

ABSTRACT
Hemagglutinin (HA) of influenza virus must be activated by proteolysis before the virus can become infectious. Previous studies indicated that HA cleavage is driven by membrane-bound or extracellular serine proteases in the respiratory tract. However, there is still uncertainty as to which proteases are critical for activating HAs of seasonal influenza A viruses (IAVs) in humans. This study focuses on human KLK1 and KLK5, 2 of the 15 serine proteases known as the kallikrein-related peptidases (KLKs). We find that their mRNA expression in primary human bronchial cells is stimulated by IAV infection. Both enzymes cleaved recombinant HA from several strains of the H1 and/or H3 virus subtype in vitro, but only KLK5 promoted the infectivity of A/Puerto Rico/8/34 (H1N1) and A/Scotland/20/74 (H3N2) virions in MDCK cells. We assessed the ability of treated viruses to initiate influenza in mice. The nasal instillation of only the KLK5-treated virus resulted in weight loss and lethal outcomes. The secretion of this protease in the human lower respiratory tract is enhanced during influenza. Moreover, we show that pretreatment of airway secretions with a KLK5-selective inhibitor significantly reduced the activation of influenza A/Scotland/20/74 virions, providing further evidence of its importance. Differently, increased KLK1 secretion appeared to be associated with the recruitment of inflammatory cells in human airways regardless of the origin of inflammation. Thus, our findings point to the involvement of KLK5 in the proteolytic activation and spread of seasonal influenza viruses in humans.
IMPORTANCE Influenza A viruses (IAVs) cause acute infection of the respiratory tract that affects millions of people during seasonal outbreaks every year. Cleavage of the hemagglutinin precursor by host proteases is a critical step in the life cycle of these viruses. Consequently, host proteases that activate HA can be considered promising targets for the development of new antivirals. However, the specific proteases that activate seasonal influenza viruses, especially H3N2 viruses, in the human respiratory tract have remain undefined despite many years of work. Here we demonstrate that the secreted, extracellular protease KLK5 (kallikrein-related peptidase 5) is efficient in promoting the infectivity of H3N2 IAV in vitro and in vivo. Furthermore, we found that its secretion was selectively enhanced in the human lower respiratory tract during a seasonal outbreak dominated by an H3N2 virus. Collectively, our data support the clinical relevance of this protease in human influenza pathogenesis.
KEYWORDS: influenza virus, proteases
INTRODUCTION
Human influenza A and B viruses cause influenza, a highly contagious respiratory illness that affects millions of people worldwide during seasonal epidemics. The subtype of an influenza A virus (IAV) depends on the antigenic properties of its hemagglutinin (HA) and neuraminidase (NA) glycoproteins. There are currently 18 HA subtypes (H1 to H18) and 11 NA subtypes (N1 to N11) known in birds and mammals. Seasonal infections in humans are caused mostly by strains of two virus subtypes, H1N1 and H3N2.
Influenza A virus infection is initiated by the binding of virus HA to sialic acid receptors on the surface of host cells; the human-adapted subtypes bind preferably to 2,6-linked sialic acid. The bound virus enters the host cell by endocytosis and then moves through the endosomal network. The acidic environment in the endosome triggers conformational changes in HA that expose the fusion peptide, which leads to virus-endosome fusion. The fusion of the virus and endosomal membranes results in the release of the virus genome and associated proteins into the cytosol. From there, they are trafficked to the nucleus, where they replicate (reviewed in reference 1). HA is initially synthesized as a precursor, HA0, that is subsequently cleaved by host proteases to give the mature HA1/HA2 form, a complex of two disulfide-linked subunits. This proteolytic activation of HA is essential for the fusion of the virus envelope with endosomal membranes and, hence, for the production of infective virus. The HAs of all human seasonal influenza virus strains have a single arginine at their cleavage sites, and several trypsin-like serine proteases from human bronchial/tracheal epithelial cells can activate monobasic HA in vitro. These serine proteases include several members of the type II transmembrane serine protease (TTSP) family (2), TMPRSS2, TMPRSS2 isoform 1, TMPRSS4, matriptase, DESC1, MSPL, and HAT (human airway trypsin-like protease) (3–5), and two secreted kallikrein-related proteases (KLKs), namely, KLK5 and KLK12 (6). The blood proteases plasmin, urokinase, plasma kallikrein, and thrombin also cleave and activate HA in vitro (7), but there is still some uncertainty as to which proteases support the multicycle replication of seasonal IAVs in the human respiratory system. Although TMPRSS2 is a marker of susceptibility to severe A (H1N1)pdm09 influenza virus (8), there is no clinical data indicating the involvement of a specific protease in the activation of seasonal IAV. However, in mice, TMPRSS2 has been identified as being essential for the spread of H1N1 IAV (9–11) and of H3N2 IAV in association with TMPRSS4 (12).
The kallikrein-related peptidase family comprises 15 secreted serine proteases (KLK1 to KLK15). Most KLKs are produced by the epithelia of the upper and lower respiratory tracts (nose, paranasal sinuses, larynx, trachea, and bronchial tree) and by the submucosal glands of the main airways (13, 14). However, little is known of their expression or function under pathophysiological conditions. We have examined the expressions of 9 KLK genes in human airway epithelia reconstituted in vitro and infected with IAV. As the amounts of the mRNAs encoding KLK1 and KLK5 were selectively increased following IAV infection, we explored the contribution of these proteases to seasonal flu. They usually cleave their substrates after a basic residue, with a preference for Arg. We therefore investigated their ability to cleave and activate HAs from several strains of H1N1 and H3N2 viruses in vitro and in vivo. Finally, we analyzed samples of human tracheal aspirates (TAs) to determine whether the secretion of KLK1 and KLK5 in the lower respiratory tract was dysregulated during seasonal influenza.
RESULTS
Selective induction of KLK1 and KLK5 mRNAs in human bronchial epithelial cells during influenza virus infection.
Airway epithelial cells are both the target of influenza virus and where KLKs are synthesized. We checked to see whether viral infection modulated the expressions of certain KLK-encoding genes. As a preliminary study showed that KLK2, KLK3, KLK4, KLK6, KLK12, and KLK15 are poorly expressed in normal lung tissue, we focused on the genes encoding the other 9 KLK genes. We infected human bronchial epithelia reconstituted in vitro with influenza A/Scotland/20/74 (H3N2) virus, and the mRNA expression levels of KLK1, -5, -7, -8, -9, -10, -11, -13, and -14 were analyzed by reverse transcription-quantitative PCR (RT-qPCR) (Fig. 1). IAV infection significantly increased the amounts of mRNAs encoding KLK1 and KLK5 and decreased the amounts of KLK13 and KLK14 mRNAs. The level of KLK9 also tended to decrease (P = 0.082). These data indicate that IAV infection differentially regulates the gene expression of members of the KLK family in the epithelium of the lower respiratory tract. Because the expression levels of KLK1 and KLK5 were increased by IAV, we looked at the possibility that these serine proteases could be beneficial for the virus.
FIG 1.
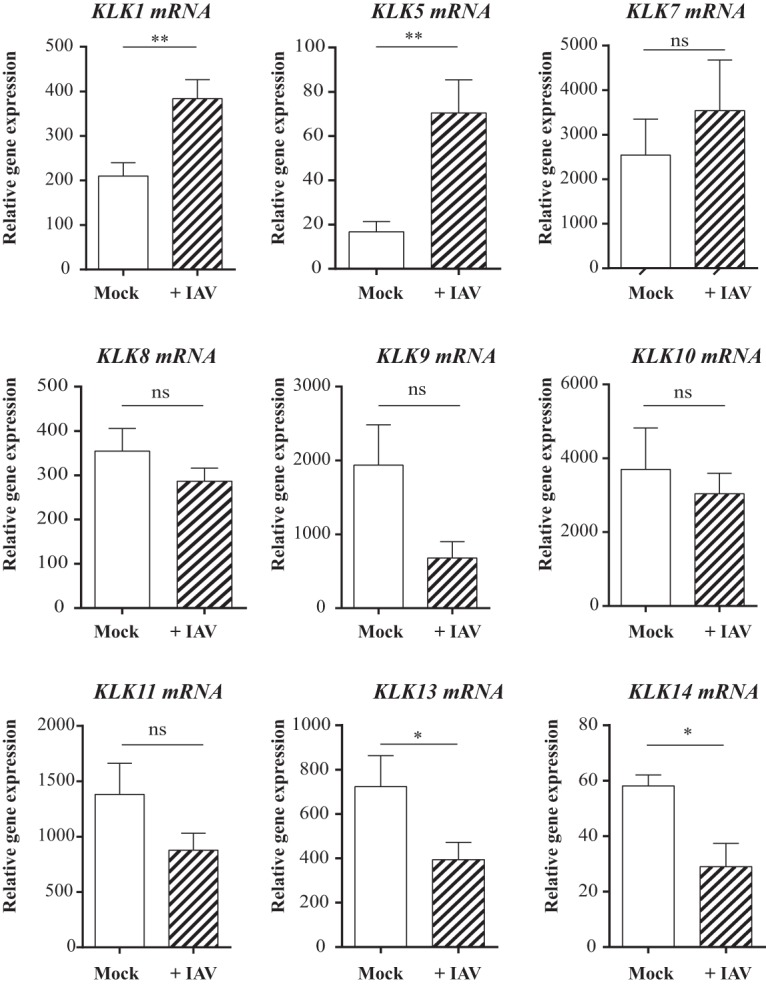
Influence of IAV infection on KLK gene expression in reconstituted human bronchial epithelium. mRNAs encoding KLKs in differentiated bronchial cells cultured at the air-liquid interface that were infected with influenza A/Scotland/20/74 virus (MOI = 1) (+IAV) or not (mock) were assayed by RT-qPCR. The data represent the means ± standard errors of the means (n = 8). Statistical confidence was measured by using a Mann-Whitney U test. *, P < 0.05; **, P < 0.01; ns, nonsignificant.
Cleavage of seasonal influenza virus HA by KLK1 and KLK5.
HA is initially synthesized as a precursor, HA0, that is subsequently cleaved by host enzymes to give its functional forms, HA1 and HA2. Therefore, we compared the abilities of KLK1 and KLK5 to cleave recombinant HA precursors from five strains of subtype H1 influenza virus (Fig. 2A) and four strains of the H3 subtype (Fig. 2B). Trypsin was used as a control (not shown) because this enzyme cleaves most HAs to generate two fragments with apparent molecular masses of 55 kDa (HA1) and 25 kDa (HA2).
FIG 2.

Cleavage of HA subtypes. (A and B) SDS-PAGE and silver staining analyses of intact HA and HA cleaved by human recombinant KLK1 or KLK5. Lanes from the gel images were cut and spliced together with Adobe Photoshop CS 5.1 to aid in band comparison. The recombinant HAs were from H1 subtype strains A/Brisbane/59/07 (Br/59/07), A/California/07/09 (Ca/07/09), A/New Caledonia/20/99 (NC/20/99), A/Puerto Rico/8/34 (PR/8/34), and A/Solomon Island/03/06 (SI/03/06) (A) and from H3 subtype strains A/Aichi/2/68 (Ai/2/68), A/Brisbane/10/07 (Br/10/07), A/Wisconsin/67/05 (Wi/67/05), and A/Wyoming/3/03 (A/Wy/3/03) (B). (C) Efficiency of HA cleavage by KLKs. The bar graph depicts the amount of the HA1 fragment generated by KLK cleavage of all the HAs examined. The values were determined by densitometry analysis in 3 distinct silver staining experiments (means ± standard errors of the means). (D) Alignment of the sequences in the vicinity of the proteolytic cleavage site of HAs.
Of the 9 HA precursors, only 4 (A/Brisbane/59/07, A/California/07/09, A/Puerto Rico/8/34 [A/PR/8/34], and A/Solomon Island/3/06) were significantly digested by KLK1. However, the fragmentation patterns of several precursors contained bands in addition to those of the HA1 and HA2 fragments. Thus, KLK1 can cleave these HAs at other sites. Conversely, KLK5 specifically generated HA1 and HA2 fragments from all HAs, although their production rates varied. We find that KLK5 most actively cleaved HAs from H1 subtype strains A/Brisbane/59/07, A/California/07/09, A/Puerto Rico/8/34, and A/Solomon Island/3/06 and from H3 subtype strain A/Wisconsin/67/05. Despite their preference for cleavage after an arginine residue, the spectra of HA cleavage by KLK1 and KLK5 in vitro appeared to differ. KLK1 seemed to prefer the H1 subtype, while KLK5 efficiently cleaved both the H1 and H3 subtypes (Fig. 2C). This is consistent with previously reported observations that all KLKs have unique specificity profiles due to subtle differences in their active sites (15). Each peptidase also seemed to prefer particular strains within the H1 and/or H3 subtype. The amino acid sequences close to the cleavage site (Fig. 2D) do not seem to be responsible for these preferences because some HAs that had the same sequences were cleaved very differently. This is clearly illustrated by the A/New Caledonia/20/99 and A/Solomon Islands/03/06 HAs; they were not cleaved at the same rate by KLK1, although the amino acid sequences around their cleavage sites are identical. Other factors, such as the three-dimensional (3D) structure of each HA, must influence cleavage preference.
KLK5 promotes the infectivity of seasonal influenza A viruses.
We checked the abilities of KLK1 and KLK5 to make noninfectious influenza A virions infective in order to examine the functional consequences of HA cleavage. The virions were first incubated with trypsin, KLK1, or KLK5. The treated virions were then incubated with mammalian cells, and the amounts of influenza virus nucleoprotein in the cells were detected by flow cytometry (Fig. 3A and B). Both trypsin and KLK5 activated the A/Scotland/20/74 (H3N2) virus, resulting in infected cells. Pretreatment of the virions with KLK1 did not increase cell infection; infection was the same as that produced by untreated viruses. These observations were confirmed by immunocytochemistry (Fig. 3C) and RT-qPCR (Fig. 3D).
FIG 3.

Activation of H1N1 and H3N2 influenza viruses by KLKs. (A to C) MDCK cells were infected with A/Scotland/20/74 (H3N2) particles that had been treated by incubation with either trypsin or specific KLKs for 1 h at 37°C. The resulting MDCK cells were collected and stained for influenza virus nucleoprotein with anti-nucleoprotein-FITC (NP-FITC) mouse IgG1. FSC-A, forward scatter area. (A and B) Representative profiles of influenza virus nucleoprotein expression (A) and fluorescence data (B) were acquired by using a MACS Quant flow cytometer. (C) Immunofluorescence staining results. The influenza virus nucleoprotein was detected with anti-nucleoprotein-FITC antibody (green), and nuclei were stained with Hoechst 33258 (blue). (D and E) MDCK cells were infected with influenza A/Scotland/20/74 (H3N2) (D) or influenza A/Puerto Rico/8/34 (H1N1) (E) virus particles that had been incubated with TPCK-treated trypsin or a KLK for 2 h at 37°C. The resulting MDCK cells were collected, and the mRNA encoding the influenza virus nucleoprotein was assayed by RT-qPCR. Results were expressed as percentages of values obtained with trypsin-treated virus. Means ± standard errors of the means are shown (n = 12). (F) MDCK cells were infected with influenza A/Scotland/20/74 virus particles that had been pretreated with supernatants (n = 8) of human bronchial epithelial cells (16HBE14o−) in the absence (+ supernatant) or in the presence (+ supernatant + Inh) of a selective KLK5 inhibitor for 16 h at 37°C. Proportions of infected MDCK cells were determined by flow cytometry using an anti-NP-FITC antibody. Results are expressed as fold changes of values obtained with the untreated virus (control [Ctrl]). Data were analyzed by one-way ANOVA followed by Tukey's multiple-comparison test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Uncleaved influenza A/PR/8/34 (H1N1) HA virions were also treated with the proteases, and the nucleoprotein (NP) mRNA that accumulated in infected Madin-Darby canine kidney (MDCK) cells was quantified by RT-qPCR. Treatment of A/PR/8/34 virions with KLK5 slightly stimulated NP mRNA production, but incubation with KLK1 had no effect (Fig. 3E). Thus, KLK5 can activate H1 and H3 subtype IAVs, increasing their in cellulo infectivity. This protease activates H3 subtypes preferentially. In contrast, KLK1 did not promote the multicycle replication of influenza viruses used in this study.
To determine whether KLK5 naturally secreted by bronchial cells may activate influenza virus, we incubated influenza A/Scotland/20/74 virions with supernatants of unchallenged 16HBE14o− cells containing around 60 ng/ml of KLK5. MDCK cells were then infected with the treated virions. As shown in Fig. 3F, treatment of the virions with the 16HBE14o− supernatant increased cell infection; this effect was significantly reduced when the virions were incubated with the supernatant pretreated with SFTI-FCHR, a synthetic inhibitor of KLK5. This molecule does not inhibit previously identified HA-activating soluble proteases such as plasma kallikrein, plasmin, thrombin, and matriptase and denotes a greater preference for KLK5 than for trypsin (Ki = 6.2 ± 0.2 versus 31.5 ± 1.4).
KLK5-treated H3N2 virus initiates viral infection in mice.
Although MDCK cells are commonly used to characterize the infectivity of influenza viruses, they are not the natural host cells of these viruses. We therefore examined the abilities of viruses treated with KLK1 or KLK5 to trigger acute pneumonia in mouse models. C57BL/6 mice were inoculated intranasally with influenza A/PR/8/34 (H1N1) or influenza A/Scotland/20/74 (H3N2) viruses that had been incubated with trypsin, human KLK1, or human KLK5 or left untreated (controls), and their weights and survival rates were monitored (Fig. 4). Neither unactivated H1N1 virus nor KLK-treated H1N1 virus caused any significant weight loss or death, but trypsin-treated virus triggered acute pneumonia, with a death rate of 50% (Fig. 4A). H3N2 virus pretreated with human KLK1 did not modify the weight loss or survival of mice; its effect was the same as that of the unactivated H3N2 virus (Fig. 4B). Conversely, mice infected with KLK5-treated H3N2 viruses lost weight from the third day postinfection, in parallel with the weight loss caused by trypsin-treated viruses. Significantly more mice inoculated with trypsin-treated or KLK5-treated H3N2 viruses died than did mice infected with the unactivated virus.
FIG 4.
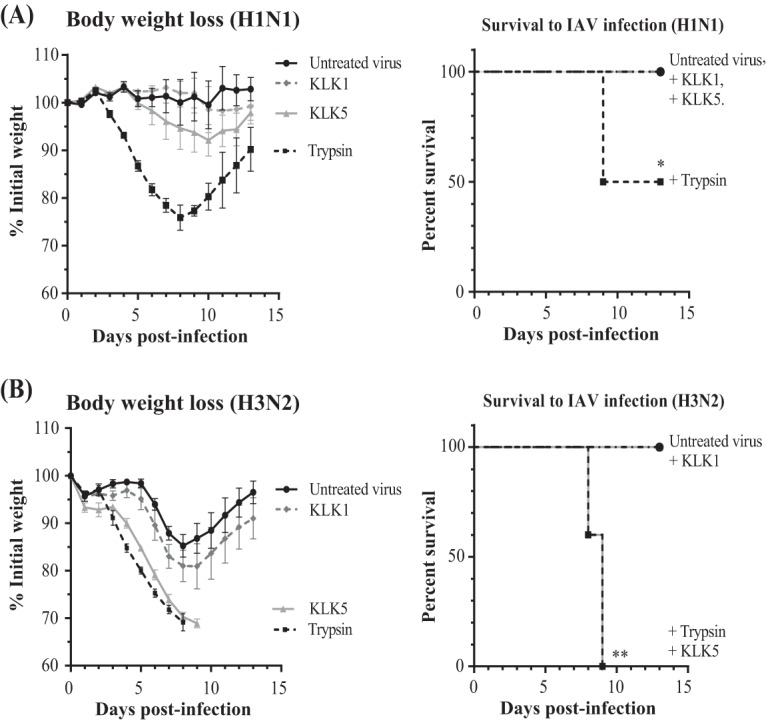
Effect of KLK-activated viruses in mice. Shown are body weight kinetics and survival of mice infected with viruses that had been treated with TPCK-treated trypsin, human KLK1, or human KLK5. Male C57BL/6 mice (n = 6 per group) were inoculated intranasally with influenza A/Puerto Rico/8/34 (H1N1) (A) or influenza A/Scotland/20/74 (H3N2) (B) virus. *, P < 0.05; **, P < 0.01 (for treated versus untreated mice, as determined by Kaplan-Meier and log rank [Mantel-Cox] tests).
Influenza and secretion of KLK1 and KLK5 in the human lower respiratory tract.
We measured the amounts of KLK1 and KLK5 in the secretions of the lower respiratory tract of influenza virus-infected humans in order to assess the impact of infection on the secretion of these proteases. We analyzed tracheal aspirate samples from 17 patients in an intensive care unit as a result of 2014-2015 seasonal influenza and aspirate samples from 18 patients hospitalized in the intensive care unit for noninfectious reasons. The intensities of inflammation, as assessed by the number of total cells and myeloperoxidase (MPO), interleukin-6 (IL-6), and IL-8 concentrations, were similar in the two groups. All the samples contained significant amounts of KLK1 and KLK5, but the KLK5 concentration in the tracheal aspirate samples from flu patients was significantly elevated (Fig. 5A). The molecular pattern of KLK5 in aspirate samples was then analyzed by Western immunoblotting. We found two main immunoreactive bands, one at about 55,000 Da and the other at 35,000 Da. The upper band is consistent with a form of KLK5 covalently complexed with a protease inhibitor, while the lower band corresponds to free, possibly active, KLK5. By normalizing the values to the total protein concentration present in each lane, immunoblotting revealed that the concentration of free KLK5 was selectively increased in flu patients (Fig. 5C), modifying the ratio of free to complexed KLK5 (Fig. 5D). Finally, tracheal aspirate samples from noninfected patients were pretreated with the vehicle or with a KLK5-selective inhibitor and then incubated with influenza A/Scotland/20/74 virions. Figure 5E shows that pretreatment of aspirates with SFTI-FCHR significantly reduced infection of MDCK cells. Collectively, these observations suggest that IAV infection likely disrupts the balance between KLK5 and its inhibitor(s) in the lower respiratory tract and that the residual activity of KLK5 can cause the proteolytic activation of influenza virus.
FIG 5.
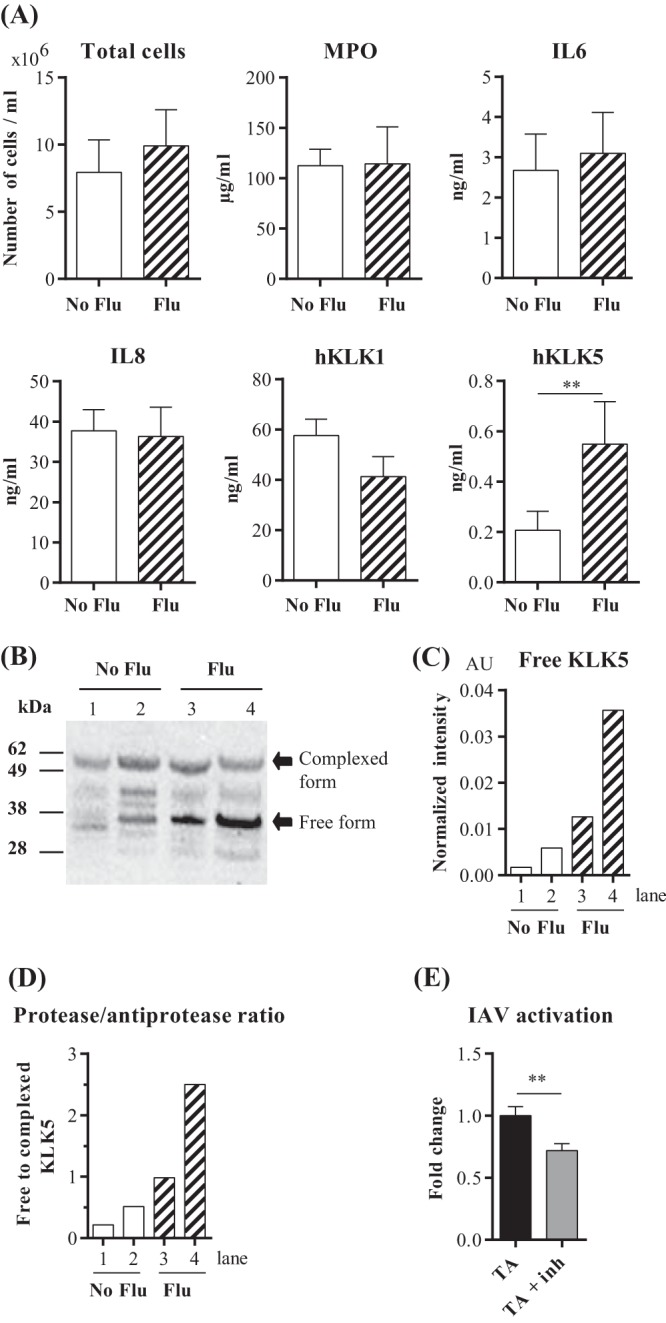
KLKs in lower respiratory tract secretions. (A) Total cell numbers and concentrations of myeloperoxidase, inflammatory mediators (IL-6 and IL-8), and KLKs were measured in tracheal aspirate samples from control patients (No Flu) (n = 18) and influenza patients (Flu) (n = 17) in an intensive care unit. Means ± standard errors of the means are shown. **, P < 0.01, as determined by a Mann-Whitney U test. (B) Western blot analysis of KLK5 in tracheal aspirates from patients in an intensive care unit. Lanes from the gel images were cut and spliced together with Adobe Photoshop CS 5.1 to aid in band comparison. Numbers at the left refer to molecular masses, in kilodaltons. (C) Densitometric analysis of the Western blot data shown in panel B. The values (AU, arbitrary units) represent the signal intensity normalized against the total protein amount in each lane assessed by Ponceau S staining after membrane transfer. (D) Ratio of free to bound KLK5. (E) MDCK cells were infected with influenza A/Scotland/20/74 virus particles that had been pretreated with supernatants (n = 9) of human tracheal aspirates (TA) in the absence or in the presence of a selective KLK5 inhibitor (TA + Inh) for 16 h at 37°C. Proportions of infected MDCK cells were determined by flow cytometry using an anti-NP-FITC antibody. Results are expressed as fold changes of values obtained without an inhibitor. Data were analyzed by a Mann-Whitney U test (**, P < 0.01).
KLK1 and KLK5 are secreted by two distinct cell compartments.
The cells responsible for the KLKs in inflamed lungs have yet to be clearly identified. KLKs seem to be produced by the surface epithelium and the submucosal glands of airways (14), but KLKs have also been detected in circulating neutrophils (16). We first examined the distribution of KLK1 and KLK5 in immune cells in tracheal aspirates by flow cytometry. Neutrophils and T and B lymphocytes all contained substantial amounts of KLK1, but it was less abundant in macrophages and NK cells (Fig. 6A). Because neutrophils are the major immune cells in inflamed airways, we then looked for a correlation between the concentrations of KLK1 in the aspirates and the neutrophil abundance estimated by measuring the MPO concentration. We found a significant positive correlation for KLK1 (Fig. 6A), indicating that neutrophils are the main source of KLK1 in inflamed airways. In contrast, there was little KLK5 in immune cells, and there was no correlation between the amounts of KLK5 and MPO in the tracheal aspirates (Fig. 6B). Thus, immune cells contribute little to the KLK5 secreted into inflamed airways, and the main source of KLK5 during IAV infection is probably the airway mucosa. This assumption is supported by the induction of KLK5 in human bronchial epithelial cells (hBECs) infected with IAV (Fig. 6C). Our data indicate that infection of the human lower respiratory tract by IAV stimulates the synthesis and secretion of the IAV-activating protease KLK5 by the airway epithelium.
FIG 6.
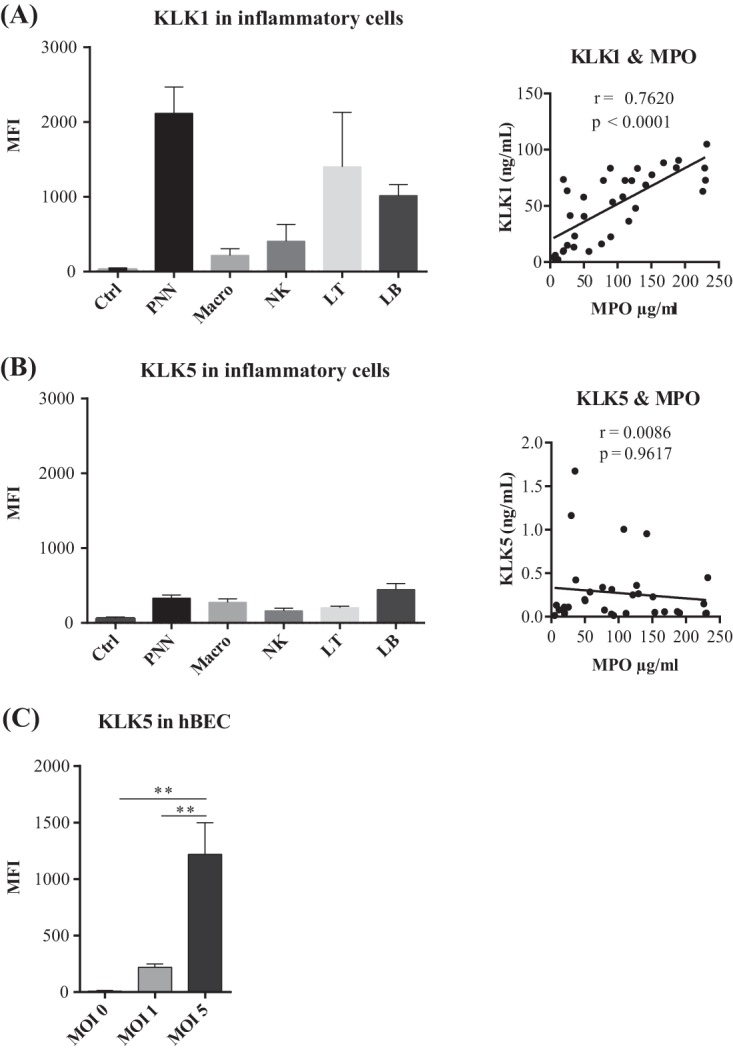
Sources of the KLKs secreted into inflamed lower respiratory tracts. (A and B, left) Flow cytometric analysis of KLK1 and KLK5 expression in immune cells in tracheal aspirates (Ctrl, negative-control cells; PNN, polynuclear neutrophils; Macro, macrophages; NK, NK cells; LT, T lymphocytes; LB, B lymphocytes; MFI, mean fluorescence intensity). All values are means ± standard errors of the means from 3 independent experiments. (Right) Correlation between KLK and MPO concentrations in tracheal aspirate samples. Correlation was tested by a Spearman rank test. (C) Flow cytometric analysis of KLK5 expression in differentiated bronchial cells cultured at the air-liquid interface and infected with influenza A/Scotland/20/74 virus at different MOIs. Fluorescence data were acquired by using a MACS Quant flow cytometer; means ± standard errors of the means are shown (n = 4). Data were analyzed by one-way ANOVA followed by Tukey's multiple-comparison test (**, P < 0.01).
DISCUSSION
Numerous groups have reported that various members of the TTSPs are responsible for activating several seasonal influenza viruses (3, 4, 10, 17, 18). Bottcher-Friebertshauser et al. used MDCK cells to show that TMPRSS2 cleaves HA within cells, while HAT cleaves HA at the cell surface (19). All these finding suggest that HA cleavage is cell associated, but there is still no direct clinical evidence for the cell-associated activation of seasonal influenza virions in the human respiratory system. A recent report showing that the TMPRSS2 gene is a marker of susceptibility to severe 2009 pandemic A(H1N1) influenza virus indicates that TMPRSS2 contributes to influenza virus pathogenesis in humans (8). In contrast, soluble activating proteases have been found in the secretions of the upper respiratory tract (20, 21), and several groups have attempted to identify them. Secreted forms of TMPRSS2 and HAT have been observed, but their catalytic activities are uncertain (19, 20, 22). Conversely, Hamilton et al. showed that the secreted catalytic domain of matriptase cleaved and activated HA from H1 subtype influenza virus in vitro (23). Hamilton and Whittaker also identified human KLK5 and human KLK12 as possible activators of seasonal influenza viruses (6). At present, 3 human KLKs (KLK1, KLK5, and KLK12) have been examined for their ability to cleave HA and activate influenza A viruses. Collectively, data from previous studies and our present findings clearly demonstrate that these KLKs are highly selective and do not cleave HA from the H1 and H3 influenza A virus subtypes with the same efficiency. Human KLK1 cleaves H1 subtype HAs best, and KLK5 cleaves a wide range of both H1 and H3 subtype HAs, while KLK12 cleaves H1 and H2 subtype HAs most efficiently. This may reflect, at least in part, the distinct specificity of each KLK for substrate recognition motifs. However, the amino acid sequence flanking the cleavage site does not appear to be the sole determinant of HA cleavage preference. Indeed, the efficiency with which HAs that have the same amino acid residues in the vicinity of the cleavage site were hydrolyzed varied. The crystal structures of HA0s from different HA subtypes indicate that the cleavage site forms a loop more or less exposed to the environment. The flexibility and accessibility of this loop are believed to contribute to the differences in the susceptibilities of subtypes to proteases (24). As IAVs vary enormously, there may be subtle differences in the loop conformations of HAs of the same subtype, and this could result in interstrain variations in the cleavage efficiency. Clearly, more work is needed to determine the factors that govern the susceptibilities of closely related HAs to a given protease.
We examined viral infectivity in cellulo using MDCK cells to establish whether the cleavage of virus HA by KLK1 and KLK5 was correlated with this infectivity. Human KLK1 did not activate uninfectious H1N1 A/Puerto Rico/8/34 or H3N2 A/Scotland/20/74 virions. This contrasts with a previous report that rat Klk1 enhances virus production in MDCK cells infected with influenza A/WSN/33 (H1N1) virus particles (25). This may be because of differences in the substrate specificities of rat Klk1 and human KLK1 (26). Alternatively, the divergent results may illustrate differences in the cleavage susceptibilities of A/Puerto Rico/8/34 HA and A/WSN/33 HA. Compared to other H1N1 strains, A/WSN/33 HA does not have the typical cleavage motif but possesses a tyrosine at the P2 position (Fig. 3D). Such a Ser-Tyr mutation enhances plasmin-mediated cleavage and virus activation of naturally occurring H1N1 influenza viruses (27). It is possible that this mutation has similar consequences for KLK1-mediated viral activation. Our results also indicate that KLK5 activates HAs of both the H1 and H3 subtypes so that KLK5-treated viruses can infect MDCK cells. However, KLK5 activates H3N2 influenza A/Scotland/20/74 virus more efficiently than it activates H1N1 influenza A/Puerto Rico/8/34 virus. This preference for the H3 subtype is consistent with a previous report showing that KLK5 produced greater fusogenic activity with H3 subtype HAs than with HAs of the H1 and H2 subtypes (6). In contrast, KLK12 cleaves H1 and H2 subtype HAs most efficiently. It does not promote the infectivity of A/Scotland/20/74 (H3N2) virions (data not shown), confirming the report by Hamilton et al. that it does not activate HAs of the H3 subtype.
Our next step was to examine the ability of treated viruses to promote influenza virus infection in vivo in mice. We confirmed that KLK1 did not functionally activate A/Puerto Rico/8/34 and A/Scotland/20/74 viruses belonging to the H1 and H3 subtypes. Acute pneumonia was observed in mice instilled with influenza A/Scotland/20/74 viruses that had been activated by KLK5. Weight loss and mortality were then identical to those obtained with trypsin-activated viruses. Thus, human KLK5 causes H3 subtype viruses to become very infective in the respiratory tract. Because KLK5 treatment of virions impacted only the first round of infection, endogenous proteases were therefore involved in subsequent viral spread and pathogenesis. TMPRSS2 appears to be essential for influenza H1N1 virus to be pathogenic in mice but not for H3N2 virus pathogenicity (9). H1N1 virus replication was suppressed in the respiratory tract of TMPRSS2−/− mice, while H3N2 virus replication was only marginally affected (11). It was recently proposed that TMPRSS4 plus TMPRSS2 activate H3N2 viruses in mice (12). We now need to know whether some endogenous kallikreins are involved in the replication and pathogenesis of H3 subtype influenza viruses in mice.
Influenza pneumonia resulted in the hospitalization of a significant number of patients in intensive care units in Europe during the 2014-2015 winter dominated by a human influenza A (H3N2) virus (28). Seventeen intubated patients suffering from acute influenza and 18 patients hospitalized in the intensive care unit for other reasons (controls) were enrolled in this study. We quantified the KLK1 and KLK5 concentrations in tracheal aspirate samples collected from these patients. Both KLKs were present in secretions, but the concentration of only KLK5 was significantly increased in influenza patients. This was not due to differences in the intensity of inflammation because we measured similar amounts of total cells, MPO, IL-6, and IL-8 in both groups. The augmented amount of luminal KLK5 is probably due to increased KLK5 production and secretion by airway epithelial cells in response to viral infection, as indicated by our experiments using primary cultures of bronchial cells. This mechanism might amplify both virus activation and subsequent spread, since we have shown that KLK5 that is naturally secreted can functionally activate noninfectious viral particles. However, pretreatment of respiratory tract secretions with a KLK5-selective inhibitor did not completely abolish the activation of influenza A/Scotland/20/74 virions, suggesting that other secreted proteases might contribute to IAV activation in the lower respiratory tract. Their identity, however, remains to be determined.
Human KLK1 seems limited in activating seasonal viruses, unlike KLK5, and influenza virus infection does not appear to specifically increase its secretion in human airways. KLK1 secreted into inflamed airways is derived mainly from luminal neutrophils. Thus, all proinflammatory factors (and not only IAV) promoting the recruitment of neutrophils may cause the KLK1 concentration to increase. This is consistent with our observation that all the patients, both controls and influenza sufferers, with similar degrees of lung inflammation had similar KLK1 concentrations. Some reports associated KLK1 with lung dysfunctions in inflammatory diseases such as asthma (29) and chronic bronchitis (30). We now need to determine whether KLK1 plays a role in acute pneumonia, whether or not pneumonia is induced by influenza virus.
In conclusion, we find that the secreted, extracellular protease KLK5 is very efficient in promoting the infectivity of H3N2 seasonal influenza virus. The selective amplification of KLK5 secretion during infection further supports the clinical relevance of this protease in human influenza pathogenesis.
MATERIALS AND METHODS
Animals, cells, viruses, and reagents.
Male C57BL/6 mice were purchased from Janvier Labs (Le Genest-Saint-Isle, France). Mice were housed under pathogen-free conditions at the animal facility of F. Rabelais University and handled according to guidelines of the European Animal Care and Use Committee (Ethical Comity agreement CEEA VdL 2012-12-6). Animals were kept in a 12-h-light–12-h-dark cycle with food and water ad libitum.
Normal primary human bronchial epithelial cells (hBECs) and media were purchased from Lonza (Basel, Switzerland). The cells were propagated in bronchial epithelial cell growth medium (BEGM) basal medium and differentiated by culture at the air-liquid interface (ALI) in bronchial ALI (B-ALI) differentiation basal medium. The human bronchial cell line (16HBE14o−) was a generous gift from D. Gruenert (University of California, San Francisco, CA, USA). NCI-H292 cells (ATCC/LGC Standards, Teddington, UK) and 16HBE14o− cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and 50 U/ml penicillin-streptomycin. MDCK cells (ATCC/LGC Standards) were maintained in minimal essential medium (MEM) supplemented with 10% FBS.
Mouse-adapted influenza A/Puerto Rico/8/34 (H1N1) and influenza A/Scotland/20/74 (H3N2) viruses were gifts from the Pasteur Institute (Paris, France). TPCK (tosyl phenylalanyl chloromethyl ketone)-treated trypsin was obtained from Worthington (Lakewood, NJ, USA). Human kallikrein 1 was obtained from Novoprotein, and human kallikrein 5 was obtained from R&D Systems (Minneapolis, MN, USA). All influenza A virus HAs used in this study were obtained from Sino Biological Inc. (Beijing, China). The enzyme-linked immunosorbent assay (ELISA) kits used were a human KLK1 (hKLK1) kit from Cloud-Clone Corp. (Houston, TX, USA) and hKLK5, hMPO, hIL-6, and hIL-8 kits from R&D Systems (Minneapolis, MN, USA). The KLK5 inhibitor SFTI-FCHR was provided by Jonathan Harris (Queensland University of Technology [QUT], Brisbane, Australia).The inhibition assays were performed as previously described (31).
Tracheal aspirate collection.
Aspirates were collected during routine tracheal suctioning of mechanically ventilated patients. Tracheal aspirate samples were collected from a total of 17 flu patients hospitalized during the 2014-2015 influenza season in the CHRU Hospital of Tours and from 18 individuals not infected with influenza virus. This study was approved by French national bioethics authorities (CPP-37 2012-R21). Informed written consent was obtained from each participant. Samples were dissociated in 3 volumes of phosphate-buffered saline (PBS) and 1 mM dithiothreitol (Sigma-Aldrich, St. Louis, MO, USA) with stirring during 1 h. After centrifugation at 790 × g during 10 min, the supernatant was picked up and stored at −80°C until use. Total cells were counted on a Malassez cell and analyzed by flow cytometry. Soluble proteins from TA samples were electrophoresed on NuPAGE Novex 4 to 12% Bis-Tris gels (Invitrogen, Carlsbad, CA, USA). KLK5 was analyzed by Western blotting using an anti-KLK5 rabbit antibody (catalog number 168340; Abcam, Cambridge, UK) and a goat anti-rabbit IgG secondary antibody (Jackson ImmunoResearch, Suffolk, UK).
Infection of hBECs.
The apical cell surface of an hBEC-ALI culture was washed twice with PBS. The influenza virus (A/Scotland/20/74) inoculum was added to the apical surface of hBECs at different multiplicities of infection (MOIs) (MOIs of 0, 1, and 5), and the cells were incubated for 24 h at 37°C. The mRNA expression of KLKs was analyzed by RT-qPCR, and the intracellular KLK5 content was assessed by flow cytometry.
Hemagglutinin cleavage assay.
To determine whether KLKs are able to cleave HA, influenza A virus HAs were incubated with KLKs or with TPCK-treated trypsin in 100 mM Tris (pH 8) at 37°C for 4 h. The HA/protease ratio was 10:1. Samples were subsequently analyzed by electrophoresis on 4 to 12% Bis-Tris protein gels and silver staining (Invitrogen, Carlsbad, CA, USA). Densitometry analysis was performed by using Multi Gauge V3.0 (Fujifilm, Bois d'Arcy, France).
Virus infection assay.
Noninfectious virions were obtained from a single round of replication in HEK-293T cells, which lack proteases that are able to cleave HA and activate influenza virions (6). HEK-293T cells were incubated with 1 ml of active viruses (MOI = 1) for 1 h at 37°C. The culture medium was removed, and cells were washed twice with PBS and incubated for 24 h in 9 ml of DMEM supplemented 50 U/ml penicillin-streptomycin. The viral suspension was centrifuged for the withdrawal of detached cells and stored at −80°C until use. Virus titers were determined by using a modified plaque assay method, as previously described (32). Influenza A virions with uncleaved HA were incubated with 200 nM KLK1 or KLK5 or with 40 nM trypsin for 2 h at 37°C. MDCK cells were infected with 200 μl of the protease-activated virus inoculum for 1 h at 37°C. At 7 h postinfection (p.i.), cells were lysed for RNA preparation and viral nucleoprotein qPCR.
16HBE14o− cell medium (without FBS) or bronchial aspirate samples were pretreated with the vehicle or with SFTI-FCHR (10 μM) for 1 h at 37°C and then incubated with uninfectious influenza A/Scotland/20/74 virions for 16 h at 37°C. MDCK cells were infected with 200 μl of the virus inoculum for 1 h at 37°C. At 16 h postinfection, cells were stained with a fluorescein isothiocyanate (FITC)-conjugated anti-influenza A virus NP antibody (Abcam, Cambridge, UK) for flow cytometry analysis.
Viral nucleoprotein immunofluorescence staining.
MDCK cells were grown in chamber slides (Labtex, Huddersfield, UK) before infection and washed twice with PBS. The virus inoculum was then incubated with the cells for 4 h. MDCK cells were fixed by using 4% paraformaldehyde for 15 min at room temperature (RT) and then permeabilized in PBS–0.1% Triton X-100 for 10 min at RT. Following a 1-h saturation step with PBS–1% bovine serum albumin (BSA)–0.1% Tween 20, cells were stained with the FITC-conjugated anti-NP antibody. The nuclei were stained with Hoechst 33258 (Invitrogen).
Mouse influenza A virus infection.
Influenza A/Puerto Rico/8/34 and A/Scotland/20/74 mouse-adapted virions were activated by treatment with either KLKs or trypsin as described above. Before infection, 9- to 11-week-old mice were anesthetized by intraperitoneal injection of ketamine-xylazine (50 mg/kg of body weight and 10 mg/kg, respectively). Infection was performed by the intranasal application of 40 μl of the virus suspension in PBS (600 PFU/mice). Subsequently, survival and body weight loss were monitored until day 13 p.i. In addition to mice that were found dead, mice with weight loss of more than 30% were euthanized and recorded as dead.
RNA preparation and real-time PCR amplification.
Total RNAs from MDCK cells, hBEC-ALI cultures, or mouse lung tissues were extracted by using the NucleoSpin RNA kit (Macherey-Nagel, Düren, Germany), including a step of genomic DNA digestion with DNase. Single-strand cDNA was synthesized from 1 μg total RNA from each sample with the High Capacity cDNA reverse transcription kit (Life Technologies SAS, Saint Aubin, France). For analysis of the viral nucleoprotein mRNA, the reverse transcription step was performed with an oligo(dT)12–18 primer (Invitrogen). mRNA levels were determined by quantitative real-time PCR with a LightCycler 480 instrument (Roche Diagnostics, Meylan, France). The mRNA concentration was normalized to the level of acidic ribosomal phosphoprotein mRNA (Rplp0). The primer pairs used are described in Table 1 and were obtained from Eurofins Genomics (Ebersberg, Germany). PCR was carried out by using 20 ng of reverse-transcribed total RNA as the template, 0.2 μM (each) forward and reverse primers, and 5 μl SYBR Premix Ex Taq (2×) (TaKaRa Bio Europe, Saint-Germain-en-Laye, France) in a final volume of 10 μl. Each reaction was performed in triplicate in white 384-well plates. The thermal protocol consisted of an initial denaturation step at 95°C for 30 s followed by 40 cycles of denaturation at 95°C for 5 s and primer annealing and extension at 60°C for 20 s. Melting curves were generated for each amplified cDNA to check the specificity of the reactions.
TABLE 1.
Primers useda
| Primer target | Primer orientation | Sequence (5′–3′) | Amplicon length (bp) | GenBank accession no. |
|---|---|---|---|---|
| hKLK1 | FOR | GACACCTGGAAGGTGGCAAAGA | 150 | NM_002257.2 |
| REV | CATAAGACAGCACTCTGACGGC | |||
| hKLK5 | FOR | CGTCCCACTAAAGATGTCAGACC | 133 | NM_001077492.1 |
| REV | TCAAGCACTGGAGGACCTTAGG | |||
| hKLK7 | FOR | GGACCCAGATGTGACCTT | 87 | NM_001243126.1 |
| REV | TCCTTGTAAACCTTCGTGC | |||
| hKLK8 | FOR | TACTCTGTGGCGGTGTCCTTGT | 149 | NM_144505.1 |
| REV | TGGGATGGACTGAACCACAGGT | |||
| hKLK9 | FOR | TCAACCTCAGCCAGACCTGTGT | 126 | NM_012315.1 |
| REV | TCTCCAGGATGCTGATGTTGGC | |||
| hKLK10 | FOR | GGACCCCGAAGCCTATG | 249 | NM_002776.4 |
| REV | CCTGAGCCCTGGTGGTA | |||
| hKLK11 | FOR | CAGGATCATCAAGGGGTTCG | 269 | NM_001136032.2 |
| REV | CATTGCGGTGGTCTTTGTTG | |||
| hKLK13 | FOR | CCAACTTCGCTCAGATGAGGAG | 159 | NM_015596.1 |
| REV | CAGGAGACGATGCCATACAGTG | |||
| hKLK14 | FOR | GAGTGTCAGGCTGGGGAACTA | 146 | NM_022046.4 |
| REV | AACTCCTGCACAGACCATGCC | |||
| Viral nucleoprotein | FOR | CTCTTGTTCGCACCGGAATG | 401 | NC_002019.1 |
| REV | GGCTACGGCAGGTCCATAC | |||
| Rplp0 | FOR | CACTGAGATCAGGGACATGTTG | 77 | NM_053275.3 |
| REV | CTTCACATGGGGCAATGG |
FOR, forward; REV, reverse.
Flow cytometry analysis.
Cells from tracheal aspirate samples were stained in PBS–2 mM EDTA–5% FBS with a dead cell marker (Molecular Probes, Eugene, OR, USA) and anti-CD45-, anti-CD15-, anti-CD56-, anti-CD16-, anti-CD14-, anti-CD11c-, anti-CD3-, and anti-CD19-conjugated antibodies (BD Biosciences, Le Pont de Claix, France) for phenotyping. Analysis of intracellular antigens was performed on cells that were fixed and permeabilized by using the Cytofix/Cytoperm kit from BD Biosciences (Le Pont de Claix, France) according to the manufacturer's recommendations. MDCK cells infected by influenza A viruses were stained with anti-influenza A virus NP antibody conjugated with FITC (Abcam) or with the appropriate isotype control for 1 h at 4°C in PBS with 2 mM EDTA and 5% FBS. KLK1 was detected in cells from tracheal aspirate samples by using FITC-conjugated anti-KLK1 (catalog number 1963R; Bioss, Woburn, MA, USA) incubated for 30 min at 4°C. For KLK5 staining, the permeabilized cells were incubated with an anti-KLK5 antibody (catalog number 168340; Abcam, Cambridge, UK) for 1 h at RT and then with a goat anti-rabbit IgG secondary antibody (Alexa Fluor 546 conjugate) for 30 min at 4°C. Appropriate isotype controls, NCI-H292 cells (deficient in KLK1) and unstimulated hBECs (deficient in KLK5), were used to determine the specificity of the antibodies used. Flow cytometric analyses were performed by using a MACS Quant analyzer (Miltenyi Biotec) and VenturiOne software (AppliedCytometry).
Statistical analysis.
Statistical analysis was performed by using GraphPad Prism (Prism 6 for Windows). Statistical significance between the different values was analyzed by a Mann-Whitney U test or one-way analysis of variance (ANOVA) followed by Tukey's multiple-comparison test according to the number of parameters and groups being compared. A P value of 0.05 was considered significant.
ACKNOWLEDGMENTS
UMR INSERM 1100 is funded in part by MedImmune. Financial support was provided by the Région Centre-Val de Loire (grant BPCO-LYSE). The funders had no role in study design or data collection and interpretation.
We thank Chrystophe Aubert, Benjamin Plante, and Elodie Theyssandier for animal handling and the staff of the intensive care unit of Bretonneau Hospital (CHRU Tours) for their support. Owen Parkes checked the English text.
REFERENCES
- 1.Hamilton BS, Whittaker GR, Daniel S. 2012. Influenza virus-mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses 4:1144–1168. doi: 10.3390/v4071144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bugge TH, Antalis TM, Wu Q. 2009. Type II transmembrane serine proteases. J Biol Chem 284:23177–23181. doi: 10.1074/jbc.R109.021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zmora P, Blazejewska P, Moldenhauer AS, Welsch K, Nehlmeier I, Wu Q, Schneider H, Pohlmann S, Bertram S. 2014. DESC1 and MSPL activate influenza A viruses and emerging coronaviruses for host cell entry. J Virol 88:12087–12097. doi: 10.1128/JVI.01427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zmora P, Moldenhauer AS, Hofmann-Winkler H, Pohlmann S. 2015. TMPRSS2 isoform 1 activates respiratory viruses and is expressed in viral target cells. PLoS One 10:e0138380. doi: 10.1371/journal.pone.0138380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton BS, Whittaker GR. 2013. Cleavage activation of the human-adapted influenza virus subtypes by kallikrein-related peptidases 5 and 12. J Biol Chem 288:17399–17407. doi: 10.1074/jbc.M112.440362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheiblauer H, Reinacher M, Tashiro M, Rott R. 1992. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis 166:783–791. doi: 10.1093/infdis/166.4.783. [DOI] [PubMed] [Google Scholar]
- 8.Cheng Z, Zhou J, To KK, Chu H, Li C, Wang D, Yang D, Zheng S, Hao K, Bosse Y, Obeidat M, Brandsma CA, Song YQ, Chen Y, Zheng BJ, Li L, Yuen KY. 2015. Identification of TMPRSS2 as a susceptibility gene for severe 2009 pandemic A(H1N1) influenza and A(H7N9) influenza. J Infect Dis 212:1214–1221. doi: 10.1093/infdis/jiv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pohlmann S, Schughart K. 2013. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog 9:e1003774. doi: 10.1371/journal.ppat.1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakai K, Ami Y, Tahara M, Kubota T, Anraku M, Abe M, Nakajima N, Sekizuka T, Shirato K, Suzaki Y, Ainai A, Nakatsu Y, Kanou K, Nakamura K, Suzuki T, Komase K, Nobusawa E, Maenaka K, Kuroda M, Hasegawa H, Kawaoka Y, Tashiro M, Takeda M. 2014. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol 88:5608–5616. doi: 10.1128/JVI.03677-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarnow C, Engels G, Arendt A, Schwalm F, Sediri H, Preuss A, Nelson PS, Garten W, Klenk HD, Gabriel G, Bottcher-Friebertshauser E. 2014. TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 influenza A virus in mice. J Virol 88:4744–4751. doi: 10.1128/JVI.03799-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn N, Bergmann S, Kosterke N, Lambertz RL, Keppner A, van den Brand JM, Pohlmann S, Weiss S, Hummler E, Hatesuer B, Schughart K. 2016. The proteolytic activation of (H3N2) influenza A virus hemagglutinin is facilitated by different type II transmembrane serine proteases. J Virol 90:4298–4307. doi: 10.1128/JVI.02693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw JL, Diamandis EP. 2007. Distribution of 15 human kallikreins in tissues and biological fluids. Clin Chem 53:1423–1432. doi: 10.1373/clinchem.2007.088104. [DOI] [PubMed] [Google Scholar]
- 14.Petraki CD, Papanastasiou PA, Karavana VN, Diamandis EP. 2006. Cellular distribution of human tissue kallikreins: immunohistochemical localization. Biol Chem 387:653–663. doi: 10.1515/BC.2006.084. [DOI] [PubMed] [Google Scholar]
- 15.Debela M, Beaufort N, Magdolen V, Schechter NM, Craik CS, Schmitt M, Bode W, Goettig P. 2008. Structures and specificity of the human kallikrein-related peptidases KLK 4, 5, 6, and 7. Biol Chem 389:623–632. doi: 10.1515/BC.2008.075. [DOI] [PubMed] [Google Scholar]
- 16.Lizama AJ, Andrade Y, Colivoro P, Sarmiento J, Matus CE, Gonzalez CB, Bhoola KD, Ehrenfeld P, Figueroa CD. 2015. Expression and bioregulation of the kallikrein-related peptidases family in the human neutrophil. Innate Immun 21:575–586. doi: 10.1177/1753425914566083. [DOI] [PubMed] [Google Scholar]
- 17.Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, Steffen I, Choi SY, Park Y, Schneider H, Schughart K, Pohlmann S. 2010. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J Virol 84:10016–10025. doi: 10.1128/JVI.00239-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beaulieu A, Gravel E, Cloutier A, Marois I, Colombo E, Desilets A, Verreault C, Leduc R, Marsault E, Richter MV. 2013. Matriptase proteolytically activates influenza virus and promotes multicycle replication in the human airway epithelium. J Virol 87:4237–4251. doi: 10.1128/JVI.03005-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bottcher-Friebertshauser E, Freuer C, Sielaff F, Schmidt S, Eickmann M, Uhlendorff J, Steinmetzer T, Klenk HD, Garten W. 2010. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J Virol 84:5605–5614. doi: 10.1128/JVI.00140-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kesic MJ, Meyer M, Bauer R, Jaspers I. 2012. Exposure to ozone modulates human airway protease/antiprotease balance contributing to increased influenza A infection. PLoS One 7:e35108. doi: 10.1371/journal.pone.0035108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kesic MJ, Hernandez M, Jaspers I. 2012. Airway protease/antiprotease imbalance in atopic asthmatics contributes to increased influenza A virus cleavage and replication. Respir Res 13:82. doi: 10.1186/1465-9921-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC, Raitano AB, Jakobovits A. 2001. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res 61:1686–1692. [PubMed] [Google Scholar]
- 23.Hamilton BS, Gludish DW, Whittaker GR. 2012. Cleavage activation of the human-adapted influenza virus subtypes by matriptase reveals both subtype and strain specificities. J Virol 86:10579–10586. doi: 10.1128/JVI.00306-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu X, Shi Y, Gao F, Xiao H, Wang M, Qi J, Gao GF. 2012. Insights into avian influenza virus pathogenicity: the hemagglutinin precursor HA0 of subtype H16 has an alpha-helix structure in its cleavage site with inefficient HA1/HA2 cleavage. J Virol 86:12861–12870. doi: 10.1128/JVI.01606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leu CH, Yang ML, Chung NH, Huang YJ, Su YC, Chen YC, Lin CC, Shieh GS, Chang MY, Wang SW, Chang Y, Chao J, Chao L, Wu CL, Shiau AL. 2015. Kallistatin ameliorates influenza virus pathogenesis by inhibition of kallikrein-related peptidase 1-mediated cleavage of viral hemagglutinin. Antimicrob Agents Chemother 59:5619–5630. doi: 10.1128/AAC.00065-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fogaca SE, Melo RL, Pimenta DC, Hosoi K, Juliano L, Juliano MA. 2004. Differences in substrate and inhibitor sequence specificity of human, mouse and rat tissue kallikreins. Biochem J 380:775–781. doi: 10.1042/bj20031047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tse LV, Marcano VC, Huang W, Pocwierz MS, Whittaker GR. 2013. Plasmin-mediated activation of pandemic H1N1 influenza virus hemagglutinin is independent of the viral neuraminidase. J Virol 87:5161–5169. doi: 10.1128/JVI.00210-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hammond N, Gusbi N, Sosa P, Fitzner J, Besselaar T, Vandemaelea K, Zhang W. 2015. Review of the 2014-2015 influenza season in the northern hemisphere. Wkly Epidemiol Rec 90:281–296.26050269 [Google Scholar]
- 29.Christiansen SC, Proud D, Sarnoff RB, Juergens U, Cochrane CG, Zuraw BL. 1992. Elevation of tissue kallikrein and kinin in the airways of asthmatic subjects after endobronchial allergen challenge. Am Rev Respir Dis 145:900–905. doi: 10.1164/ajrccm/145.4_Pt_1.900. [DOI] [PubMed] [Google Scholar]
- 30.Casalino-Matsuda SM, Monzon ME, Conner GE, Salathe M, Forteza RM. 2004. Role of hyaluronan and reactive oxygen species in tissue kallikrein-mediated epidermal growth factor receptor activation in human airways. J Biol Chem 279:21606–21616. doi: 10.1074/jbc.M309950200. [DOI] [PubMed] [Google Scholar]
- 31.de Veer SJ, Swedberg JE, Akcan M, Rosengren KJ, Brattsand M, Craik DJ, Harris JM. 2015. Engineered protease inhibitors based on sunflower trypsin inhibitor-1 (SFTI-1) provide insights into the role of sequence and conformation in Laskowski mechanism inhibition. Biochem J 469:243–253. doi: 10.1042/BJ20150412. [DOI] [PubMed] [Google Scholar]
- 32.Matrosovich M, Matrosovich T, Garten W, Klenk HD. 2006. New low-viscosity overlay medium for viral plaque assays. Virol J 3:63. doi: 10.1186/1743-422X-3-63. [DOI] [PMC free article] [PubMed] [Google Scholar]