogen). Samples were acquired on an LSRII cytometer (BD Biosciences, Mississauga, ON, Canada), and data analyses were performed using FlowJo vX.0.7 (Tree Star, Ashland, OR, USA).
Measurement of serum or A32-mediated ADCC responses was performed using a previously described FACS-based ADCC assay (11, 17, 21, 32, 33, 37, 51–55). Briefly, infected primary CD4+ T cells were stained with viability (AquaVivid; Invitrogen) and cellular (cell proliferation dye eFluor670; eBioscience) markers and used as target cells. Uninfected, autologous PBMC effector cells from healthy donors, stained with another cellular marker (cell proliferation dye eFluor450; eBioscience), were then mixed at an effector/target (E/T) ratio of 10:1 in 96-well V-bottom plates (Corning) for 5 to 6 h with a 1:1,000 final concentration of plasma or 5 μg/ml of the A32 monoclonal antibody in the presence of 5 μg/ml of purified anti-human CD314 (NKG2D) (139-NK-50; R&D Systems) or matched IgG isotype control to appropriate wells. After the 5- to 6-h coincubation period, cells were fixed in a 2% phosphate-buffered saline (PBS)-formaldehyde solution. Samples were acquired on an LSRII cytometer (BD Biosciences). Infected cells were identified by intracellular p24 staining as described above. The percentage of ADCC was calculated with the following formula: (percentage of p24+ cells in targets plus effectors) − (percentage of p24+ cells in targets plus effectors plus serum or A32)/(percentage of p24+ cells in targets), as previously reported (37). Of note, in some samples some negative cytotoxicity was observed. This is due to the killing of gp120-coated uninfected bystander CD4+ T cells, which are present in the culture of HIV-1-infected cells and affects ADCC calculation and was previously reported in detail elsewhere (54).
Statistical analyses.
Statistics were analyzed using GraphPad Prism version 6.01 (GraphPad, San Diego, CA, USA). P values of <0.05 were considered significant: *, P < 0.05; **, P < 0.01, ***, P < 0.001; ****, P < 0.0001.
ACKNOWLEDGMENTS
We thank Florencia Pereyra, Bruce D. Walker, and all investigators who have contributed to The International HIV Controllers Study (www.hivcontrollers.org), funded by the Mark and Lisa Schwarz Foundation, the Bill and Melinda Gates Foundation, and the AIDS Healthcare Foundation; the CRCHUM Flow Cytometry Platform for technical assistance as well as Mario Legault for cohort coordination; and all subjects for their participation and collaboration.
This work was supported by a Canada Foundation for Innovation Program Leader grant, CIHR foundation grant 352417, and amfAR innovation grant 109343-59-RGRL, with support from FAIR to A.F. and by the FRQS AIDS and Infectious Diseases Network. A.F. and M.A.B. are supported by the Canada Research Chairs program. J.R. is the recipient of CIHR Fellowship Award 135349. N.A. is the recipient of a King Abdullah scholarship for higher education from the Saudi Government. M.S.P. is a recipient of a CIHR Fellowship Award. D.E.K. is supported by a Research Scholar Career Award of the Quebec Health Research Fund (FRQS). This study was also supported by NIH AI100645 and AI100663 Center for HIV/AIDS Vaccine Immunology and Immunogen Design (CHAVI-ID).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We have no conflicts of interest to report.
REFERENCES
- 1.Guy B, Kieny MP, Riviere Y, Le Peuch C, Dott K, Girard M, Montagnier L, Lecocq JP. 1987. HIV F/3′ orf encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature 330:266–269. doi: 10.1038/330266a0. [DOI] [PubMed] [Google Scholar]
- 2.Foster JL, Denial SJ, Temple BR, Garcia JV. 2011. Mechanisms of HIV-1 Nef function and intracellular signaling. J Neuroimmune Pharmacol 6:230–246. doi: 10.1007/s11481-011-9262-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J. 1995. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270:988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 4.Zaunders JJ, Geczy AF, Dyer WB, McIntyre LB, Cooley MA, Ashton LJ, Raynes-Greenow CH, Learmont J, Cooper DA, Sullivan JS. 1999. Effect of long-term infection with nef-defective attenuated HIV type 1 on CD4+ and CD8+ T lymphocytes: increased CD45RO+CD4+ T lymphocytes and limited activation of CD8+ T lymphocytes. AIDS Res Hum Retrovir 15:1519–1527. doi: 10.1089/088922299309801. [DOI] [PubMed] [Google Scholar]
- 5.Miller MD, Feinberg MB, Greene WC. 1994. The HIV-1 nef gene acts as a positive viral infectivity factor. Trends Microbiol 2:294–298. doi: 10.1016/0966-842X(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 6.Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391:397–401. doi: 10.1038/34929. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz O, Dautry-Varsat A, Goud B, Marechal V, Subtil A, Heard JM, Danos O. 1995. Human immunodeficiency virus type 1 Nef induces accumulation of CD4 in early endosomes. J Virol 69:528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cerboni C, Neri F, Casartelli N, Zingoni A, Cosman D, Rossi P, Santoni A, Doria M. 2007. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J Gen Virol 88:242–250. doi: 10.1099/vir.0.82125-0. [DOI] [PubMed] [Google Scholar]
- 9.Mwimanzi P, Markle TJ, Martin E, Ogata Y, Kuang XT, Tokunaga M, Mahiti M, Pereyra F, Miura T, Walker BD, Brumme ZL, Brockman MA, Ueno T. 2013. Attenuation of multiple Nef functions in HIV-1 elite controllers. Retrovirology 10:1. doi: 10.1186/1742-4690-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toyoda M, Ogata Y, Mahiti M, Maeda Y, Kuang XT, Miura T, Jessen H, Walker BD, Brockman MA, Brumme ZL, Ueno T. 2015. Differential ability of primary HIV-1 Nef isolates to down-regulate HIV-1 entry receptors. J Virol 89:9639–9652. doi: 10.1128/JVI.01548-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alsahafi N, Ding S, Richard J, Markle T, Brassard N, Walker B, Lewis GK, Kaufmann DE, Brockman MA, Finzi A. 2015. Nef Proteins from HIV-1 elite controllers are inefficient at preventing antibody-dependent cellular cytotoxicity. J Virol 90:2993–3002. doi: 10.1128/JVI.02973-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smalls-Mantey A, Connors M, Sattentau QJ. 2013. Comparative efficiency of HIV-1-infected T cell killing by NK cells, monocytes and neutrophils. PLoS One 8:e74858. doi: 10.1371/journal.pone.0074858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wren LH, Stratov I, Kent SJ, Parsons MS. 2013. Obstacles to ideal anti-HIV antibody-dependent cellular cytotoxicity responses. Vaccine 31:5506–5517. doi: 10.1016/j.vaccine.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 14.Ward JP, Bonaparte MI, Barker E. 2004. HLA-C and HLA-E reduce antibody-dependent natural killer cell-mediated cytotoxicity of HIV-infected primary T cell blasts. AIDS 18:1769–1779. doi: 10.1097/00002030-200409030-00005. [DOI] [PubMed] [Google Scholar]
- 15.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671. doi: 10.1016/S1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 16.Gunn BM, Alter G. 2016. Modulating antibody functionality in infectious disease and vaccination. Trends Mol Med doi: 10.1016/j.molmed.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veillette M, Coutu M, Richard J, Batraville LA, Dagher O, Bernard N, Tremblay C, Kaufmann DE, Roger M, Finzi A. 2015. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J Virol 89:545–551. doi: 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Bredow B, Arias JF, Heyer LN, Gardner MR, Farzan M, Rakasz EG, Evans DT. 2015. Envelope glycoprotein internalization protects human and simian immunodeficiency virus infected cells from antibody-dependent cell-mediated cytotoxicity. J Virol 89:10648–10655. doi: 10.1128/JVI.01911-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT. 2014. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 111:6425–6430. doi: 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarez RA, Hamlin RE, Monroe A, Moldt B, Hotta MT, Rodriguez Caprio G, Fierer DS, Simon V, Chen BK. 2014. HIV-1 Vpu antagonism of tetherin inhibits antibody-dependent cellular cytotoxic responses by natural killer cells. J Virol 88:6031–6046. doi: 10.1128/JVI.00449-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richard J, Sindhu S, Pham TN, Belzile JP, Cohen EA. 2010. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 115:1354–1363. doi: 10.1182/blood-2009-08-237370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward J, Davis Z, DeHart J, Zimmerman E, Bosque A, Brunetta E, Mavilio D, Planelles V, Barker E. 2009. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog 5:e1000613. doi: 10.1371/journal.ppat.1000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward J, Bonaparte M, Sacks J, Guterman J, Fogli M, Mavilio D, Barker E. 2007. HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 110:1207–1214. doi: 10.1182/blood-2006-06-028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogli M, Mavilio D, Brunetta E, Varchetta S, Ata K, Roby G, Kovacs C, Follmann D, Pende D, Ward J, Barker E, Marcenaro E, Moretta A, Fauci AS. 2008. Lysis of endogenously infected CD4+ T cell blasts by rIL-2 activated autologous natural killer cells from HIV-infected viremic individuals. PLoS Pathog 4:e1000101. doi: 10.1371/journal.ppat.1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomescu C, Mavilio D, Montaner LJ. 2015. Lysis of HIV-1-infected autologous CD4+ primary T cells by interferon-alpha-activated NK cells requires NKp46 and NKG2D. AIDS 29:1767–1773. doi: 10.1097/QAD.0000000000000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell EM, Planelles V, Barker E. 2010. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 8:397–409. doi: 10.1016/j.chom.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matusali G, Potesta M, Santoni A, Cerboni C, Doria M. 2012. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J Virol 86:4496–4504. doi: 10.1128/JVI.05788-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis ZB, Sowrirajan B, Cogswell A, Ward JP, Planelles V, Barker E. 2016. CD155 on HIV-infected cells is not modulated by HIV-1 Vpu and Nef but synergizes with NKG2D ligands to trigger NK cell lysis of autologous primary HIV-infected cells. AIDS Res Hum Retrovir 33:93–100. doi: 10.1089/aid.2015.0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni AG, Paranjape RS, Thakar MR. 2014. Higher expression of activating receptors on cytotoxic NK cells is associated with early control on HIV-1C multiplication. Front Immunol 5:222. doi: 10.3389/fimmu.2014.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons MS, Richard J, Lee WS, Vanderven H, Grant MD, Finzi A, Kent SJ. 2016. NKG2D acts as a co-receptor for natural killer cell-mediated anti-HIV-1 antibody-dependent cellular cytotoxicity. AIDS Res Hum Retrovir 32:1089–1096. doi: 10.1089/aid.2016.0099. [DOI] [PubMed] [Google Scholar]
- 32.Richard J, Veillette M, Brassard N, Iyer SS, Roger M, Martin L, Pazgier M, Schon A, Freire E, Routy JP, Smith AB III, Park J, Jones DM, Courter JR, Melillo BN, Kaufmann DE, Hahn BH, Permar SR, Haynes BF, Madani N, Sodroski JG, Finzi A. 2015. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proc Natl Acad Sci U S A 112:E2687–E2694. doi: 10.1073/pnas.1506755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding S, Veillette M, Coutu M, Prevost J, Scharf L, Bjorkman PJ, Ferrari G, Robinson JE, Sturzel C, Hahn BH, Sauter D, Kirchhoff F, Lewis GK, Pazgier M, Finzi A. 2015. A highly conserved residue of the HIV-1 gp120 inner domain is important for antibody-dependent cellular cytotoxicity responses mediated by anti-cluster A antibodies. J Virol 90:2127–2134. doi: 10.1128/JVI.02779-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, Fouts TR, Gallo RC, DeVico AL, Lewis GK. 2013. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc Natl Acad Sci U S A 110:E69–E78. doi: 10.1073/pnas.1217609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanier LL. 2008. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norman JM, Mashiba M, McNamara LA, Onafuwa-Nuga A, Chiari-Fort E, Shen W, Collins KL. 2011. The antiviral factor APOBEC3G enhances the recognition of HIV-infected primary T cells by natural killer cells. Nat Immunol 12:975–983. doi: 10.1038/ni.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richard J, Veillette M, Batraville LA, Coutu M, Chapleau JP, Bonsignori M, Bernard N, Tremblay C, Roger M, Kaufmann DE, Finzi A. 2014. Flow cytometry-based assay to study HIV-1 gp120 specific antibody-dependent cellular cytotoxicity responses. J Virol Methods 208:107–114. doi: 10.1016/j.jviromet.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, Neil S, Pickering S, Schneider DK, Piechocka-Trocha A, Walker BD, Thomas R, Shaw GM, Hahn BH, Keele BF, Lifson JD, Carrington M. 2016. HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe 19:686–695. doi: 10.1016/j.chom.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee WS, Parsons MS, Kent SJ, Lichtfuss M. 2015. Can HIV-1-specific ADCC assist the clearance of reactivated latently infected cells? Front Immunol 6:265. doi: 10.3389/fimmu.2015.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.International HIV Controllers Study, Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O'Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DM, Vine S, Addo MM, et al. . 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A, Kadie CM, Allen TM, Pereyra F, Heckerman D, Walker BD, Brockman MA. 2011. Reduced replication capacity of NL4-3 recombinant viruses encoding reverse transcriptase-integrase sequences from HIV-1 elite controllers. J Acquir Immune Defic Syndr 56:100–108. doi: 10.1097/QAI.0b013e3181fe9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miura T, Brockman MA, Brumme ZL, Brumme CJ, Pereyra F, Trocha A, Block BL, Schneidewind A, Allen TM, Heckerman D, Walker BD. 2009. HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J Virol 83:140–149. doi: 10.1128/JVI.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, Ueda P, Lu Z, Cohen D, Wrin T, Petropoulos CJ, Rosenberg ES, Walker BD. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis 197:563–571. doi: 10.1086/526786. [DOI] [PubMed] [Google Scholar]
- 44.Miura T, Brockman MA, Brumme CJ, Brumme ZL, Carlson JM, Pereyra F, Trocha A, Addo MM, Block BL, Rothchild AC, Baker BM, Flynn T, Schneidewind A, Li B, Wang YE, Heckerman D, Allen TM, Walker BD. 2008. Genetic characterization of human immunodeficiency virus type 1 in elite controllers: lack of gross genetic defects or common amino acid changes. J Virol 82:8422–8430. doi: 10.1128/JVI.00535-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ueno T, Motozono C, Dohki S, Mwimanzi P, Rauch S, Fackler OT, Oka S, Takiguchi M. 2008. CTL-mediated selective pressure influences dynamic evolution and pathogenic functions of HIV-1 Nef. J Immunol 180:1107–1116. doi: 10.4049/jimmunol.180.2.1107. [DOI] [PubMed] [Google Scholar]
- 46.Robinson JE, Holton D, Elliott S, Ho DD. 1992. Distinct antigenic sites on HIV gp120 identified by a panel of human monoclonal antibodies. J Cell Biochem Suppl 16E:Q449. [Google Scholar]
- 47.Fontaine J, Chagnon-Choquet J, Valcke HS, Poudrier J, Roger M, Montreal Primary HIV Infection and Long-Term Non-Progressor Study Groups. 2011. High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood 117:145–155. doi: 10.1182/blood-2010-08-301887. [DOI] [PubMed] [Google Scholar]
- 48.Fontaine J, Coutlee F, Tremblay C, Routy JP, Poudrier J, Roger M, Montreal Primary HIV Infection and Long-Term Non-Progressor Study Groups. 2009. HIV infection affects blood myeloid dendritic cells after successful therapy and despite nonprogressing clinical disease. J Infect Dis 199:1007–1018. doi: 10.1086/597278. [DOI] [PubMed] [Google Scholar]
- 49.Peretz Y, Ndongala ML, Boulet S, Boulassel MR, Rouleau D, Cote P, Longpre D, Routy JP, Falutz J, Tremblay C, Tsoukas CM, Sekaly RP, Bernard NF. 2007. Functional T cell subsets contribute differentially to HIV peptide-specific responses within infected individuals: correlation of these functional T cell subsets with markers of disease progression. Clin Immunol 124:57–68. doi: 10.1016/j.clim.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 50.Kamya P, Boulet S, Tsoukas CM, Routy JP, Thomas R, Cote P, Boulassel MR, Baril JG, Kovacs C, Migueles SA, Connors M, Suscovich TJ, Brander C, Tremblay CL, Bernard N, Canadian Cohort of HIV Infected Slow Progressors. 2011. Receptor-ligand requirements for increased NK cell polyfunctional potential in slow progressors infected with HIV-1 coexpressing KIR3DL1*h/*y and HLA-B*57. J Virol 85:5949–5960. doi: 10.1128/JVI.02652-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Batraville LA, Richard J, Veillette M, Labbe AC, Alary M, Guedou F, Kaufmann DE, Poudrier J, Finzi A, Roger M. 2014. Short communication: anti-HIV-1 envelope immunoglobulin Gs in blood and cervicovaginal samples of Beninese commercial sex workers. AIDS Res Hum Retrovir 30:1145–1149. doi: 10.1089/aid.2014.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding S, Verly MM, Princiotto A, Melillo B, Moody T, Bradley T, Easterhoff D, Roger M, Hahn BH, Madani N, Smith Iii AB, Haynes BF, Sodroski JMD, Finzi A. 2017. Small molecule CD4-mimetics sensitize HIV-1-infected cells to ADCC by antibodies elicited by multiple envelope glycoprotein immunogens in non-human primates. AIDS Res Hum Retrovir 33:428–431. doi: 10.1089/aid.2016.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gooneratne SL, Richard J, Lee WS, Finzi A, Kent SJ, Parsons MS. 2015. Slaying the Trojan horse: natural killer cells exhibit robust anti-HIV-1 antibody-dependent activation and cytolysis against allogeneic T cells. J Virol 89:97–109. doi: 10.1128/JVI.02461-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richard J, Veillette M, Ding S, Zoubchenok D, Alsahafi N, Coutu M, Brassard N, Park J, Courter JR, Melillo B, Smith AB III, Shaw GM, Hahn BH, Sodroski J, Kaufmann DE, Finzi A. 2016. Small CD4 mimetics prevent HIV-1 uninfected bystander CD4 + T cell killing mediated by antibody-dependent cell-mediated cytotoxicity. EBioMedicine 3:122–134. doi: 10.1016/j.ebiom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tolbert WD, Gohain N, Veillette M, Chapleau JP, Orlandi C, Visciano ML, Ebadi M, DeVico AL, Fouts TR, Finzi A, Lewis GK, Pazgier M. 2016. Paring down HIV Env: design and crystal structure of a stabilized inner domain of HIV-1 gp120 displaying a major ADCC target of the A32 region. Structure 24:697–709. doi: 10.1016/j.str.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]