

Abstract
Hepatocyte cell death is a characteristic indication in the development of non-alcoholic steatohepatitis (NASH); however, the underlying mechanism is still unclear. In this study, we examined the potential mechanism(s) involved in the development of liver injury using a methionine-choline deficient (MCD) diet feeding NASH model. Male C57BL6/J mice were fed MCD and methionine-choline sufficient (MCS) diet for two weeks before being killed. Our results showed that MCD diet feeding resulted in fatty liver and liver injury, evidenced by increased hepatic triglyceride (TG), plasma alanine aminotransferases and hepatic thiobarbituric acid reactive substances levels in MCD-fed mice. Furthermore, we found that MCD diet feeding caused remarkable suppression of hepatic extracellular signal-regulated kinases (ERK) 1/2 activation and increased transforming growth factor (TGF)-beta1 levels in plasma and the liver tissue. In vitro investigations showed that intracellular MEK/ERK1/2 activation status played a critical role in the determination of sensitivity of hepatocytes to TGF-beta1-induced cell death. HepG2 cells, otherwise resistant to TGF-beta1 killing due to high level of ERK1/2 activation, was sensitized by U0126, a specific MEK/ERK1/2 inhibitor, to TGF-beta1 cytotoxicity. H4IIEC3 cells, which have lower level of constitutive ERK1/2 activity, are sensitive to TGF-beta1-induced cell death. Lastly, we demonstrated that administration of epidermal growth factor, a strong ERK1/2 activator, to MCD-fed mice attenuated liver injury without affecting hepatic TG accumulation. Our findings demonstrated that hepatic ERK1/2 inactivation aggravates TGF-beta1-induced hepatotoxicity, which may contribute, at least in part, to the initiation of liver injury in NASH.
Keywords: methionine, NASH, ERK1/2, TGF-beta1
Introduction
Non-alcoholic fatty liver disease is now regarded as the most common cause of abnormal liver function worldwide.1–3 The disease spectrum is characterized by simple steatosis (neutral fat accumulation), non-alcoholic steatohepatitis (NASH, featured by inflammation and hepatocyte cell death) and eventually, in some individuals, progressing to late-stage fibrosis, leading to liver failure.4 While simple hepatic steatosis is generally considered to be benign, NASH is a potentially serious condition with poor prognosis. NASH patients are at great risk of progressing to cirrhosis and liver failure, or to hepatocellular carcinoma.4–7 The pathogenesis of NASH has not yet been clearly defined; however, one generally accepted theory is the ‘two-hit’ hypothesis, wherein the first hit involves the development of hepatic steatosis, rendering the liver more susceptible to a second undefined hit, resulting in more severe liver damage.6 The factors driving progression from steatosis to steatohepatitis remain to be fully understood, and the currently well-established factors mainly include oxidative stress and proinflammatory cytokines.4,8
Methionine-choline deficient (MCD) diet feeding is a well-established nutritional model for NASH in that it induces similar liver histological changes to human NASH, including hepatic steatosis, lobular inflammation, hepatocyte cell death and pericelluar fibrosis.4,9,10 Elevated transforming growth factor (TGF)-beta1 expression is one of the major features associated with NASH in MCD diet feeding model.11,12 Since TGF-beta1 is a critical fibro-genetic factor and plays a central role in the development of fibrosis/cirrhosis in the late stage of NASH, elevated TGF-beta1 expression has long been used as an indicator of the onset of fibrogenic process. Accumulating evidence suggest that in addition to its fibrogenic action, TGF-beta1 is also a strong apoptosis inducer and its signaling is closely related with acute liver injury.13,14 However, evidence with regard to its potential implication in liver injury in the early stage of NASH is scarce.
The extracellular signal-regulated kinases-1/2 (ERK1/2) are members of the mitogen-activated protein kinase (MAPK) family and play critical roles in cellular proliferation, survival, differentiation and homeostasis.15,16 It is well documented that MEK/ERK1/2 pathway suppression inhibits hepatic regeneration following partial hepatectomy or chemical-induced liver injury.17,18 Several recent studies, including ours, have demonstrated that suppressed hepatic ERK1/2 activation was paralleled with liver injury in mice chronically exposed to alcohol diet,19–21 implicating the dysregulation of the hepatic ERK1/2 signaling pathway in ethanol-related liver disease. In the current experiment, we investigated the effects of MCD diet feeding on hepatic ERK1/2 activation in mice and its potential implication in early-stage liver injury in this animal model of NASH. We found that MCD diet feeding for two weeks caused liver injury, which was paralleled with hepatic ERK1/2 suppression and increased TGF-beta1 levels in plasma and the liver tissue. An in vitro study demonstrated that ERK1/2 suppression aggregated TGF-beta1-induced hepatocyte cell death. Our results suggest that ERK1/2 suppression, together with TGF-beta1 overproduction, may play a critical role in the development of liver injury in this NASH model.
Materials and methods
Animals and treatments
Male C57BL/6 mice weighing 25 ± 0.5 g (means ± SD) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed in the animal quarters at the University of Illinois, Chicago, and the studies were approved by the Institutional Animal Care and Use Committee, which is certified by the American Association of Accreditation of Laboratory Animal Care. All mice were initially housed in conventional conditions and fed standard diet and water ad libitum at the animal facility for one week before experiments began. Thereafter, the mice were divided into three groups (n = 8/group) and started on one of three treatments: methionine-choline sufficient (MCS) diet, MCD diet and MCD diet with administration of epidermal growth factor (MCD + EGF). In the MCD + EGF group, EGF administration was initiated at the end of the first week of feeding via subcutaneous injection (5 μg/mouse/d). Food intake and body weight were recorded daily. Two weeks later, the mice were killed and plasma and liver tissue samples were harvested for assays.
Cells and culture conditions
HepG2 cells, a human hepatoma cell line, and H4IIEC3 cells, a rat hepatoma cell line, were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and were cultured in Dulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovine serum, 2 mmol/L glutamine, 5 U/mL penicillin and 50 μg/mL streptomycin at 378°C in a humidified O2/CO2 (19:1) atmosphere.
Plasma alanine aminotransferases assay
Plasma alanine aminotransferases (ALT) assays were performed with commercially available kits (Infinity, Thermo Electron, Melbourne, Australia).
Histological examination
At the time of killing, the liver was harvested and small pieces were fixed immediately in 10% buffered formalin. After paraffin embedding, 5-μm sections were deparaffinized in xylene and were rehydrated through a series of decreasing concentrations of ethanol. Sections were stained with hematoxylin–eosin. Photomicrographs were taken on a Nikon Eclipse E600 microscope (Fryer Company, Cincinnati, OH, USA) equipped with a digital camera (Diagnostic Instruments, Sterling Heights, MI, USA).
Measurement of liver triglyceride
Hepatic fat accumulation was determined by measuring total hepatic triglyceride (TG) content. Liver tissues (~80 mg) were homogenized in 1.0 mL NaCl (50 mmol/L) solution and hepatic total lipids were extracted overnight in 10 mL heptane:isopropanol (3:2) mixture at 48C. Hepatic TG content was determined by enzymatic colorimetric methods using a commercially available kit (Infinity, Thermo Electron).
Lipid peroxidation assay
The liver thiobarbituric acid reactive substances (TBARS) levels, as an index of lipid peroxidation, were determined by measurement of the purple color generated by the reaction of thiobarbituric acid (TBA) with malondialdehyde in spectrophotometry. Each liver tissue was homogenized (10% w/v) in 25 mmol/L Tris–HCl pH 7.4, and centrifuged at 12,500 g for 15 min. For the malondialdehyde assay, 2.5 mL of TBA solution (10%) was added to 0.5 mL of the supernatant followed by heating in a boiling water bath for 15 min. After cooling to room temperature, the samples were centrifuged at 12,000 g for 10 min, and then 2 mL of the reaction mixture was transferred to a test tube containing 1 mL of TBA solution (0.67%). Each tube was then placed in a boiling water bath for 15 min. After cooling to room temperature, the absorbance was measured at 532 nm.
Enzyme-linked immunosorbent assay measurements of TGF-beta1
TGF-beta1 levels in plasma or in liver tissue were determined by enzyme-linked immunosorbent assay (ELISA) (Biotrak Easy ELISA, GE Healthcare, San Diego, CA, USA). Briefly, plasma was diluted in phosphate buffered saline (PBS) and the liver tissue was homogenized in radio-immunoprecipitation (RIPA) buffer. The samples were acidified with 1 mol/L HCl to activate latent TGF-beta1 to the immunoreactive form, and were neutralized with 1 mol/L NaOH. Then the prepared samples were assayed following the manufacturer’s instructions.
Lactate dehydrogenase assay
Cell death was determined by the measurement of lactate dehydrogenase (LDH) release into the culture medium. LDH activity was determined spectrophotometrically at 340 nm using a commercially available kit (Thermo scientific, Middletown, VA, USA).
Quantitative realtime reverse transcriptase-polymerase chain reaction
Total RNA from frozen liver tissue was isolated according to the manufacturer’s protocol with an additional phenol–chloroform extraction. Isolated RNA was quantified (A260) and assessed for purity by determining the A260/A280 ratio. For each sample, 1.0 μg total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit as described by the manufacturer (Applied Biosystems, Foster City, CA, USA). The cDNA (1.0 μL) was used as a template in a 25 μL polymerase chain reaction (PCR) solution containing 10.5 μL deionized water, 1 μL 10 μmol/L gene-specific primers and 12.5 μL 1× SYBR Green PCR master mix (SuperArray Bioscience, Frederick, MD, USA). PCR amplification was conducted in MicroAmp Optical 96-well reaction plates (Applied Biosystems) on an Applied Biosystems PRISM 7000 sequence detection system. No-template controls (NTCs) were included on each plate. Samples with a CT value within 2 SD of the mean CT values for the NTCs were considered below the limits of detection. The copy number of each unknown sample for each gene was standardized to a house-keeping gene (mouse or human 18S rRNA) to control for differences in RNA loading, quality and cDNA synthesis. For graphing purposes, the relative expression levels were scaled such that the expression level of the control group was equal to 1.
Western blotting detections
Liver tissues or hepatocytes were lysed in Western lysis buffer consisting of the following: 20 mmol/L Tris ● HCl, pH 7.4, 150 mmol/L NaCl, 10% glycerol, 2% Nonidet P-40, 1 mmol/L ethylenediaminetetraacetic acid, pH 8.0, 20 mmol/L sodium fluoride, 30 mmol/L sodium pyrophosphate, 0.2% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L dithiothreitol, 1 mmol/L sodium vanadate, 50 μmol/L leupeptin and 5 μmol/L aprotinin. Samples were incubated on ice with frequent vortexing for 15 min and centrifuged for 20 min at 18,000g. The protein content of each supernatant was quantified via a protein assay reagent from Bio-Rad Laboratories (Hercules, CA, USA) in accordance with the manufacturer’s instructions. Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to 0.45 μm polyvinylidene difluoride membrane (PerkinElmer Life Sciences, Covina, CA, USA). After transfer, membranes were blocked in 5% (wt/vol) non-fat dry milk in PBS-0.1% Tween 20 and probed with the antibodies specified. Horseradish peroxidase-conjugated secondary antibodies (Sigma, St Louis, MO, USA) and enhanced chemiluminescence substrate kit (PerkinElmer Life Science) were used in the detection of specific proteins.
Statistical analysis
All data were expressed as means ± SD. Statistical analysis was performed using a one-way analysis of variance and was analyzed further by Newman–Keuls test for statistical difference. Differences between treatments were considered to be statistically significant at P < 0.05.
Results
MCD diet causes fatty liver and liver injury
The pathological alterations in livers from MCS and MCD groups were evaluated by histological examination and measurements of hepatic TG content, circulating liver enzyme levels, as well as hepatic lipid peroxidation product levels. As shown in Figure 1, in comparison to the MCS group, MCD diet feeding for two weeks increased hepatic TG content (Figure 1a and b). Liver injury was induced by MCD diet feeding, which was evidenced by increased plasma ALT levels (Figure 1c) and lipid peroxidation product levels (Figure 1d) in the liver tissues of the MCD group compared with that in the MCS group.
Figure 1.
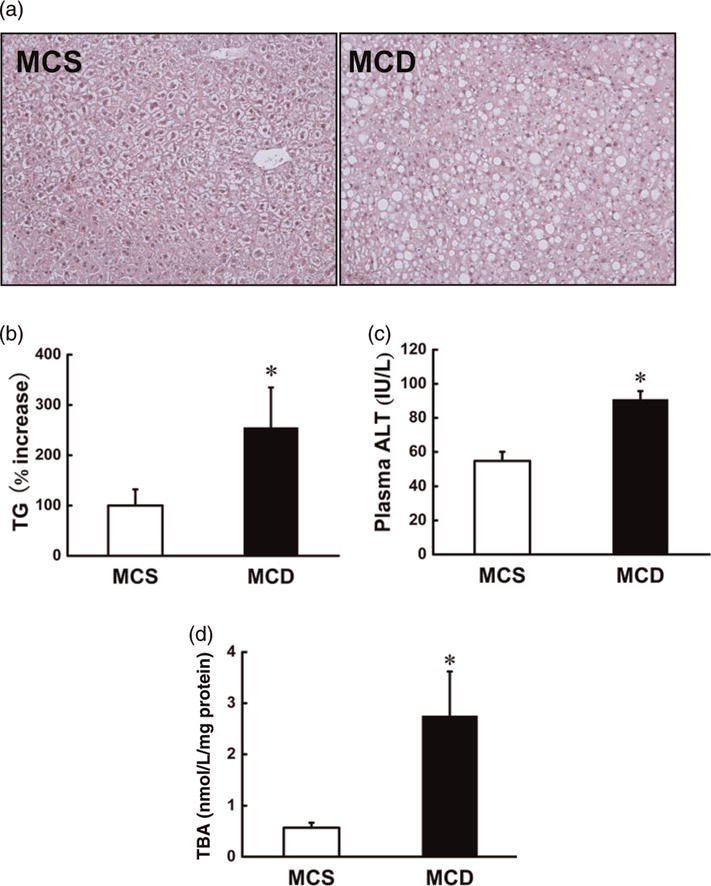
MCD diet caused hepatic fatty liver and liver injury in mice. Male C57BL/6 mice were fed with MCS or MCD diet for two weeks. (a) H&E staining. (b) Hepatic triglyceride content. (c) Plasma ALT levels. (d) Lipid peroxidation products in the liver. Data are means ± SD (n 4). *P < 0.05. MCS, methionine and choline sufficient; MCD, methionine-choline deficient; ALT, alanine aminotransferases; TG, triglyceride; TBA, thiobarbituric acid; H&E, hematoxylin–eosin. (A color version of this figure is available in the online journal)
MCD diet suppresses hepatic ERK1/2 activation
Western blot analysis was used to determine the effects of MCD diet feeding on hepatic ERK1/2 activation. As shown in Figure 2, MCD diet feeding for two weeks caused a remarkable reduction in hepatic ERK1/2 phosphorylation, while no changes in total ERK1/2 expression were observed.
Figure 2.
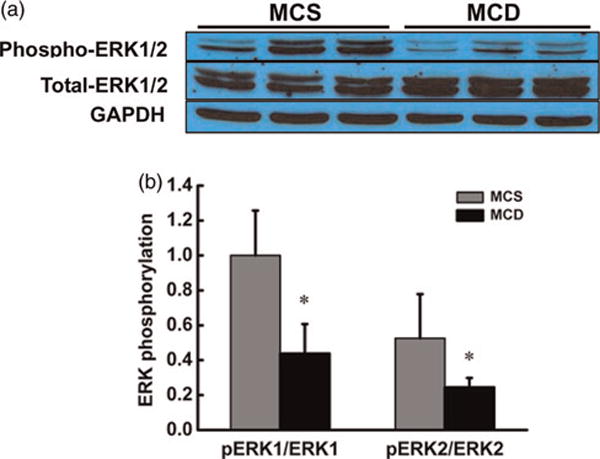
MCD diet resulted in ERK1/2 suppression in the liver. Male C57BL/6 mice were fed with MCS or MCD diet for two weeks. (a) MCD diet feeding decreased the phosphorylation levels of ERK1/2 in the liver. (b) Quantitative analysis of ERK1/2 phosphorylation in the liver, normalized by corresponding total ERK1/2 expression level. Data are means ± SD (n = 4). *P < 0.05. MCS, methionine and choline sufficient; MCD, methionine-choline deficient; ERK, extracellular signal-regulated kinase. (A color version of this figure is available in the online journal)
MCD diet increases TGF-beta1 levels in both plasma and liver tissue
TGF-beta1 gene expression and production in liver tissues and plasma were determined by quantitative realtime reverse transcriptase-polymerase chain reaction and a TGF-beta1-specific ELISA kit. As shown in Figure 3, MCD diet feeding for two weeks significantly augmented hepatic TGF-beta1 gene expression (Figure 3a) and protein abundance (Figure 3b) when compared with the MCS group. Moreover, MCD diet feeding also increased plasma TGF-beta1 level (Figure 3c).
Figure 3.
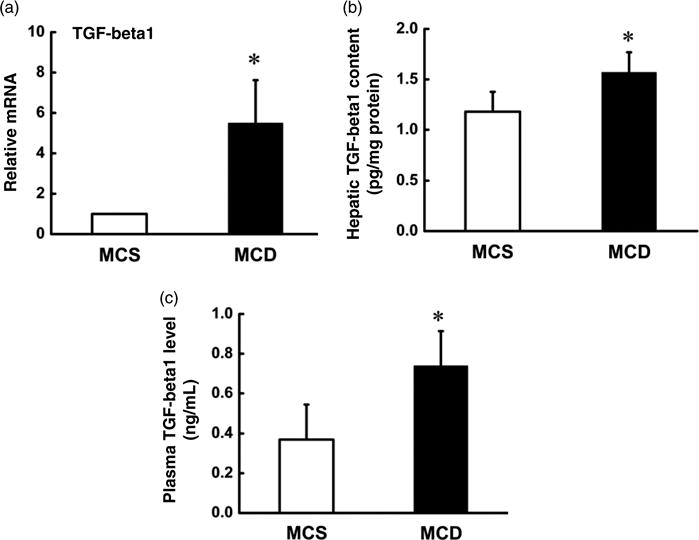
MCD diet augmented TGF-beta1 level in both liver tissue and plasma. Male C57BL/6 mice were fed with MCS or MCD diet for two weeks, and hepatic and plasma TGF-beta1 levels were assayed by ELISA and quantitative real-time RT-PCR. MCD diet elevated hepatic TGF-beta1 gene expression (a) and protein abundance (b) in liver tissue. (c) MCD diet augmented plasma TGF-beta1 level. Data are means ± SD (n 4). *P < 0.05. MCS, methionine and choline sufficient; MCD, methionine-choline deficient; TGF, transforming growth factor; ELISA, enzyme-linked immunosorbent assay; RT-PCR, reverse transcriptase-polymerase chain reaction
ERK1/2 inhibition aggravates TGF-beta1-induced cell death in hepatocytes
To determine whether ERK1/2 suppression leads to increased TGF-beta1 cytotoxicity, cell culture studies were conducted. HepG2 cells were pretreated with U0126, a specific inhibitor of MEK/ERK1/2 pathway, for two hours and then TGF-beta1 (10 ng/mL) was added to the culture medium. After 20 h, culture medium and total protein were harvested for LDH release assay and Western blotting to determine the extent of cell injury. As shown in Figure 4, TGF-beta1 alone had no effect on LDH release in comparison to the control cells and U0126 alone slightly increased LDH release; however, U0126 pretreatment significantly increased TGF-beta1-induced LDH release (Figure 4a). These observations were confirmed by Western blot assay for poly (ADP ribose) polymerase (PARP) cleavage (Figure 4b and c). To further validate the critical role of ERK1/2 activation in TGF-beta1-induced hepatotoxicity, H4IIEC3 cells, which have lower level of constitutive ERK1/2 activity in comparison to HepG2 cells (Figure 4d and e), were treated with TGF-beta1 (1 ng/mL) for 20 h. Our results showed that in comparison to HepG2 cells, H4IIEC3 cells were more sensitive to TGF-beta1-induced hepatotoxicity (Figure 4f).
Figure 4.
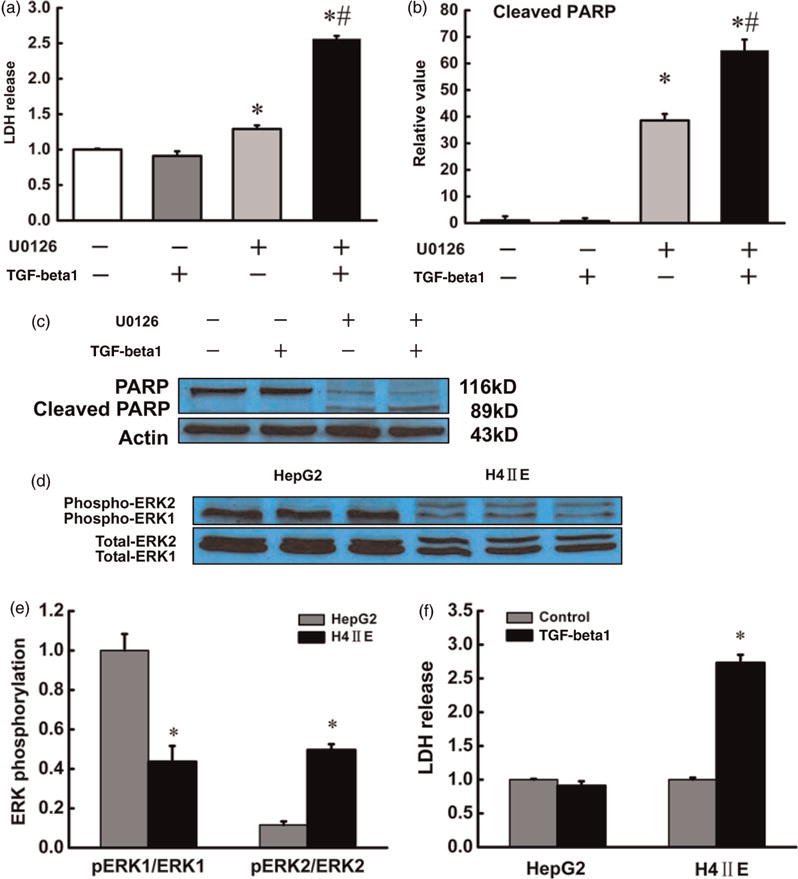
Intracellular ERK1/2 activity determined the sensitivity of hepatocytes to TGF-beta1-induced cytotoxicity. HepG2 cells were treated with vehicle, U0126 (10 μmol/L), TGF-beta1 (10 ng/mL) or U0126 plus TGF-beta1 (U0126 was pretreated for 2 h) for 20 h. The culture media were collected for LDH release assay and the total protein in adherent cells was exacted for PARP immunoblotting assay. (a), (b) and (c) MEK/ERK1/2 inhibition aggravated TGF-beta1 cytotoxicity. All values are denoted as means ± SD from three or more independent batches of cells, *P < 0.05 compared with the untreated group; #P < 0.05 compared with U0126 along treated group. Additionally, immunoblotting and LDH assay were conducted to compare ERK1/2 activation and susceptibility to TGF-beta1-induced cytotoxicity between HepG2 cells and H4IIEC3 cells. Lower ERK1/2 activity in H4IIEC3 cells (d and e) was concomitant with a higher LDH release in response to TGF-beta1 (1 ng/mL) challenging (f) compared with that in HepG2 cells. All values are denoted as means ± SD from three or more independent batches of cells, *P < 0.05. ERK, extracellular signal-regulated kinase; TGF, transforming growth factor; LDH, lactate dehydrogenase; PARP, poly (ADP ribose) polymerase. (A color version of this figure is available in the online journal)
4-Hydroxynonenal suppresses ERK1/2 activation and aggravates TGF-beta1 hepatotoxicity
Mechanisms leading to ERK1/2 suppression by MCD diet feeding are multifactorial, among which 4-hydroxynonenal (4-HNE), a major product of lipid peroxidation, has been reported to suppress ERK1/2 activation in hepatocytes.19 Since MCD diet feeding for two weeks was associated with increased lipid peroxidation (Figure 1c), we examined if 4-HNE represented a potential mechanism involved in ERK1/2 suppression and sensitization to TGF-beta1 hepatotoxicity in this model. In line with previous reports, 4-HNE dose-dependently decreased the phosphorylation levels of ERK1/2 in HepG2 cells (Figure 5a). In association with ERK1/2 suppression, 4-HNE pretreatment aggravated TGF-beta1-induced cell injury, evidenced by significant higher LDH release in comparison to 4-HNE alone treatment (Figure 5b).
Figure 5.
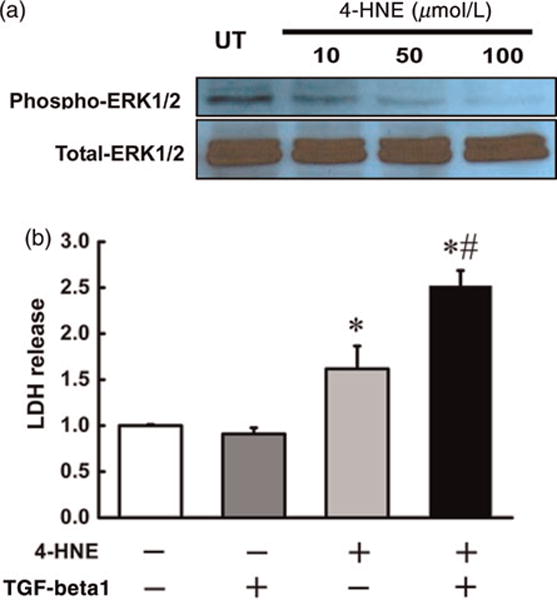
4-HNE suppressed ERK1/2 activity and sensitized hepatocytes to TGF-beta1-inducd cytotoxicity in HepG2 cells. Immunoblotting and LDH measurements were applied to examine the effect of 4-HNE on the cytotoxicity in hepatocytes. (a) 4-HNE (10 μmol/L to 100 μmol/L) decreased the phosphorylation levels of ERK1/2 in a concentration-dependant way in HepG2 cells. (b) 4-HNE (50 μmol/L) pretreatment aggravated TGF-beta1-induced LDH release in HepG2 cells. All values are denoted as means ± SD from three or more independent batches of cells, *P < 0.05 compared with the untreated group; #P < 0.05 compared with 4-HNE-treated group. TGF, transforming growth factor; LDH, lactate dehydrogenase; ERK, extracellular signal-regulated kinase; 4-HNE, 4-hydroxynonenal. (A color version of this figure is available in the online journal)
EGF alleviates MCD diet-induced liver injury
To test if prevention of ERK1/2 suppression can alleviate liver injury in MCD-fed mice, EGF, a strong ERK1/2 activator (Figure 6a), was administrated to MCD-diet-fed animals via daily subcutaneous injection during the second week of feeding. As shown in Figure 6, EGF administration decreased circulating ALT levels in MCD diet mice (Figure 6b) without affecting hepatic TG levels (Figure 6c).
Figure 6.
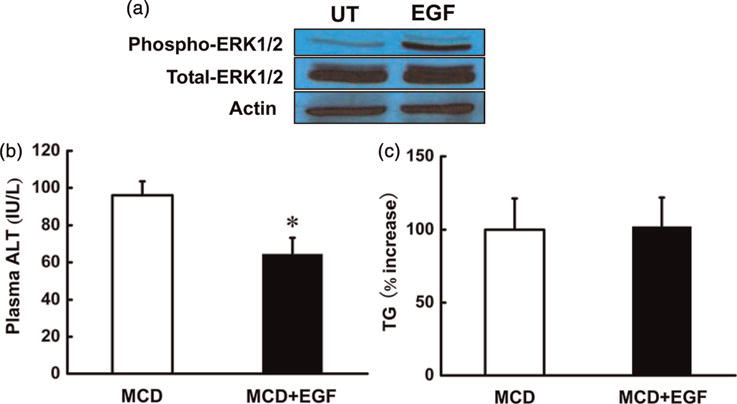
EGF attenuated liver injury in MCD-diet-fed mice. Male C57BL/6 mice were fed with MCD diet for two weeks. At the end of the first week of feeding, the animals were daily treated with either EGF (5 μg/mouse, subcutaneously) in 10 mmol/L acetic acid in sterile saline or vehicle for another week. EGF, a potent ERK1/2 activator in hepatocytes (a), lowered plasma ALT level in MCD diet mice (b). Meanwhile, EGF injection for one week had no effect on TG content in the liver of MCD diet mice (c). Data are means ± SD (n = 6). *P < 0.05. MCD, methionine-choline deficient; ALT, alanine aminotransferases; EGF, epidermal growth factor; TG, triglyceride. (A color version of this figure is available in the online journal)
Discussion
Feeding mice or rats with MCD diet is a classical dietary model for the study of pathogenesis of NASH, a liver disease characterized by steatosis, liver injury and lobular inflammation. The mechanisms involved in early-stage liver injury in mice-fed MCD diet are multiple and remain to be well-defined. In the present study, we showed that after a two-week feeding, the plasma ALT levels and hepatic TG contents were significantly elevated in MCD-diet-fed mice. These pathological alterations were associated with suppressed hepatic ERK1/2 phosphorylation and increased TGF-beta1 gene expression and protein concentrations in both the liver and plasma. In vitro investigations revealed that suppression of MEK/ERK1/2 pathway aggravated TGF-beta1-induced cell death in HepG2 cells. Moreover, H4IIEC3 cells, rat hepatoma cells with a lower level of constitutive ERK1/2 activity, were more sensitive to TGF-beta1 killing, suggesting that ERK1/2 activation plays a critical role in TGF-beta1 cytotoxicity. Further study with EGF administration showed that the ERK1/2 activator attenuated liver injury in MCD-fed mice without affecting fat accumulation. Our results suggest that ERK1/2 suppression and TGF-beta1 overproduction may be synergistically involved in the observed early-stage liver injury in this model.
The MAPKs constitute a family of serine/threonine kinases that include ERK1/2, c-Jun N-terminal kinase (JNK) and p38 MAPK. These enzymes are positively regulated by a hierarchy of kinases and inactivated by phosphatases. ERK1/2 is activated by a number of factors including growth factors and cytokines, and regulate a number of critical cellular functions including proliferation, differentiation and survival.15,16 ERK1/2 activation is critical for hepatocyte resistance to cell death. The blocking of MEK/ERK1/2 pathway resulted in the sensitization of hepatocytes to apoptosis from previously non-toxic concentrations of menadione.22,23 Moreover, ERK1/2 activation conferred the resistance of primary rat hepatocytes to cell death induced by oxidative stress. In contrast, pharmacological inhibition of ERK1/2 led to a significant reduction in hepatocyte survival in the presence of H2O2.24 However, the studies with regard to the effects of MCD diet feeding on ERK1/2 activation are scarce. In the current study, we showed that MCD diet resulted in ERK1/2 suppression in a short feeding period, implying that ERK1/2 suppression may be critically involved in the early-stage liver injury in this model.
It has been well-recognized that TGF-beta1 induces growth inhibition and apoptosis in hepatocytes both in vivo and in vitro. Physiologically, TGF-beta1-induced apoptosis in hepatocytes is finely regulated and could be correlated with its function as a tumor suppressor; however, an exaggerated apoptotic pathway of TGF-beta1 in the liver under certain pathological conditions may contribute to the pathogenesis of liver injury and liver diseases.25,26 This notion was supported by several previous studies. For instance, blocking TGF-beta1 signaling pathway by ectopic expression of Smad7 in hepatocytes efficiently inhibited CCl4-dependent liver damage and fibrogenesis in mice.27 In mice with acute hepatitis, TGF-beta receptor siRNA reduced the apoptotic cells in the liver, prevented liver cell damage and declined the ALT level in plasma.28,29 Also, TGF-beta1 ribbon-type antisense oligonucleotide effectively prevented liver fibrosis.30 Elevated TGF-beta1 expression is one of the major features associated with non-alcoholic fatty liver disease in the MCD diet feeding model. In late disease stage, TGF-beta1 signaling contributes to stellate cell activation and (myo)fibroblast generation, the latter being responsible for ECM deposition and scar formation.27,31 Since TGF-beta1 is a critical fibro-genetic factor and plays a central role in the development of fibrosis/cirrhosis in the late stage of NASH, elevated TGF-beta1 expression has long been used as an indicator of the onset of fibrogenic process. However, increased gene expression of TGF-beta1 in the liver of mice-fed MCD could be detected as early as after the first-week of MCD diet feeding,12 in parallel with the development of liver injury, implying that this cytokine may also critically be implicated in the initiation of hepatocyte cell death. This notion was supported by a recent clinical trial showing that patients with fatty liver disease had elevated plasma TGF-beta1 levels.32 To characterize the potential involvement of TGF-beta1 in early NASH, we first measured its gene expression in the liver tissue and protein abundance in the liver and plasma. MCD diet feeding for two weeks led to significant increase in hepatic TGF gene expression and protein concentrations in both liver tissue and plasma. We then examined if suppressed hepatic ERK1/2 phosphorylation could sensitize hepatocytes to TGF-beta1 cytotoxicity. Using cell-culture systems, we demonstrated that activation of ERK1/2 pathway played a critical role in the determination of sensitivity of hepatocytes to TGF cytotoxicity. Namely, inhibition of MEK/ERK1/2 pathway aggravated TGF killing. These in vitro observations suggested that modulation of hepatic MEK/ERK1/2 pathway may be an ideal therapeutic choice for NASH. This notion was validated by our subsequent study showing that EGF, a strong ERK1/2 activator, conferred protective effects on the liver of mice-fed MCD diet without affecting hepatic TG accumulation.
The mechanisms underlying ERK1/2 pathway suppression by MCD diet feeding remain to be fully elucidated. One potential mechanism is lipid peroxidation. In line with previous reports, we showed that MCD diet feeding was associated with increased lipid peroxidation in the liver, evidenced by increased hepatic TBARS contents in MCD-diet-fed mice. It has been reported that 4-HNE, a major product of membrane lipid peroxidation, suppressed ERK1/2 activation.19 In the present study, similar inhibitory effects of 4-HNE on ERK1/2 activation were found in HepG2 cells. More importantly, we found that pretreatment of HepG2 cells with 4-HNE increased TGF-beta1-induced cytotoxicity, suggesting that oxidative stress could aggravate TGF-beta1-induced hepatotoxicity via suppressing ERK1/2 phosphorylation.
In conclusion, we demonstrated in this study that MCD diet feeding caused suppression of ERK1/2 activation and overproduction of TGF-beta1. Mechanistic investigations revealed that ERK1/2 suppression increased sensitivity of hepatocytes to TGF-beta1-induced cytotoxicity, suggesting that these two alterations may be synergistically implicated in the pathogenesis of early-stage liver injury in this animal model of NASH. Our results suggest that modulation of the ERK1/2 pathway and/or TGF-beta1 signaling may represent a therapeutic choice for NASH.
Acknowledgments
This study was supported by the National Institutes of Health grants K01 AA015344 and R01 AA017442 (ZS).
Footnotes
Author contributions: All authors participated in the design, interpretation of the studies and analysis of the data and review of the manuscript. ZW and TY participated in the acquisition and analysis of the data and drafting the manuscript. ZS participated in study design, analysis, and interpretation of the data, and editing the manuscript.
References
- 1.Angulo P. GI epidemiology: nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2007;25:883–9. doi: 10.1111/j.1365-2036.2007.03246.x. [DOI] [PubMed] [Google Scholar]
- 2.Williams R. Global challenges in liver disease. Hepatology. 2006;44:521–6. doi: 10.1002/hep.21347. [DOI] [PubMed] [Google Scholar]
- 3.Ong JP, Younossi ZM. Epidemiology and natural history of NAFLD and NASH. Clin Liver Dis. 2007;11:1–16. doi: 10.1016/j.cld.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 5.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–31. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 6.Day CP, James OF. Steatohepatitis: a tale of two ‘hits’? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 7.Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, Musso A, De Paolis P, Capussotti L, Salizzoni M, Rizzetto M. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134–40. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 8.Mehta K, Van Thiel DH, Shah N, Mobarhan S. Nonalcoholic fatty liver disease: pathogenesis and the role of antioxidants. Nutr Rev. 2002;60:289–93. doi: 10.1301/002966402320387224. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–74. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 10.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–96. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 11.Koca SS, Bahcecioglu IH, Poyrazoglu OK, Ozercan IH, Sahin K, Ustundag B. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation. 2008;31:91–8. doi: 10.1007/s10753-007-9053-z. [DOI] [PubMed] [Google Scholar]
- 12.Tomita K, Oike Y, Teratani T, Taguchi T, Noguchi M, Suzuki T, Mizutani A, Yokoyama H, Irie R, Sumimoto H, Takayanagi A, Miyashita K, Akao M, Tabata M, Tamiya G, Ohkura T, Hibi T. Hepatic AdipoR2 signaling plays a protective role against progression of nonalcoholic steatohepatitis in mice. Hepatology. 2008;48:458–73. doi: 10.1002/hep.22365. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol. 2005;166:1029–39. doi: 10.1016/s0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y, Inoue K. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology. 2002;35:49–61. doi: 10.1053/jhep.2002.30083. [DOI] [PubMed] [Google Scholar]
- 15.Cobb MH, Hepler JE, Cheng M, Robbins D. The mitogen-activated protein kinases, ERK1 and ERK2. Semin Cancer Biol. 1994;5:261–8. [PubMed] [Google Scholar]
- 16.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–76. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 17.Duguay L, Coutu D, Hetu C, Joly JG. Inhibition of liver regeneration by chronic alcohol administration. Gut. 1982;23:8–13. doi: 10.1136/gut.23.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wands JR, Carter EA, Bucher NL, Isselbacher KJ. Inhibition of hepatic regeneration in rats by acute and chronic ethanol intoxication. Gastroenterology. 1979;77:528–31. [PubMed] [Google Scholar]
- 19.Sampey BP, Stewart BJ, Petersen DR. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J Biol Chem. 2007;282:1925–37. doi: 10.1074/jbc.M610602200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Yao T, Song Z. Chronic alcohol consumption disrupted cholesterol homeostasis in rats: down-regulation of low-density lipoprotein receptor and enhancement of cholesterol biosynthesis pathway in the liver. Alcohol Clin Exp Res. 2010;34:471–8. doi: 10.1111/j.1530-0277.2009.01111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeon JE, Califano S, Xu J, Wands JR, De La Monte SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology. 2003;38:703–14. doi: 10.1053/jhep.2003.50368. [DOI] [PubMed] [Google Scholar]
- 22.Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology. 2003;37:1405–13. doi: 10.1053/jhep.2003.50233. [DOI] [PubMed] [Google Scholar]
- 23.Conde de la Rosa L, Schoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–29. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 24.Rosseland CM, Wierød L, Oksvold MP, Werner H, Ostvold AC, Thoresen GH, Paulsen RE, Huitfeldt HS, Skarpen E. Cytoplasmic retention of peroxide-activated ERK provides survival in primary cultures of rat hepatocytes. Hepatology. 2005;42:200–7. doi: 10.1002/hep.20762. [DOI] [PubMed] [Google Scholar]
- 25.Cain K, Freathy C. Liver toxicity and apoptosis: role of TGF-beta1, cytochrome c and the apoptosome. Toxicol Lett. 2001;120:307–15. doi: 10.1016/s0378-4274(01)00283-1. [DOI] [PubMed] [Google Scholar]
- 26.Freathy C, Brown DG, Roberts RA, Cain K. Transforming growth factor-beta(1) induces apoptosis in rat FaO hepatoma cells via cytochrome c release and oligomerization of Apaf-1 to form a approximately 700-kd apoptosome caspase-processing complex. Hepatology. 2000;32:750–60. doi: 10.1053/jhep.2000.18329. [DOI] [PubMed] [Google Scholar]
- 27.Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, Ueberham E, Gebhardt R, Kanzler S, Geier A, Breitkopf K, Weng H, Mertens PR. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135:642–59. doi: 10.1053/j.gastro.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 28.Mizuguchi Y, Yokomuro S, Mishima T, Arima Y, Shimizu T, Kawahigashi Y, Takizawa T, Tajiri T. Therapeutic use of short hairpin RNA in acute liver failure. J Nippon Med Sch. 2007;74:74–6. doi: 10.1272/jnms.74.74. [DOI] [PubMed] [Google Scholar]
- 29.Mizuguchi Y, Yokomuro S, Mishima T, Arima Y, Shimizu T, Kawahigashi Y, Kanda T, Yoshida H, Takizawa T, Tajiri T. Short hairpin RNA modulates transforming growth factor beta signaling in life-threatening liver failure in mice. Gastroenterology. 2005;129:1654–62. doi: 10.1053/j.gastro.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Doh KO, Jung HK, Moon IJ, Kang HG, Park JH, Park JG. Prevention of CCl4-induced liver cirrhosis by ribbon antisense to transforming growth factor-beta1. Int J Mol Med. 2008;21:33–9. [PubMed] [Google Scholar]
- 31.McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497–502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- 32.Tarantino G, Conca P, Riccio A, Tarantino M, Di Minno MN, Chianese D, Pasanisi F, Contaldo F, Scopacasa F, Capone D. Enhanced serum concentrations of transforming growth factor-beta1 in simple fatty liver: is it really benign? J Transl Med. 2008;6:72. doi: 10.1186/1479-5876-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]