

ABSTRACT
Newcastle disease virus (NDV) is an oncolytic virus being developed for the treatment of cancer. Following infection of a human ovarian cancer cell line (OVCAR3) with a recombinant low-pathogenic NDV, persistent infection was established in a subset of tumor cells. Persistently infected (PI) cells exhibited resistance to superinfection with NDV and established an antiviral state, as demonstrated by upregulation of interferon and interferon-induced genes such as myxoma resistance gene 1 (Mx1) and retinoic acid-inducing gene-I (RIG-I). Viruses released from PI cells induced higher cell-to-cell fusion than the parental virus following infection in two tumor cell lines tested, HT1080 and HeLa, and remained attenuated in chickens. Two mutations, one in the fusion (F) protein cleavage site, F117S (F117S), and another in hemagglutinin-neuraminidase (HN), G169R (HN169R), located in the second sialic acid binding region, were responsible for the hyperfusogenic phenotype. F117S improves F protein cleavage efficiency, facilitating cell-to-cell fusion, while HN169R possesses a multifaceted role in contributing to higher fusion, reduced receptor binding, and lower neuraminidase activity, which together result in increased fusion and reduced viral replication. Thus, establishment of persistent infection in vitro involves viral genetic changes that facilitate efficient viral spread from cell to cell as a potential mechanism to escape host antiviral responses. The results of our study also demonstrate a critical role in the viral life cycle for the second receptor binding region of the HN protein, which is conserved in several paramyxoviruses.
IMPORTANCE Oncolytic Newcastle disease virus (NDV) could establish persistent infection in a tumor cell line, resulting in a steady antiviral state reflected by constitutively expressed interferon. Viruses isolated from persistently infected cells are highly fusogenic, and this phenotype has been mapped to two mutations, one each in the fusion (F) and hemagglutinin-neuraminidase (HN) proteins. The F117S mutation in the F protein cleavage site improved F protein cleavage efficiency while the HN169R mutation located at the second receptor binding site of the HN protein contributed to a complex phenotype consisting of a modest increase in fusion and cell killing, lower neuraminidase activity, and reduced viral growth. This study highlights the intricate nature of these two mutations in the glycoproteins of NDV in the establishment of persistent infection. The data also shed light on the critical balance between the F and HN proteins required for efficient NDV infection and their role in avian pathogenicity.
KEYWORDS: NDV, Newcastle disease virus, fusion, paramyxovirus, persistent infection
INTRODUCTION
Newcastle disease virus (NDV) is a negative-stranded RNA virus belonging to the Paramyxoviridae family which infects avian species, with results ranging from subtle to fatal disease. The severity of the infection is dependent on the virulence of the virus in its host avian species: velogenic (highly virulent), mesogenic (moderately virulent), or lentogenic (avirulent). Mesogenic and velogenic NDVs have potent oncolytic properties whereby they efficiently infect, replicate, and kill tumor cells while sparing normal cells (1–3). The susceptibility of tumor cells to NDV or other oncolytic viruses is likely attributed to changes that have arisen during oncogenesis, including defects in host interferon pathways resulting in an ineffective antiviral response and a concomitant increase in susceptibility to viral infection (4, 5). In a previous study, we generated a candidate oncolytic NDV based on a mesogenic NDV 73T strain by reducing avian pathogenicity without compromising the oncolytic potency of the virus (6).
NDV contains two membrane glycoproteins, hemagglutinin-neuraminidase (HN) and the fusion (F) protein, which are required for viral attachment, entry, and release. The F protein is synthesized as an inactive precursor (F0) which must be proteolytically cleaved by host cell proteases during transport to the cell surface to yield F1 and F2 proteins, which enable fusion of the viral envelope with the host cell plasma membrane. The presence of multiple basic amino acid residues in the F protein cleavage site (FPCS) of mesogenic and velogenic NDV strains results in very efficient F protein cleavage that drives virulence in chickens. In contrast, viruses with a single basic amino acid residue at the FPCS require exogenous proteases for their growth in tissue culture and are also nonvirulent in their host species in vivo (7). During viral infection, the binding of the HN protein to its cognate receptor, cellular sialic acid, causes a conformational change that facilitates the specific homotypic interaction between F and HN, triggering fusion of the viral envelope with the host cell membrane (7–9). After the release of the viral ribonucleoprotein complex into the cytoplasm, the nucleocapsid protein (NP), phosphoprotein (P), and large (L) protein polymerase complex initiate viral RNA genome transcription and replication, viral protein synthesis, and finally virion assembly/egress mediated through interaction between the matrix protein (M) and F/HN glycoproteins (10). To circumvent antiviral responses mediated by interferon (IFN), many paramyxoviruses, including NDV, encode an interferon antagonist protein. The NDV V protein, synthesized from gene editing of P, targets host cellular RNA helicase MDA-5 to inhibit IFN production (11).
NDV preferentially infects and kills tumor cells via apoptosis (10, 12, 13). Similar to other RNA viruses such as measles virus (14), Sendai virus (SeV) (15), mumps virus (16), vesicular stomatitis virus (17, 18), parainfluenza virus type 3 (PIV3) (19), and respiratory syncytial virus (RSV) (20), NDV can establish persistent infection under some circumstances, as reported previously (15, 21, 22). For establishment of persistent infection in vitro, there is normally a fine dynamic equilibrium between virus infection and viral clearance by host innate immune responses. Viruses isolated from persistently infected (PI) cells have genomic mutations leading to the generation of temperature-sensitive mutants or defective interfering particles (DIPs) (also termed defective viral genomes [DVGs]), as shown for Sendai virus (23), measles virus (14), and RSV (24). The PI cells display some of the following phenotypes: reduced viral release (21), resistance to superinfection (25), and resistance to exogenous IFN (26). Studies of persistent NDV infection conducted in the 1960s and 1970s were limited in describing the characteristics of PI cultures and could not explain the molecular mechanism(s) that caused establishment of persistent infection due to a lack of viral genomic sequence information and the lack of a reverse genetics system to dissect genetic determinants. A recent study reported that a velogenic NDV strain can establish persistent infection in a colorectal cancer cell line (27). The virus causing persistent infection was shown to exhibit a smaller plaque morphology and reduced ability to kill other cancer cells, but the genetic determinants of these phenotypes were not studied.
In this study, we describe the generation of a persistently infected tumor cell line following NDV infection. We demonstrate that the PI cells are in a constant antiviral state due to constitutive expression of a low level of IFN. Additionally, the virus recovered from the PI cells is highly fusogenic. The mutations responsible for this phenotype were identified to be in both the F and HN proteins, and their contribution to the hyperfusogenic phenotype and pathogenicity in chickens is described.
RESULTS
NDV establishes persistent infection in an ovarian cancer cell line.
NDV selectively replicates and kills a number of human cancer cell lines in vitro (13, 28, 29). The ovarian cancer cell line OVCAR3 was susceptible to NDV infection, and the majority of the cells died from viral infection. However, a small percentage of cells were found to survive viral infection several days after infection with a recombinant NDV (rNDV) containing a green fluorescent protein (GFP) transgene, rNDV-GFP, (6) at a low multiplicity of infection (MOI). The surviving cells expressed GFP without obvious cytopathic effect (CPE) and were termed PI cells. The PI cells were maintained in cell culture, along with uninfected OVCAR3 cells (control cells), and passaged when the cells reached confluence. After continuous passage of the PI cells for about 2 months (around passage 10), up to 105 PFU/ml of infectious virus could be recovered from cell culture supernatants. A small percentage of cells (1 to 5%) maintained expression of GFP in early passages. Upon further passaging, there was a gradual loss of GFP expression, and by about passage 25, the viral titer decreased to 103 to 104 PFU/ml of culture medium. This level of viral productivity was maintained up to passage 40.
Characteristics of persistently infected OVCAR3 cells.
Although the PI cells were able to produce low levels of recoverable virus up to 104 PFU/ml, the lack of GFP expression in the majority of the cells implied that a large percentage of cells were uninfected but resistant to infection or infected but lost GFP expression. To examine whether the GFP-negative cells within the PI cell culture could be infected, the PI cells and control cells were infected with rNDV-GFP. As shown in Fig. 1A, a majority of the control cells were infected with rNDV-GFP (MOI of 3), as indicated by GFP-positive cells (top right panel). In contrast, the PI cells had a constitutively low level of GFP expression that did not increase upon superinfection with rNDV-GFP, suggesting that PI cells are resistant to superinfection (Fig. 1A, bottom panels). Next, the ability of the PI cells to support replication of rNDV-GFP was examined. Viral titers from PI cells reinfected with rNDV-GFP at an MOI of 0.001 remained low, from 50 PFU/ml on day 0 to 133 PFU/ml on day 4, in this study (Fig. 1B). The control cells supported virus growth, with peak titers on day 2 postinfection (p.i.) at ∼105 PFU/ml. The viability of PI cells and control cells upon infection with rNDV-GFP at MOIs of 0.01, 0.1, and 1 was measured (Fig. 1C). At all MOIs tested, the PI cells were viable, in contrast to the control cells whose viability decreased with increasing MOI (with only 8% cells alive at an MOI of 1 on day 3 postinfection). The PI cells were also resistant to superinfection with a heterologous virus, vesicular stomatitis virus (data not shown). Thus, the PI cells constitutively release a low level of virus and are resistant to superinfection by either homologous or heterologous virus.
FIG 1.

PI cells were resistant to superinfection with NDV. (A) Control cells or PI cells were either mock infected or infected with rNDV-GFP at an MOI of 3. At 24 h p.i., GFP expression was monitored by GFP fluorescence. (B) Control cells or PI cells were infected at an MOI of 0.001 in triplicates, their supernatants were collected from day 0 to day 4, and titers were determined by plaque assay. (C) Control cells or PI cells were either mock infected or infected at an increasing MOI in triplicates for 3 days. The percentage of dead cells relative to mock cells is plotted as means ± standard errors of the means. Data shown are from one study representative of two independent experiments.
PI cells exhibit an antiviral state.
The PI cells were next examined for their antiviral status by examining IFN levels and IFN-induced protein expression. As depicted in Fig. 2, PI cells constitutively produced a low level of type I IFN-β. As expected, depletion of IFN by addition of anti-IFN-β antibody to PI cells reduced their antiviral state, making them more susceptible to NDV superinfection. The control cells had a very low level of IFN-β without infection but produced a high level of IFN-β (>300 pg/ml) upon infection with rNDV-GFP. Addition of 1,000 U of anti-IFN-β antibody to the control cells 24 h prior to NDV infection reduced IFN-β production to a level similar to that of uninfected control cells. PI cells and superinfected persistent cells had similarly low levels of IFN-β, at approximately 70 pg/ml. Viral titers from untreated and anti-IFN-β antibody-treated PI cells were compared with those of the control cells infected with rNDV-GFP. On day 2 postinfection, the control cells produced viral titers of ∼2 × 104 PFU/ml, and the viral titer increased 2-fold in the presence of an anti-IFN-β antibody (blue versus red bars in control cells). In contrast, PI cells produced viral titers of ∼1 × 103 PFU/ml, which remained unchanged upon NDV reinfection. Addition of anti-IFN-β antibody to PI cells increased viral production by 14-fold to a titer of 1.4 × 104 PFU/ml, indicating an increased susceptibility to infection upon neutralization of IFN-β.
FIG 2.

Neutralization of IFN-β in PI cells. Control cells or PI cells were pretreated with antibody against IFN-β at 24 h prior to infection with rNDV-GFP. Supernatants were harvested at 48 h p.i. and tested for their levels of IFN-β by ELISA (top). Titers of these supernatants were determined by plaque assay (bottom). The data shown are means of triplicates. Error bars represent standard errors of the means.
Next, the expression levels of IFN-induced genes and viral genes were examined by Western blotting (Fig. 3A). Retinoic acid-inducing gene I (RIG-I), a cytosolic viral RNA sensor that initiates the signals required for inducing an effective antiviral response (30), was significantly increased in control cells that were infected with NDV but was constitutively expressed in PI cells with and without NDV superinfection. A similar expression pattern was observed for other antiviral proteins examined: melanoma differentiation associated factor 5 (MDA5), another RIG-I-like receptor (RLR) which also senses viral RNA (vRNA) but with some distinct as well as redundant signals as RIG-I (31), and myxoma resistance gene 1 (Mx1), an interferon-stimulated gene (ISG) induced early during an IFN response that acts to inhibit viral replication. Expression of a phosphorylated form of interferon regulatory factor 3, pIRF3, a key inducer of IFN-β and the ISGs (32–35), was induced upon viral infection of control cells. pIRF3 was not detected in the PI cells because the PI cells were already expressing IFN and ISGs at high levels, and pIRF3 gets activated only transiently until IFN-β and ISGs are made (32, 33). As expected, IRF3, the inactive latent form of pIRF3, was expressed to similar levels in infected and uninfected control cells and in PI cells with and without superinfection. The PI cells expressed low levels of NDV proteins F, HN, NP, and M compared to control cell levels, which did not increase upon superinfection with rNDV-GFP (Fig. 3B). The ratio of HN/NP of superinfected persistent cells was 0.16 versus 0.24 for infected control cells (Fig. 3B). The presence of low levels of IFN and the associated ISGs in the PI cell culture may contribute to an antiviral state resulting in resistance of PI cells to superinfection. In addition, the expression of low levels of viral glycoproteins on PI cells might render them less visible to the host immune system and enable a persistent infection.
FIG 3.

PI cells were at an antiviral state. (A) PI cells or control cells were either mock infected or infected with rNDV-GFP at an MOI of 1. At 24 h p.i., cell lysates were harvested and tested for expression of ISGs as indicated. (B) Viral gene expression to detect F, HN, NP, and M proteins. The actin, NP, and M blots contain 20% of the input amount of lysate used for the F and HN blots. The data and images shown are representative of at least two independent experiments. The HN-to-NP ratio of each virus is indicated.
Virus from PI cells was more fusogenic.
Since the PI cells produced a low level of virus in the presence of constitutively expressed IFN, the virus from PI cells may have evolved to adapt to this type of selective immune pressure. Virus collected from PI cell supernatants after 12 to 30 passages was termed NDVpi. Growth of NDVpi was compared with that of rNDV-GFP in HT1080 cells, a human fibrosarcoma cancer cell line highly sensitive to NDV-mediated cell killing (Fig. 4A). NDVpi grew to a titer of 0.75 × 103 PFU/ml in HT1080 cells while the titer of rNDV-GFP was approximately 250-fold higher at 1.88 × 105 PFU/ml. However, microscopic examination of infected HT1080 cells revealed the presence of strikingly large syncytia in NDVpi-infected cultures (Fig. 4B). NDVpi also did not replicate well in OVCAR3 cells, with a peak titer of less than 103 PFU/ml (data not shown). To examine genetic changes that might have occurred in these persistently infecting viruses, NDVpi from PI cell passages 12 and 18 (NDVpi-P12, and NDVpi-P18, respectively) were purified by two rounds of plaque isolation, and the viral genomic sequences of three representative plaque isolates were determined. Among the mutations found in the three isolates, several were unique to each virus and thus might have occurred randomly; thus, these were not investigated further. Interestingly, there were seven mutations that were common among all three plaque isolates (Table 1): three in NP (N47A, M130I, and A154S), one in F (F117S), one in HN (G169R), and two in L (I425V and S1098F). Since the hyperfusogenic phenotype was observed in the three plaque-purified viruses in HT1080, HeLa, and Vero cells (data not shown), the common mutations in the F and HN glycoproteins were considered to have most likely contributed to the hyperfusogenic phenotype of NDVpi (36). The F117S mutation in the F (F117S) protein is located at the F protein cleavage site, and the G169R mutation in the HN (HN169R) protein is located at the second sialic acid binding region of the HN protein (37, 38).
FIG 4.

NDVpi was fusogenic compared to rNDV-GFP. (A) NDVpi and the parental virus rNDV-GFP were used to infect fresh HT1080 cells at an MOI of 0.001. The supernatants from HT1080 cells were harvested daily for 3 days. Viral titers were measured by plaque assay. (B) GFP fluorescence images of HT1080 cells infected with either NDVpi or rNDV-GFP at an MOI of 0.001 at 24 h p.i.
TABLE 1.
Mutations commonly observed in three virus isolates that cause persistent infectionsa
| Gene | Nucleotide change | Amino acid position | Amino acid change |
|---|---|---|---|
| NP | A261C | 47 | Asp → Ala |
| G511A/T | 130 | Met → Ile | |
| G581T | 154 | Ala → Ser | |
| F | T5649C | 117 | Phe → Ser |
| HN | G7672A | 169 | Gly → Arg |
| L | A10607G | 425 | Ile → Val |
| C12627T | 1098 | Ser → Phe |
Full-length genomes of three plaque-purified virus isolates were sequenced, and the mutations that were conserved in all three isolates are listed. Mutations in F and HN characterized in this study are italicized.
The individual contributions of the F117S and HN169R mutations and their combined effect on the fusion phenotype were examined by transiently expressed F and HN proteins harboring these mutations in a dual split-protein (DSP) reporter assay that quantitatively measures cell-to-cell fusion. The F and HN sequences used here were derived from rNDV-GFP as described previously (6). As shown in Fig. 5A, coexpression of F and HN resulted in a 4-fold increase in the cell-to-cell fusion activity compared to that of the empty vector (EV) control, whereas HN169R improved fusion by 3-fold, 12-fold more than the EV control. F117S paired with HN or HN169R increased fusion activity by 27-fold or 39-fold, respectively. These data indicated that F117S and HN169R each contributed to the hyperfusogenic phenotype and that the combined effects of the two mutations were synergistic. The fusion phenotype was also evaluated visually in 293T cells transfected with a bicistronic expression plasmid encoding GFP and F or HN or their mutant versions (Fig. 5B). Similar to the data obtained from the quantitative fusion assay, expression of F117S or HN169R alone slightly increased cell-to-cell fusion compared to the level with the parental F and HN sequences, while coexpression of F117S and HN169R proteins had a maximal effect on syncytium formation. Additionally, lysates from cells expressing different HN protein variants were examined for neuraminidase (NA) activity (Fig. 5C). The HN169R mutation caused a 3-fold reduction in NA activity compared to HN, implicating a role for residue 169 in NA enzymatic activity.
FIG 5.

The HN169R mutation caused higher fusion than HN. (A) A fusion assay was performed on 293T cells transfected with the indicated plasmids that coexpress enhanced GFP. (B) GFP images were taken at 24 h posttransfection. (C) An NA assay was performed on 293T cells transfected using the indicated HN version of the plasmids. Data are representative of two or three independent experiments.
Contribution of the F117S and HN169R mutations to viral phenotype.
To address the impact of the F and HN mutations on virus phenotype, rNDVs incorporating either the single F117S (rNDV-F117S) or HN169R (rNDV-HN169R) change or both changes (rNDV-F117S/HN169R) were generated by reverse genetics. The F and HN gene sequences of these recombinant NDVs were confirmed by sequencing. The 73T backbone containing the granulocyte-macrophage colony-stimulating factor (GM-CSF) transgene described in Cheng et al. (6) was termed rNDV. The single and double mutant viruses were next compared with rNDV for their phenotypes in HT1080 and HeLa cells (Fig. 6). In HT1080 cells, rNDV-F117S and rNDV-F117S/HN169R caused larger syncytia than rNDV and rNDV-HN169R. But in HeLa cells, syncytia were obvious only in rNDV-F117S/HN169R-infected cells (Fig. 6A). Thus, the double mutant was the most effective in inducing cell-to-cell fusion, while the F117S and HN169R mutation-containing viruses varied in their potential to cause syncytia depending on the cell type. Next, the ability of these rNDVs to induce cell killing in HT1080 and HeLa cells was determined (Fig. 6B). The HT1080 cell line is more sensitive to NDV-mediated cell death than HeLa cells. In HT1080 cells, the single mutants, rNDV-F117S and rNDV-HN169R, or the double mutant, rNDV-F117S/HN169R, showed higher cell killing than rNDV, with rNDV-F117S/HN169R being the highest inducer of cell death. In HeLa cells, rNDV-F117S and rNDV-F117S/HN169R but not rNDV-HN169R had higher cell killing than rNDV. Thus, similar to the data shown in Fig. 6A, the single mutants varied in their abilities to kill the two tumor cells, while the double mutant consistently induced greater cell death than the single mutants or rNDV.
FIG 6.

Characterization of recombinant viruses with F117S and HN1169R mutations. Recombinant viruses that contained either F117S or HN169R or both were used to infect HT1080 cells or HeLa cells. (A) Bright-field images were acquired with an EVOS microscope at 14 h p.i. in cells infected at an MOI of 1. (B) Cell killing was measured using a CellTiter-Glo assay on day 2 postinfection at an MOI of 0.1. ns, not significant.
The kinetics of viral growth of the rNDV-F117S, rNDV-HN169R, and rNDV-F117S/HN169R viruses in HT1080 and HeLa cells were compared with those of rNDV. As shown in Fig. 7, rNDV-F117S had growth kinetics and peak titers on day 2 similar to those of rNDV in both cell types when cells were infected at a low MOI of 0.001 to allow for multistep growth cycles. rNDV-F117S grew to 2.8 × 105 PFU/ml in HT1080 cells and to 1.7 ×106 PFU/ml in HeLa cells, which was 2- to 3-fold higher than the titers obtained for rNDV. In contrast, rNDV-HN169R grew only to a low viral titer in both HT1080 (1.6 × 103 PFU/ml) and HeLa cells (1.3 × 104 PFU/ml). rNDV-F117S/HN169R had slow growth kinetics and the lowest titers in both cell lines. Poor growth of the double mutant was more pronounced in HT1080 cells than in HeLa cells, reflecting the increased cell death observed with the double mutant compared to that in the single mutants, as shown in Fig. 6B. Analysis of viral proteins in infected HT1080 and HeLa cells by Western blotting (Fig. 8A) showed that F117S improved the F protein cleavage efficiency, with the dominant cleaved F1 subunit detected in the cells infected with rNDV-F117S and rNDV-F117S/HN169R. Total F protein expression (F0 plus F1) in cells infected with rNDV-HN169R and rNDV-F117S/HN169R was reduced compared to that observed in rNDV-infected HeLa and HT1080 cells. rNDV-F117S had levels of total F protein expression similar to those of rNDV in both cell lines. Relative to rNDV, rNDV-HN169R-infected HeLa and HT1080 cells had overall lower levels of viral proteins in both cell lines. The double mutant rNDV-F117S/HN169R had a protein expression profile similar to that of rNDV with the exception of more efficient F protein cleavage.
FIG 7.

Growth characteristics of recombinant viruses with F117S and HN1169R mutations. Recombinant viruses that contained either F117S or HN169R or both were used to infect HT1080 or HeLa cells. Viral supernatants were harvested at day 0 to day 3, and titers were determined by plaque assay. Data shown represent means ± standard errors from three replicate samples.
FIG 8.

Characterization of recombinant viruses with F117S and HN169R mutations. Recombinant viruses that contained either F117S or HN1169R or both were used to infect HT1080 cells or HeLa cells. (A) Cell lysates were harvested from cells infected at an MOI of 1 at 9 h postinfection. Viral gene expression was detected by Western blotting. The data shown are representative of at least three experiments. (B) An NA assay was performed by harvesting lysates from infected cells at 24 h p.i.
Impact of HN169R on HN protein function.
The effect of HN169R mutation on the hemagglutinin (HA) and neuraminidase (NA) activity of the HN protein was examined in infected HT180 and HeLa cells (Fig. 8B). rNDV-F117S-infected cells had NA activity comparable to that of rNDV-infected cells. However, the NA activity of rNDV-HN169R in infected cells was greatly reduced compared to that of rNDV (∼23-fold reduction in HT1080 and ∼15-fold reduction in HeLa cells). The NA activity of rNDV-F117S/HN169R in infected cells was much lower than that of rNDV but higher than that of rNDV-HN169R in both cell lines, which was partly reflected by the larger amount of HN protein produced in rNDV-F117S/HN169R-infected cells than in rNDV-HN169R-infected cells (Fig. 8A). It is also likely that HN169 impacts the interaction of F and HN on the cell surface while the rNDV-F117S/HN169R double mutant provides a counterbalancing act provided by the F117S.
The receptor binding ability of the four viruses amplified from HeLa cells was examined by hemagglutination of chicken red blood cells (cRBCs). At the same starting titer of 4 × 107 PFU/ml, rNDV and rNDV-F117S had the same HA titer of 16, while the HA titer of NDV-HN169R was 4-fold lower and the HA titer of the double mutant rNDV-F117S/HN169R was not measurable (data not shown). Thus, the 169R mutation in the HN protein reduced neuraminidase and receptor binding activity.
The impact of F and HN mutations on viral pathogenicity.
Recombinant NDV (rNDV) used for in vitro passage contained a GFP transgene in the P-M intergenic region in addition to the modified F cleavage site and a 198-nucleotide (nt) insertion in the HN-L intergenic region as described previously (6). rNDV is attenuated in chickens with an intracerebral pathogenicity index (ICPI) of 0.13 (Table 2). Mutations that occurred in persistently infecting viruses did not change viral pathogenicity, as plaque-purified viral isolates recovered from passage 12 (rNDVpi-P12) and passage 18 (rNDVpi-P18) remained attenuated in chickens, with ICPI values of 0.0 and 0.1, respectively. The F and HN mutations were further evaluated for their impact on viral pathogenicity using recombinant viruses bearing single or double mutations. rNDV-F117S alone significantly increased viral pathogenicity, with an ICPI value of 1.05. However, viruses with a single HN mutation or double F and HN mutations had low pathogenicity, with ICPI values of 0.20 or 0.25, respectively (Table 2). These data confirmed that the F protein cleavage efficiency is an important virulence factor; the G169R mutation in the HN protein counteracted the virulent nature of the F protein, explaining the nonvirulent nature of the persistently infecting viruses isolated from viral passage.
TABLE 2.
Chicken pathogenicity of persistently infecting isolates and recombinants with a substitution in F and/or HN
| Virus | FPCSa | HN residue 169 | ICPIb |
|---|---|---|---|
| rNDV | HNRTKR/F117 | G | 0.13 |
| NDVpi-P12 | HNRTKR/S117 | R | 0.0 |
| NDVpi-P18 | HNRTKR/S117 | R | 0.1 |
| rNDV-F117S | HNRTKR/S117 | G | 1.05 |
| rNDV-G169R | HNRTKR/F117 | R | 0.20 |
| rNDV-F117S/G169R | HNRTKR/S117 | R | 0.25 |
FPCS, F protein cleavage sequence. The substituted residue is in boldface.
Intracerebral pathogenicity index (ICPI) of each virus was determined in 1-day-old pathogen-free chicks. A strain with an ICPI value of <0.7 is attenuated and not considered as a select agent.
DISCUSSION
In this study, we have described the establishment of a persistent NDV infection in an ovarian cancer cell line, OVCAR3. The PI cell line constitutively produced a low level of virus in the presence of small amounts of IFN, reflecting a fine balance between viral replication and innate immune surveillance. The PI cells exhibited a heightened antiviral state, as evidenced by constitutive expression of type I IFN and IFN-induced antiviral proteins such as RIG-I, MDA5, and Mx1 (Fig. 2 and 3). Activation of IRF3 by RIG-I has been shown to be essential for preventing establishment of viral persistence for Sendai virus (SeV) (39), which is different from what we observed for NDV persistent infection since the control cells are capable of IRF3 activation, as demonstrated by pIRF3 expression (Fig. 3A). Our attempts to establish persistent infections in several other human cancer cell lines such as OAW28, CAL27, FaDu, and PE/CA PJ15 were futile, indicating that not all cancer cells are susceptible to persistent NDV infection (data not shown). The persistently infecting virus from passage 18 had a very low titer in OVCAR3 cells, suggesting its replication deficiency in general. The establishment of persistent infection requires changes of not only virus but also host cell adaptation to the persistently infecting viruses. Persistent infections occur only under certain conditions where the cells are unable to completely clear a virus infection. This might be the case for some human cancer cells that have defects in IFN production and/or IFN response pathways (40). Based on the data shown in Fig. 2 and 3, the parental OVCAR3 cells (control cells) were capable of producing IFN and inducing several ISGs examined. Therefore, the contribution of IFN and individual ISGs in the establishment of persistent infection appears complex and has yet to be determined. It is also likely that other cellular pathway defects might contribute to the establishment of persistent infection.
Persistent infections by RNA viruses such as measles virus, human parainfluenza virus, SeV, and dengue virus have been associated with the development of DIPs or DVGs (14, 24, 41). DIPs are usually produced during viral replication at a high MOI in vitro and lack essential genes to sustain viral replication in the absence of a helper virus (42). By using a PCR methodology used to detect copy-back DIPs in measles and SeV (14, 23), we could detect very few or no copy-back DIPs in persistently NDV-infected cells (data not shown). Thus, DIPs do not appear to play a role in NDV persistent infection, and the persistent infection was established most likely due to the virus escaping an IFN-mediated antiviral response.
Virus from NDV PI cells had a hyperfusogenic phenotype due to the changes found in both the F and HN glycoproteins, F117S and HN169R. Highly fusogenic variants of paramyxoviruses from persistently infected cells have been reported previously. In a simian virus 5 (SV5) persistent infection of mouse fibroblast cells that are capable of producing and responding to IFN, 5 to 20% of the cells were persistently infected at any given point of time. Upon culture of these PI cells, fusogenic variants were identified which were capable of responding to IFN but spread more efficiently than the parental virus in the absence of IFN. Additionally, these fusogenic persistently infecting variants did not have mutations in the F protein, and the HN sequence was not reported (43). Bioselection by in vivo passage of the NDV MTH87 strain in human xenograft-bearing mice resulted in several isolates with increased oncolytic activity and increased fusogenicity. The isolate with highest fusogenicity had changes in the HN protein (T118A, N119K, E259A, and E495K) but not in the F protein (44), indicating a contribution of HN mutations to the fusion phenotype as observed in our study. In a study of persistent SeV infection, Nishio et al. identified several changes in the L (45), M (46), or P (47) protein that contributed to persistent infection and to a temperature-sensitive phenotype. Knockout of the accessory C protein of measles virus led to the generation of more copy-back DIPs than generated by the parental virus (14). In our study, the NDV V protein, which is an IFN antagonist, contained a mutation (T164A) in one persistently infecting isolate but was not observed in other isolates and thus may not play a role in the establishment of persistent infection. There were about 25 additional unique sense mutations observed in the entire viral genome and the GFP transgene (which explains the loss of GFP expression) among the three viral plaque isolates from PI cells. These mutations were not unexpected because single-stranded RNA viruses like NDV lack proofreading ability during viral replication. The mutations that were commonly found in all three isolates were expected to most likely contribute to the establishment of persistent infection. Due to the hyperfusogenic nature of all three of the virus isolates from persistent infection, we focused on characterizing the mutations in F and HN which are sufficient and necessary for inducing fusion. The importance of mutations found in the other genes for establishing persistent NDV infection was not investigated in this study.
The role of the individual mutations in the F and HN proteins on viral phenotype provides insights into the intricate relationship between these two integral membrane proteins. The F117S change at the FPCS improved F protein cleavage efficiency and fusion phenotype. The impact of the HN169R mutation appears more complex since it resulted in a modest increase in fusion, reduced F and HN protein expression, and decreased neuraminidase and hemagglutination activity. The double mutant rNDV-F117S/HN169R containing both mutations found in the persistently infecting virus isolated from OVCAR3 cells exhibited hyperfusion and reduced replication but high cell-killing capabilities despite having reduced HA and NA activities. The replication defect with rNDV-HN169R was more pronounced in HT1080 cells than in HeLa cells (Fig. 7), in line with the higher sensitivity of HT1080 cells to NDV-mediated cell killing. Although the persistently infecting virus with the identified F117S and HN169R mutations persisted in PI cells that constitutively make IFN, there was no statistically significant difference in the amount of type I IFN produced by the different recombinant viruses with a single F/HN mutation or double mutations (data not shown). These data indicate that the F and HN mutations do not affect IFN production and that IFN levels are not the cause of the defective replication of rNDV-HN169R. HeLa cells are known to produce a very low level of IFN upon NDV infection and fail to induce ISGs unless prestimulated by treatment with either IFN-α or IFN-γ (6, 48, 49). HT1080 can produce IFN but is unable to form the IFN-stimulated gene factor 3 (ISGF-3) complex required for a sustained antiviral effect (28). Thus, the differences in viral replication observed with these two cell lines may not be attributed to the differences in their interferon pathways.
NDV HN 169 residue has been previously identified to be important for the fusion activity in a transient expression system through scanning alanine mutagenesis (47). The HN G169A mutation increased fusion by 60% and decreased the NA activity by 25% although the mutation was an alanine instead of arginine discovered in our study. Based on structural and functional studies, NDV HN is known to contain two sialic acid binding sites (Fig. 9A): the primary binding site encompasses both the NA active site and receptor binding activity (50), while the second site (site II) was shown to possess receptor binding activity only (38) through a network of polar interactions mediated by residues 156, 169, 170, and 515 to 519, as shown in Fig. 9B, and has been implicated in fusion function (51). Residue G169 is located at the HN dimer interface, which is part of a hydrophobic pocket containing L552 and F553 from one monomer and F156, V517, and L561 of the other monomer that together make up site II (38, 52). The G169 residue also makes a main-chain polar contact to the sialic acid ligand. The G169R mutation could drastically affect the site II binding pocket by sterically blocking interaction with the ligand and by altering the binding pocket electrostatics. F553 at site II was also shown to be important for NA activity and fusion promotion in a transfection-based fusion assay, highlighting the importance of site II in both fusion promotion and NA activity (37). A previous study explored the impact of changes at HN residue R516 which forms hydrogen bonds with sialic acid. Viruses containing R516S/A mutations exhibited decreased receptor binding, decreased fusion, and decreased replication but had wild-type NA activity (51). This is different from our finding with the HN169R mutation, which impacted not only viral receptor binding but also NA and fusion activity, altogether resulting in poor viral replication. The G169R mutation located at the HN dimer interface could affect surface charge and impact the primary sialic acid binding site (Fig. 9C). Blocking of the second sialic acid binding site by a C-terminal extension of the HN protein in a low-virulence NDV Ulster strain has been shown to be associated with its dependence on exogenous trypsin for in vitro replication and its low-virulence phenotype (52). In addition to NDV, PIV1 and human PIV3 (hPIV3) have also been demonstrated to contain a second sialic acid binding site, suggesting a potentially conserved mechanism among certain paramyxoviruses (53, 54). Additional biochemical investigation is needed to fully explain the effects of the G169R and second sialic acid binding site on HN protein functions and the interaction of HN with the F protein in the viral fusion phenotype of NDV. Interestingly, although the F117S mutation at the F protein cleavage site increased F protein cleavage efficiency and viral pathogenicity, rNDV F117S/HN169R with the double F and HN mutations remained attenuated in a chicken host, which was consistent with the low-pathogenicity nature of the persistently infecting viral isolates. These data further underscore the importance of the F and HN functional balance in the NDV viral replication cycle. Any effect on viral pathogenicity of mutations identified in the F protein cleavage site and the HN protein should be closely monitored during the oncolytic NDV virotherapy development process.
FIG 9.
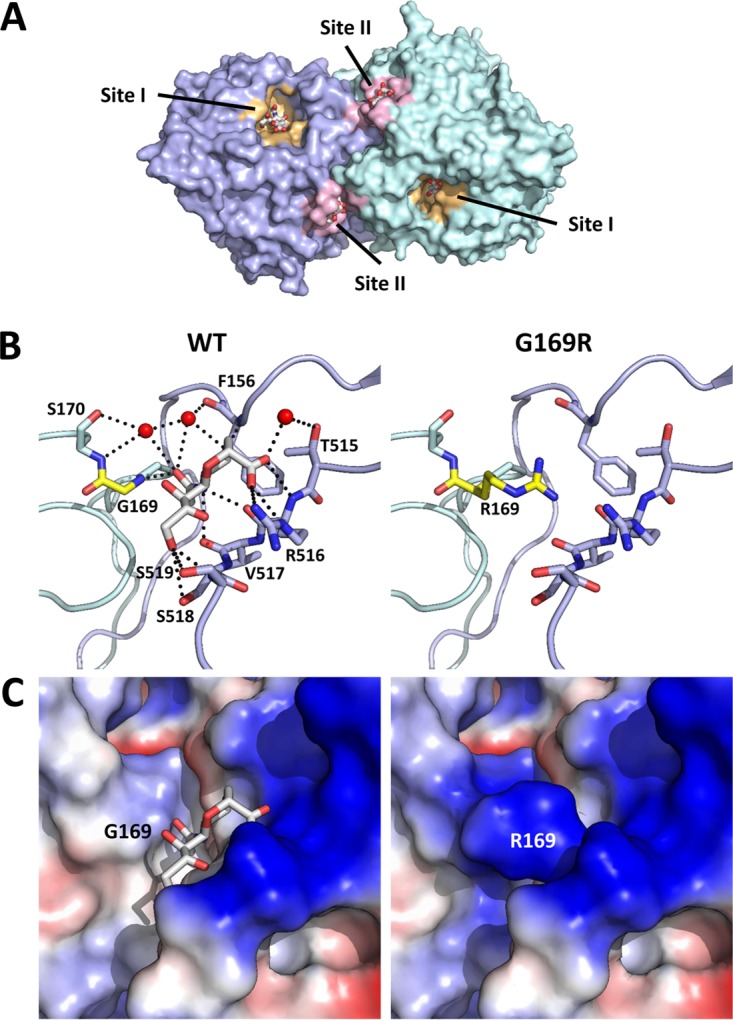
The HN G169R mutation disrupts site II ligand binding. (A) Surface representation of HN shows residue 169 (yellow) located in site II (pink) at the dimer interface and distal to site I (orange). Residues of site I and site II binding pockets within 6 Å of the respective ligands are colored similarly. A comparison of site II binding pocket polar interactions (B, dashed black lines) to ligand and surface charge properties (C) between the wild type and the model of the G169R mutation is shown. HN monomeric units are colored individually, and residue 169 is shown in yellow in panel B. Site I and site II ligands (gray) as well as site II sialic acid polar contact residues in panel B are shown as sticks with oxygen and nitrogen atoms shown in red and blue, respectively. Waters directly involved in coordinating ligand residue contacts in panel B are shown as red spheres. Negative and positive charges in panel C are shown in blue and red, respectively. PDB accession number 1USR (38) was used for the HN structure and to model the structure of the G169R mutation, which was generated in PyMol.
In summary, we have shown that NDV can establish persistent infection in an ovarian tumor cell line, and we have characterized the residues that contribute to persistent infection establishment and the fusion phenotype required for viral spread between cells in persistently infected cultures. Further studies enabling a better understanding of the role of the HN169R mutation in contributing to virus biology would greatly aid in unraveling the intricate interaction between the F and HN proteins of NDV. It has yet to be determined whether NDV can establish persistent infection in its natural avian host. NDV may not be able to establish persistent infection in humans where there is a strong antiviral response elicited by the normal cells enabling efficient viral clearance. Studies like ours would be beneficial in understanding the natural mutational course of NDV under selection pressure.
MATERIALS AND METHODS
Cell lines.
NIH-OVCAR3 cells were obtained from the ATCC and maintained in complete RPMI 1640 medium supplemented with 10% gamma-irradiated fetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml penicillin-streptomycin (P-S), and 0.01 mg/ml bovine insulin (Sigma). Vero cells, HT1080 cells, and HeLa cells were also obtained from the ATCC and maintained as follows: Vero cells were grown in Dulbecco's modified essential medium (DMEM) supplemented with 5% FBS, 2 mM l-glutamine, P-S, and 10 mM nonessential amino acids (NEAA); HT1080 cells were grown in minimum essential medium (MEM) with Earle's balanced salt solution (EBSS), supplemented with 2 mM l-glutamine, 10% heat-inactivated FBS, P-S, and 10 mM NEAA; HeLa cells were grown in MEM supplemented with 2 mM l-glutamine, 10% heat-inactivated FBS, and P-S.
Establishment of persistently infected OVCAR3 cells.
PI cells were made by infecting NIH-OVCAR3 cells with rNDV-GFP at an MOI of 0.01 in a T25 flask. When the majority of cells were detached on day 5, the culture flask was washed, the medium was replaced with fresh complete medium (same as NIH-OVCAR3 medium), and the culture was maintained until a subconfluent monolayer was formed. The control cells were maintained in a similar way, except that they were split when they became confluent. Viruses from PI OVCAR3 cells were cloned by plaque assay on Vero cells. Single plaques were picked from infected Vero cells and amplified for genomic sequencing analysis.
Viruses.
rNDV that contains the GM-CSF transgene (termed 73T-R-198 in [6]) and rNDV-GFP viruses have been previously published (6). rNDV-F117S, rNDV-HN169R, and rNDV-F117S/HN169R were made by introducing the corresponding mutations in rNDV using a QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The NDV antigenomic cDNA clones with introduced mutations were used for generating rNDV mutants using a reverse genetics system established previously (6). The F and HN genes of recombinant viruses were completely sequenced to confirm the introduced mutations, and sequencing of RT-PCR-amplified cDNA was used to confirm that there were no unintended mutations.
Cell viability assay.
Cells were plated at 1 × 104 cells per well in 96-well, clear-bottomed, white-walled plates overnight prior to infection with the viruses at a given MOI. At 2 days postinfection, cell viability was assessed by measuring ATP as an indirect readout of living cells using a CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI) per the manufacturer's instructions. Mock-infected cells were set at 100% viable, and data are represented as the percentages of cells killed by deducting the percent viable from 100.
Virus growth and plaque assay to measure viral titers.
Viral growth kinetics were determined by infecting HT1080 or HeLa cells with the corresponding viruses at an MOI of 0.001 in Opti-MEM (Thermo Fisher Scientific, Waltham, MA), and virus supernatants were harvested daily. Viral titers were determined by plaque assay and enumerated by immune staining or crystal violet dye staining (6).
Western blotting and ELISA.
Whole-cell lysates from infected cells were harvested at 24 h (Fig. 3) or 9 h (Fig. 8) by washing cells in ice-cold phosphate-buffered saline (PBS), followed by direct lysis in 2× Laemmli sample buffer (Bio-Rad Laboratories, Inc., Hercules, CA) containing β-mercaptoethanol. Lysates were heated to 100°C and separated on 4 to 20% or 8 to 16% Novex Tris-glycine SDS-PAGE gels (Thermo Fisher Scientific). Western blotting to transfer resolved proteins to polyvinylidene difluoride (PVDF) membranes was done by using a Trans-Blot Turbo transfer system (Bio-Rad Laboratories, Inc.) or wet transfer (Thermo Fisher Scientific). Antibodies to viral proteins F and HN were raised in rabbits; NP and M antibodies were raised in guinea pigs as described previously (6). Antibodies used to detect cellular proteins were rabbit anti-RIG-I, rabbit anti-MDA-5, rabbit anti-pIRF3, rabbit-IRF3 (Cell Signaling Technology), rabbit anti-Mx1 (Abcam), and mouse anti-actin (Sigma). An enzyme-linked immunosorbent assay (ELISA) for IFN-β was done using an ELISA kit (PBL Biosciences ltd) and performed as per the manufacturer's instruction.
Transient expression of F and HN proteins.
NDV F and HN genes were cloned in a dual expression plasmid containing GFP and F or HN (55), termed pVITRO2-GFP-F or pVITRO2-GFP-HN. The F used in this study contained a modification in the FPCS corresponding to 73T-R as described in Cheng et al. (6). The F117S mutation in F and G169R mutation in HN were made by site-directed mutagenesis using a QuikChange site-directed mutagenesis kit and confirmed by sequencing. F and/or HN expression plasmids were transfected into 293T cells in 12-well plates using TransIT-293 reagent (Mirus Bio LLC) as per the manufacturer's instruction.
Fusion assay.
The start codon of GFP in the dual expression plasmid was mutated to a stop codon in order to conduct the fusion assay using a dual split-protein reporter system as described in Hotard et al. (56). Plasmids DSP1–7 and DSP8–11 (57, 58) were kind gifts of Zene Matsuda (Chinese Academy of Sciences, Beijing, China). One day prior to transfection, 293T cells were plated in white-walled 96-well plates so that they would reach 50% confluence at the time of transfection. One set of cells was transfected with DSP1–7, F, and HN plasmids at a ratio of DSP/F/HN of 1:0.5:0.5 for a total of 0.1 μg of DNA/well. A second set was transfected with DSP8–11 and pVITRO2 EV in a 1:1 ratio. At 24 h posttransfection, the medium in the DSP1–7/F/HN-containing cells was replaced with 100 μl of fresh Opti-MEM containing EnduRen live cell substrate (Promega, Madison, WI). DSP8–11/empty vector (EV)-containing cells were treated similarly, harvested by pipetting, and mixed with the DSP1–7/F/HN-containing cells. A base reading of luminescence was taken at time (t) of mixing as t0, and the plates were returned to a 37°C incubator. Luminescence was measured every hour starting at 2 h postmixing using a GloMax Discover System (Promega). Each condition was done at least in triplicates, and the data are represented as fold luminescence of the value at 5 h over the value at t0 and normalized to EV-containing samples.
NA assay.
The neuraminidase(NA) activity of HN was assessed by a fluorometric assay as described previously (59), with minor modifications. Briefly, 293T cells in 12-well plates were transfected with pVITRO2-HN (wild type or the mutants). At 48 h posttransfection, the cell lysates were harvested in PBS containing 1% Triton-X. For virus infections, HeLa or HT1080 cells were infected with respective viruses at an MOI of 1 for 24 h, and cell lysates were harvested as above. Twofold serial dilutions of the lysates were made in an assay buffer containing 0.1 M sodium phosphate, pH 5.9, and 4 mM CaCl2 in triplicates. Fifty microliters of 0.2 mM 2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid (4-MU-NANA) substrate (Sigma, St. Louis, MO) was then added to the samples, and they were incubated at 37°C for 1 h, followed by the addition of 100 μl of 0.5 M glycine (pH 10.7) to stop the reaction. The fluorescence was measured by reading the plate using an excitation wavelength of 355 nm and an emission wavelength of 460 nm on a SpectraMax M Series Multi-Mode microplate reader (Molecular Devices, Sunnyvale, CA). Data are represented as relative fluorescence units (RFU) of samples after subtraction of the RFU value of no-virus/EV-transfected lysates.
HA assay.
A hemagglutination (HA) assay was performed on V-bottomed 96-well microtiter plates using chicken red blood cells (cRBCs). Fifty microliters of 2-fold serially diluted viruses was added to 50 μl of 1× PBS and incubated with 50 μl of 0.5% cRBCs for 60 min at room temperature. The HA titer was obtained by calculating the reciprocal of the highest dilution that agglutinated the cRBCs.
Chicken pathogenicity test.
The pathogenicity of the persistently infecting viruses and recombinant viruses with F and HN mutations at a titer of 2 × 107 PFU/ml was determined by the intracerebral pathogenicity index (ICPI) test in conducted in 1-day-old specific-pathogen-free (SPF) chicks at the USDA's National Veterinary Service Laboratory (NVSL; Ames, IA) (6). NDV strains with an ICPI of <0.7 are defined as not causing Newcastle disease and thus are not classified as a select agent by the USDA.
Structure representations.
All structural renderings of proteins were generated using the PyMOL molecular graphics system, version 1.5.0.4 (Schrödinger, LLC [http://www.pymol.org/]).
ACKNOWLEDGMENTS
We thank Yang He and Lily Yang for providing cultured cells, Jackie Zhao for sequence analysis, Bin Lu for helping with figure preparation, Zene Matsuda for providing the split fusion plasmids, and Nichole Hines at USDA for conducting ICPI tests. We also thank Qi Xu, Zhongying Chen, and Jonathan Travers for discussions and JoAnn Suzich for discussions and critical reviews of the manuscript.
REFERENCES
- 1.Everts B, van der Poel HG. 2005. Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther 12:141–161. doi: 10.1038/sj.cgt.7700771. [DOI] [PubMed] [Google Scholar]
- 2.Schirrmacher V, Fournier P. 2009. Newcastle disease virus: a promising vector for viral therapy, immune therapy, and gene therapy of cancer. Methods Mol Biol 542:565–605. doi: 10.1007/978-1-59745-561-9_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinkovics JG, Horvath JC. 2000. Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol 16:1–15. doi: 10.1016/S1386-6532(99)00072-4. [DOI] [PubMed] [Google Scholar]
- 4.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med 6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 5.Stojdl DF, Lichty BD, ten Oever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275. doi: 10.1016/S1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 6.Cheng X, Wang W, Xu Q, Harper J, Carroll D, Galinski MS, Suzich J, Jin H. 2016. Genetic modification of oncolytic Newcastle disease virus for cancer therapy. J Virol 90:5343–5352. doi: 10.1128/JVI.00136-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorman JJ, Nestorowicz A, Mitchell SJ, Corino GL, Selleck PW. 1988. Characterization of the sites of proteolytic activation of Newcastle disease virus membrane glycoprotein precursors. J Biol Chem 263:12522–12531. [PubMed] [Google Scholar]
- 8.Bose S, Jardetzky TS, Lamb RA. 2015. Timing is everything: fine-tuned molecular machines orchestrate paramyxovirus entry. Virology 479–480:518–531. doi: 10.1016/j.virol.2015.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu R, Palmer SG, Porotto M, Palermo LM, Niewiesk S, Wilson IA, Moscona A. 2013. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo. mBio 4:e00803–. doi: 10.1128/mBio.00803-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samal SK. 2011. Newcastle disease and related avian paramyxoviruses, p 69–114. In Samal SK. (ed), The biology of paramyxoviruses. Caister Academic Press, Poole, United Kingdom. [Google Scholar]
- 11.Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S. 2007. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 12.Alexander DJ. 2000. Newcastle disease and other avian paramyxoviruses. Rev Sci Tech 19:443–462. doi: 10.20506/rst.19.2.1231. [DOI] [PubMed] [Google Scholar]
- 13.Elankumaran S, Rockemann D, Samal SK. 2006. Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death. J Virol 80:7522–7534. doi: 10.1128/JVI.00241-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfaller CK, Mastorakos GM, Matchett WE, Ma X, Samuel CE, Cattaneo R. 2015. Measles virus defective interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA-1-like hypermutations. J Virol 89:7735–7747. doi: 10.1128/JVI.01017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawton P, Karimi Z, Mancinelli L, Seto JT. 1986. Persistent infections with Sendai virus and Newcastle disease viruses. Arch Virol 89:225–233. doi: 10.1007/BF01309891. [DOI] [PubMed] [Google Scholar]
- 16.Walker DL, Hinze HC. 1962. A carrier state of mumps virus in human conjunctiva cells. I. General characteristics. J Exp Med 116:739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westcott MM, Liu J, Rajani K, D'Agostino R Jr, Lyles DS, Porosnicu M. 2015. Interferon beta and interferon alpha 2a differentially protect head and neck cancer cells from vesicular stomatitis virus-induced oncolysis. J Virol 89:7944–7954. doi: 10.1128/JVI.00757-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youngner JS, Dubovi EJ, Quagliana DO, Kelly M, Preble OT. 1976. Role of temperature-sensitive mutants in persistent infections initiated with vesicular stomatitis virus. J Virol 19:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cole FE Jr, Hetrick FM. 1965. Persistent infection of human conjunctiva cell cultures by myxovirus parainfluenza 3. Can J Microbiol 11:513–521. doi: 10.1139/m65-068. [DOI] [PubMed] [Google Scholar]
- 20.Valdovinos MR, Gomez B. 2003. Establishment of respiratory syncytial virus persistence in cell lines: association with defective interfering particles. Intervirology 46:190–198. doi: 10.1159/000071461. [DOI] [PubMed] [Google Scholar]
- 21.Henle G, Deinhardt F, Bergs VV, Henle W. 1958. Studies on persistent infections of tissue cultures. I. General aspects of the system. J Exp Med 108:537–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thacore H, Youngner JS. 1969. Cells persistently infected with Newcastle disease virus. I. Properties of mutants isolated from persistently infected L cells. J Virol 4:244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tapia K, Kim WK, Sun Y, Mercado-Lopez X, Dunay E, Wise M, Adu M, Lopez CB. 2013. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog 9:e1003703. doi: 10.1371/journal.ppat.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Jain D, Koziol-White CJ, Genoyer E, Gilbert M, Tapia K, Panettieri RA Jr, Hodinka RL, Lopez CB. 2015. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog 11:e1005122. doi: 10.1371/journal.ppat.1005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergs VV, Henle G, Deinhardt F, Henle W. 1958. Studies on persistent infections of tissue cultures. II. Nature of the resistance to vesicular stomatitis virus. J Exp Med 108:561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thacore HR, Youngner JS. 1972. Viral ribonuclei acid synthesis by Newcastle disease virus mutants isolated from persistently infected L cells: effect of interferon. J Virol 9:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chia SL, Yusoff K, Shafee N. 2014. Viral persistence in colorectal cancer cells infected by Newcastle disease virus. Virol J 11:91. doi: 10.1186/1743-422X-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krishnamurthy S, Takimoto T, Scroggs RA, Portner A. 2006. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J Virol 80:5145–5155. doi: 10.1128/JVI.02618-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fabian Z, Csatary CM, Szeberenyi J, Csatary LK. 2007. p53-independent endoplasmic reticulum stress-mediated cytotoxicity of a Newcastle disease virus strain in tumor cell lines. J Virol 81:2817–2830. doi: 10.1128/JVI.02490-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 31.Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. 2009. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol 83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18:2986–2996. doi: 10.1128/MCB.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. 1998. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett 425:112–116. doi: 10.1016/S0014-5793(98)00210-5. [DOI] [PubMed] [Google Scholar]
- 34.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J 17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 36.Lamb RA, Kolakofsky D. 2001. Paramyxoviridae: the viruses and their replication, p 1305–1340. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 37.Takimoto T, Taylor GL, Connaris HC, Crennell SJ, Portner A. 2002. Role of the hemagglutinin-neuraminidase protein in the mechanism of paramyxovirus-cell membrane fusion. J Virol 76:13028–13033. doi: 10.1128/JVI.76.24.13028-13033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaitsev V, von Itzstein M, Groves D, Kiefel M, Takimoto T, Portner A, Taylor G. 2004. Second sialic acid binding site in Newcastle disease virus hemagglutinin-neuraminidase: implications for fusion. J Virol 78:3733–3741. doi: 10.1128/JVI.78.7.3733-3741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters K, Chattopadhyay S, Sen GC. 2008. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J Virol 82:3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moscona A. 1991. Defective interfering particles of human parainfluenza virus type 3 are associated with persistent infection in cell culture. Virology 183:821–824. doi: 10.1016/0042-6822(91)91018-C. [DOI] [PubMed] [Google Scholar]
- 42.Lopez CB. 2014. Defective viral genomes: critical danger signals of viral infections. J Virol 88:8720–8723. doi: 10.1128/JVI.00707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young DF, Didcock L, Randall RE. 1997. Isolation of highly fusogenic variants of simian virus 5 from persistently infected cells that produce and respond to interferon. J Virol 71:9333–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beier R, Hermiston T, Mumberg D. 2013. Isolation of more potent oncolytic paramyxovirus by bioselection. Gene Ther 20:102–111. doi: 10.1038/gt.2012.13. [DOI] [PubMed] [Google Scholar]
- 45.Nishio M, Nagata A, Tsurudome M, Ito M, Kawano M, Komada H, Ito Y. 2004. Recombinant Sendai viruses with L1618V mutation in their L polymerase protein establish persistent infection, but not temperature sensitivity. Virology 329:289–301. doi: 10.1016/j.virol.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 46.Nishio M, Tsurudome M, Ito M, Kawano M, Komada H, Ito Y. 2003. Characterization of Sendai virus persistently infected L929 cells and Sendai virus pi strain: recombinant Sendai viruses having Mpi protein shows lower cytotoxicity and are incapable of establishing persistent infection. Virology 314:110–124. doi: 10.1016/S0042-6822(03)00404-5. [DOI] [PubMed] [Google Scholar]
- 47.Nishio M, Nagata A, Yamamoto A, Tsurudome M, Ito M, Kawano M, Komada H, Ito Y. 2006. The properties of recombinant Sendai virus having the P gene of Sendai virus pi strain derived from BHK cells persistently infected with Sendai virus. Med Microbiol Immunol 195:151–158. doi: 10.1007/s00430-006-0012-3. [DOI] [PubMed] [Google Scholar]
- 48.Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marie I, Levy DE. 2004. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc Natl Acad Sci U S A 101:9578–9583. doi: 10.1073/pnas.0400567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blach-Olszewska Z, Halasa J, Matej H, Cembrzynska-Nowak M. 1977. Why HeLa cells do not produce interferon? Arch Immunol Ther Exp (Warsz) 25:683–691. [PubMed] [Google Scholar]
- 50.Crennell S, Takimoto T, Portner A, Taylor G. 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat Struct Biol 7:1068–1074. doi: 10.1038/81002. [DOI] [PubMed] [Google Scholar]
- 51.Bousse TL, Taylor G, Krishnamurthy S, Portner A, Samal SK, Takimoto T. 2004. Biological significance of the second receptor binding site of Newcastle disease virus hemagglutinin-neuraminidase protein. J Virol 78:13351–13355. doi: 10.1128/JVI.78.23.13351-13355.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan P, Paterson RG, Leser GP, Lamb RA, Jardetzky TS. 2012. Structure of the Ulster strain Newcastle disease virus hemagglutinin-neuraminidase reveals auto-inhibitory interactions associated with low virulence. PLoS Pathog 8:e1002855. doi: 10.1371/journal.ppat.1002855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mishin VP, Watanabe M, Taylor G, Devincenzo J, Bose M, Portner A, Alymova IV. 2010. N-linked glycan at residue 523 of human parainfluenza virus type 3 hemagglutinin-neuraminidase masks a second receptor-binding site. J Virol 84:3094–3100. doi: 10.1128/JVI.02331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porotto M, Fornabaio M, Kellogg GE, Moscona A. 2007. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J Virol 81:3216–3228. doi: 10.1128/JVI.02617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cotter CR, Jin H, Chen Z. 2014. A single amino acid in the stalk region of the H1N1pdm influenza virus HA protein affects viral fusion, stability and infectivity. PLoS Pathog 10:e1003831. doi: 10.1371/journal.ppat.1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hotard AL, Lee S, Currier MG, Crowe JE Jr, Sakamoto K, Newcomb DC, Peebles RS Jr, Plemper RK, Moore ML. 2015. Identification of residues in the human respiratory syncytial virus fusion protein that modulate fusion activity and pathogenesis. J Virol 89:512–522. doi: 10.1128/JVI.02472-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 58.Loening AM, Fenn TD, Wu AM, Gambhir SS. 2006. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng Des Sel 19:391–400. doi: 10.1093/protein/gzl023. [DOI] [PubMed] [Google Scholar]
- 59.Lu B, Zhou H, Ye D, Kemble G, Jin H. 2005. Improvement of influenza A/Fujian/411/02 (H3N2) virus growth in embryonated chicken eggs by balancing the hemagglutinin and neuraminidase activities, using reverse genetics. J Virol 79:6763–6771. doi: 10.1128/JVI.79.11.6763-6771.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]