

Abstract
Rationale
The nasopharyngeal (NP) microbiota of newborns and infants plays a key role in modulating airway inflammation and respiratory symptoms during viral infections. Premature (PM) birth modifies the early NP environment and is a major risk factor for severe viral respiratory infections. However, it is currently unknown if the NP microbiota of PM infants is altered relative to full-term (FT) individuals.
Objectives
To characterize the NP microbiota differences in preterm and FT infants during rhinovirus (RV) infection.
Methods
We determined the NP microbiota of infants 6 months to ≤2 years of age born FT (n=6) or severely PM<32 weeks gestation (n=7). We compared microbiota composition in healthy NP samples and performed a longitudinal analysis during naturally occurring RV infections to contrast the microbiota dynamics in PM versus FT infants.
Results
We observed significant differences in the NP bacterial community of PM versus FT. NP from PM infants had higher within-group dissimilarity (heterogeneity) relative to FT infants. Bacterial composition of NP samples from PM infants showed increased Proteobacteria and decreased in Firmicutes. There were also differences in the major taxonomic groups identified, including Streptococcus, Moraxella, and Haemophilus. Longitudinal data showed that these prematurity-related microbiota features persisted during RV infection.
Conclusions
PM is associated with NP microbiota changes beyond the neonatal stage. PM infants have an NP microbiota with high heterogeneity relative to FT infants. These prematurity-related microbiota features persisted during RV infection, suggesting that the NP microbiota of PM may play an important role in modulating airway inflammatory and immune responses in this vulnerable group.
INTRODUCTION
Newborns and infants are obligated nasal breathers;1,2 thus, the nasopharyngeal (NP) airway is the first line of defense and main port of entry of bacterial and viral respiratory pathogens in early life. This notion has led to the emergent and potentially powerful idea of assessing the NP microbiota of newborns and infants to underpin early-life exposures and host immune responses that determine the initiation and progression of respiratory disorders in humans.3–8 Indeed, recent studies have established that the early NP microbial composition correlates with individual nasal cytokine signatures,3 frequency and severity of upper and lower respiratory infections,4–8 as well as subsequent asthma risk.5–8 In turn, the infant NP microbiota is a reflection of early exposures (eg, breastfeeding, antibiotics, viral infections, daycare, etc), which critically shape bacterial composition and airway responses against respiratory viruses.8
The NP airway of infants born severely premature (PM) undergoes dramatically different early-life exposures in the neonatal intensive care unit (NICU). Prematurity-related challenges include nosocomial pathogens, supplemental oxygen, mechanical respiratory support, broad-spectrum antimicrobials and deprivation of the normal intrauterine environment. We have shown that, beyond the NICU stage, severely PM infants born <32 weeks gestational age (GA) have persistent abnormal nasal airway immune responses against respiratory viruses during the first 2 years of life;9,10 however, it is unknown if the NP microbiota of PM infants is also persistently different during early childhood relative to infants born at term.
In this study, we contrasted the NP microbiota of infants 6 months to ≤2 years of age born full term (FT) or severely PM. We compared the microbial composition in healthy NP samples and performed an analysis during naturally occurring rhinovirus (RV) infections to contrast the microbiome dynamics in PM versus FT infants. Our hypothesis was that prematurity leads to a distinct microbiome with unique bacterial composition and this distinct microbiota difference is maintained during viral infections, which are known to change the microbiome composition. The impact of this work is that it presents, for the first time, to the best of our knowledge, a potentially modifiable environmental factor (prematurity-related NP microbiome) that may contribute to the previously described abnormal nasal airway immune responses and high risk for severe viral respiratory infections in PM infants during their first 2 years of life.9,10 This new knowledge may ultimately lead to novel approaches to predict and prevent potentially life-threatening respiratory complications in this vulnerable population.
MATERIALS AND METHODS
Study population and nasal sampling
Two nasal washes (W1 and W2) were collected from children aged 6 months to ≤2 years enrolled in the Viral Immuno-genetic Responses of the Airways and the Lungs (VIRAL) cohort at Children’s National Medical System.9–12 The VIRAL cohort is a longitudinal study of children admitted to the hospital with suspected viral respiratory infections. W1 was taken during acute RV infection as confirmed by PCR. W2 was obtained at follow-up during the study period (1–3 months after the initial sample). W2 was negative for RV and other viruses as confirmed by respiratory virus PCR analysis used for clinical purposes. For paired samples (n=6), we included age-matched and gestational (for preterm subjects) age-matched children born at FT, defined as >37 weeks GA (n=3), or born severely PM, defined as <32 weeks GA (n=3). We also included seven additional subjects (four born FT and three born PM) in which only one time point was available. Table 1 shows all subjects included (n=13) and their baseline characteristics. We used a standard nasal lavage technique consisting of gently washing the nasal cavity with 3–4 mL sterile normal saline as previously described.9–12 Exclusion criteria included mixed viral infection or antibiotic therapy <4 weeks prior to microbiome analysis. This study was approved by the Institutional Review Board of Children’s National Health System, Washington, DC.
Table 1.
Baseline characteristics for subjects
| Full term n=6 |
Premature n=7 |
p Value | |
|---|---|---|---|
| Male, n (%) | 3 (50) | 4 (71) | 0.38 |
| Age (years), median (IQR) | 1.82 (0.8–2.5) | 1.41 (0.8–1.9) | 0.43 |
| Gestational age (weeks), median (IQR) | 38.5 (37–40) | 24 (23–27) | <0.01 |
| Black, n (%) | 2 (33) | 4 (57) | 0.42 |
Demographics for all study subjects (n=13). p Values based on Wilcoxon rank-sum test for continuous variables; χ2 test for categorical variables.
16S ribosomal RNA PCR and next-generation sequencing
Amplification of bacterial 16S ribosomal RNA (rRNA) was performed as previously described by our group.13 Briefly, the 16S small rRNA subunit was amplified using ‘universal’ primers (B27F, 5=-AGAGTTTGATCCTGGCTCAG-3=; and U1492R, 5=-GGTTACCTTGTTACGACTT-3=) with Escherichia coli 16S for numbering and a predicted product of 1466 bp. ABI 2700 thermocycler was used for amplifications with a hot-start AmpliTaq Gold 360 DNA polymerase master mix with GC enhancer (Life Technologies). An optimal loading concentration of 500 ng of gDNA was used for most samples (several amplified well with 100–250 ng). After PCR was performed, the presence of amplimers at the expected 1466 bp size was confirmed by gel electrophoresis on a 1% agarose gel stained with ethidium bromide and Agilent 2100 bioanalyzer (Agilent Technologies). PCR products were purified with Agencourt AMPure XP magnetic beads (Beckman-Coulter) and then quantified with an optical density at 260 or 280 nm (OD260/280; NanoDrop) and fluorescence staining (Qubit). Amplimers were pooled if insufficient samples were identified; samples without detectable amplimers were excluded from the analysis. Next-generation sequencing studies were conducted as per published protocols,13 using purified 16S amplimers (200 to 750 ng) prepared for long-read single-molecule sequencing (Pacific Biosystems) with a DNA Prep kit 2.0 (250 to <3 kb) followed by standard sequencing primer/polymerase steps (PacBio RS II platform and P4/C2 chemistry). Sequencing data were generated in PacBio native bas.h5 and standard FastQ formats.
Analytical methods
Raw FASTQ files were analyzed in mothur V.1.35.1 using pipelines previously described.14,15 Briefly, sequences were aligned to the SILVA_V.123 bacterial reference alignment at http://www.mothur.org. Chimeras were excluded by uchime16 and non-chimeric sequences classified according to naïve Bayesian classifiers.17 Operational taxonomic units (OTUs) clustering used a 0.03 threshold (species level). OTU sequence representatives and taxonomy were imported (BIOM format) into QIIME18 for subsequent analyses. The mothur OTU table was filtered (minimum of 2 sequences per OTU) and subsampled (rarefaction analysis, smallest sample size=1037 sequences) to eliminate sample size bias on community composition. OTU differential abundance tests19 and DESeq2 negative binomial Wald test20 were also applied for comparison.
Trees for phylogenetic diversity were built using FastTree.21 Taxonomic α-diversity was assessed as the number of observed OTUs, and by the Chao1 and Shannon indexes; phylogenetic α-diversity was determined by Faith’s index.22 Taxonomic (Bray-Curtis and Euclidean) and phylogenetic (unweighted and weighted unifrac) β-diversity metrics were compared between pairs of samples. Procrustes analysis contrasting principal coordinates analysis (PCoA) plots (weighted unifrac distances) of seven paired samples23 was performed with 10,000 Monte Carlo iterations. The α-diversity and β-diversity metrics were contrasted between and within groups using a non-parametric version of the t-test. Taxonomic and phylogenetic distances were compared among groups using the non-parametric PERMANOVA and adonis tests from the vegan R’s library.24 Significance was determined through 10,000 permutations. Finally, OTU abundance differences between sample pairs were assessed using Fisher’s exact test, while OTU abundance differences between sample groups were estimated using rarefied (analysis of variance, Kruskal-Wallis and Welch’s t-test) and non-rarefied (metagenomeSeq zero-inflated Gaussian and DESeq2 negative binomial Wald test) tests. Bonferroni or Benjamini-Hochberg false discovery rate (FDR) multiple test correction methods were applied. All analyses were performed in mothur, QIIME, STAMP25 and RStudio.26
RESULTS
A total of 19 nasal microbiomes corresponding to 13 children (6 paired samples and 7 single samples) were analyzed via PacBio sequencing of 16S rRNA amplicons. A total of 97,982 full 16S sequences ranging from 1037 to 8914 sequences per sample (mean=5157; median=5711) were obtained after quality control analyses and OTU filtering. From these data, we identified 592 OTUs (mean=53 OTUs per sample), belonging to 75 bacterial genera and 8 phyla. About 90% of the sequences corresponded to the following seven genera: Streptococcus (34.2%), Moraxella (19.3%), Staphylococcus (10.3%), Burkholderia (8.8%), Neisseria (6.2%), Haemophilus (6.1%), and Janthinobacterium (5.2%; figure 1). The taxonomic profile of the FT group showed a predominance of Streptococcus (green–brown in figure 1); other genera were present in FT during RV infection including Moraxella, Staphylococcus, Burkholderia and Haemophilus. As shown in figure 1, the group of PM infants had a heterogeneous taxonomic profile with a predominance of Moraxella (light blue) and Burkholderia (light green) mixed with Streptococcus, Neisseria, Staphylococcus, Janthinobacterium, and Haemophilus. In FT and PM, Haemophilus (dark purple) was mostly present during RV infection and it was overall more abundant in the PM group (figure 1).
Figure 1.
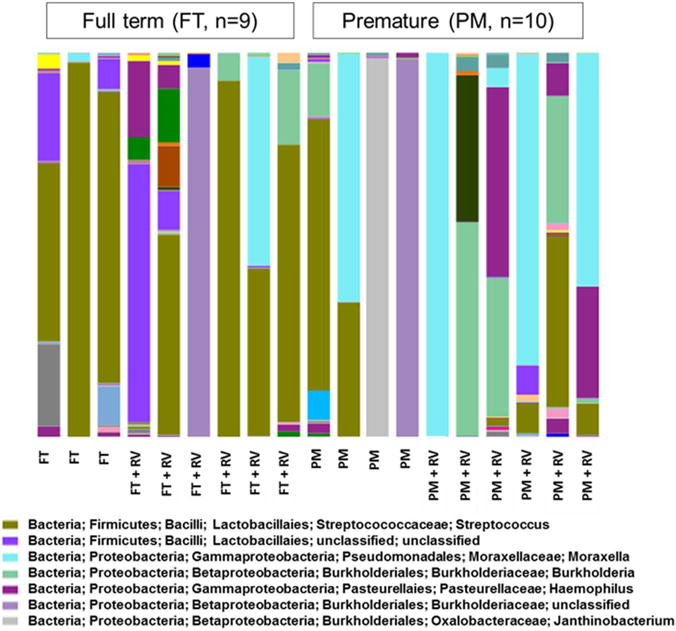
Prematurity-related signatures in global nasopharyngeal microbiota. Bars represent taxonomic profiles of 19 nasopharyngeal microbiota from 12 children (6 paired samples and 7 single samples) analyzed via PacBio sequencing of 16S rRNA amplicons. Each color represents a different genus. FT, full term; PM, premature; rRNA, ribosomal RNA; RV, rhinovirus.
Ordination analysis (PCoA—figure 2A) revealed within-group dissimilarities between FT and PM NP microbiomes. PCoA clustered FT subjects without viral infection (figure 2 red squares within ellipse) but not PM individuals due to high heterogeneity within the PM group (figure 2 scattered blue dots). These visual dissimilarities were then confirmed by the PERMANOVA and adonis tests, which showed significant differences (p<0.05) in community composition only between FT and PM (using Bray-Curtis and weighted unifrac distances). All the other comparisons resulted no significance including PCoA according to RV status.
Figure 2.
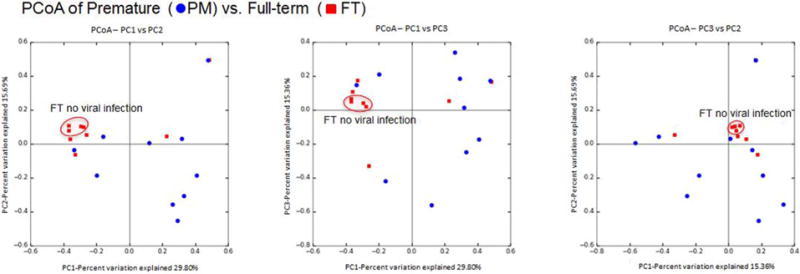
PCoA of seasonal groups (Bray-Curtis distances). These plots show dissimilarities or similarities between groups. PCoA clustered full-term subjects without viral infection (red squares within ellipse). FT, full term; PCoA, principal coordinates analysis; PM, premature.
Significant differences (p<0.05) in OTU abundance (rarefied-based and non-rarefied-based performed tests) were observed between different groups after FDR correction, including an increase in Proteobacteria and decrease in Firmicutes in the PM relative to the FT group (figure 3A). Similarly, we also detected significant differences in OTU abundance (Fisher’s exact test) between all of the six paired samples (after FDR correction; figure 3B). These differences in community composition were also manifested in the Procrustes analysis comparing the PCoA plots of microbiomes from paired samples (figure 4), which showed a great dissimilarity between pairs (M2=0.704; p<0.001). Notably, the dissimilarity between pairs, represented as the linear distance between healthy and RV samples in PCoA plots, was greater in the PM relative to the FT group (figure 4). We did not detect significant differences in α-diversity (observed OTUs, Chao1, Shannon and Faith) between any of the groups compared. Nonetheless, FT showed overall greater mean diversity than PT for all four indexes compared, while RV and non-virus comparison showed more similar estimates for the same indexes (data not shown).
Figure 3.
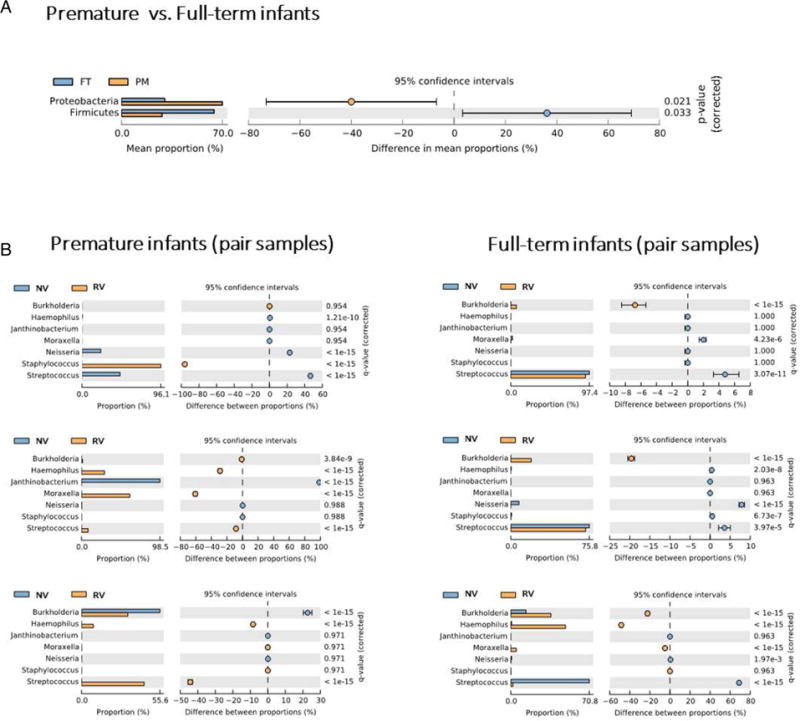
Differences in OTUs abundance. (A) Extended error bar plot showing the bacterial taxa that have a significant difference (Welch’s t-test and ANOVA; p<0.05) in proportions between groups. (B) Extended error bar plot showing proportions for the most abundant bacterial genera between sample pairs. Significance was tested by Fisher’s test (p<0.05). ANOVA, analysis of variance; FT, full term; NV, no viral infection; OUT, operational taxonomic unit; PM, premature; RV, rhinovirus.
Figure 4.
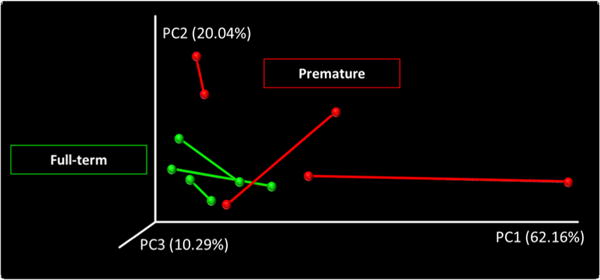
Procrustes analysis of PCoA plots of microbiota with and without RV infection. Nasopharyngeal pair samples collected from the same individual (n=6) are connected by a line that represents dissimilarity between healthy (no virus) and RV infection in the same subject. All pair samples showed dissimilarity (healthy vs RV) but dissimilarities were greater in the premature group. Red=premature; green=full term. PCoA, principal coordinates analysis; RV, rhinovirus.
DISCUSSION
The NP microbiota of newborns and infants plays a key role in modulating airway inflammation and respiratory symptoms during viral infections.8 We have shown that severely PM infants (<32 weeks GA) have abnormal nasal airway immune responses against viruses,9 10 but it is unknown if the NP microbiota of PM infants differs from that seen in FT individuals and if this difference is maintained during viral infections. Filling this important gap, in this study we found that PM infants have an NP microbiota with high within-group dissimilarity (heterogeneity) relative to FT age-matched controls. Also, our data suggest that the microbiota in PM infants is less resilient during RV infection (figure 4), suggesting that the NP microbiota of PM infants may play an important role in modulating airway inflammatory and immune responses in this vulnerable group.
There is increasing evidence demonstrating that PM infants have widespread defects in innate immunity that facilitate the inception of infections caused by bacteria, viruses and other microorganisms.27,28 In addition, prematurity appears to be associated with an immune dysregulated state characterized by abnormal secretion of proinflammatory cytokines in response to different environmental stressors.29,30 The abnormal immune responses and enhanced inflammatory responses of prematurity generate a unique host environment, which, in combination with early nosocomial exposures, results in distinct microbial communities colonizing different systems.31 32 An example is the increasingly clear link between the abnormal gut microbiota composition in PM infants and the development of necrotizing enterocolitis.32 In addition, abnormalities in the airway microbiome diversity of PM infants have also been described in association with the development of bronchopulmonary dysplasia (BPD),31 the most common chronic respiratory complication in this vulnerable population. Our current study adds another layer of complexity to the previously described abnormalities in the microbiota of PM infants, identifying significant differences in the NP microbiota of severe preterm children after NICU discharge relative to FT controls, difference that is maintained during RV infection, which is known to change the microbiota composition.7,8 These findings suggest that the PM airway microbiota is altered at baseline and can be disrupted further by relatively minor environmental challenges, such as viral illnesses.
Our taxonomic results of NP microbiota are consistent with previous data reported in infants and children.3,8 In a recent study, Teo et al8 demonstrated that the NP microbiome of infants has a simple structure dominated by six clusters: Streptococcus, Moraxella, Staphylococcus, Haemophilus, Corynebacterium and Alloiococcus (genus Dolosigranulum in some databases).8 In our study, we found NP dominance of Streptococcus and Moraxella with a scattered presence of Staphylococcus and Haemophilus. We did not identify children with NP dominance of Corynebacterium or Alloiococcus, likely because these two clusters are associated with healthy children without a history of respiratory infections;7,8 in our sample, all FT subjects had a history of RV infection. Interestingly, we found that while FT infants had NP dominance of Streptococcus (green–brown in figure 1), PM infants had a highly heterogeneous microbiota with less Streptococcus and emergence of a mixed flora including Moraxella (light blue in figure 1) and other Gram-negative bacteria (eg, Burkholderia, Neisseria and Janthinobacterium). At the phylum level, PM infants showed increased Proteobacteria (major group of Gram-negative bacteria) and decreased Firmicute (Gram-positive bacteria such as Clostridium and Lactobacillus), which is an airway microbiome signature recently described in PM newborns, particularly in those with increased BPD risk.31
The most striking feature identified in the NP microbiota of PM infants was the presence of within-group dissimilarity (high heterogeneity), suggesting that this group does not develop a stable NP microbiome in early life. In this context, it is noteworthy that recent longitudinal studies6–8 have demonstrated that by 2 months of age most humans develop a stable NP microbiota with low intrasubject variability and dominance of specific genera.8 Interestingly, the individual NP microbiome predominance reflects environmental exposures (eg, antibiotics, daycare, siblings, etc),6–8 and predicts subsequent risk to develop respiratory conditions including viral respiratory infections, pneumonia and asthma.3–8 Specifically, while NP microbiota dominated by Corynebacterium or Alloiococcus clusters is linked to less respiratory morbidity,7 8 early NP colonization with Streptococcus, Moraxella or Haemophilus is associated with an increased risk for pneumonia or bronchiolitis in early childhood.3–8 We speculate that the presence of multiple environmental challenges may prevent or delay the establishment of a stable NP microbiome in PM infants, which may play a pivotal role in the generation of prematurity-related acute and long-term respiratory complications. One of the most important environmental factors that shape the NP microbiota of infants is the presence of viral respiratory infections. Prior studies have established that RV and respiratory syncytial virus increase the abundance of Streptococcus, Moraxella and Haemophilus.7 8 33 Teo et al8 have shown that Haemophilus is rarely found in healthy NP samples of infants, but is common during and shortly after acute viral respiratory infections. In our study, we found distinct microbiota during RV infection in infants with major differences seen in the abundance of Haemophilus and Moraxella, particularly in the PM group (figure 3). Notably, the high within-group dissimilarity seen in the NP microbiome of PM infants was augmented during RV infection (figure 4), further supporting the concept that prematurity may lead to an ‘unstable’ NP microbiome in early childhood, predisposing to widespread changes in bacterial diversity changes during relatively minor ecological perturbations (viral illnesses). This may have significant pathogenic consequences because exposure to common respiratory bacteria alters the airway epithelial response to subsequent viral infection.34
A limitation of this study is that the first set of samples was obtained during a viral infection and the second set after recovery and viral clearance (negative PCR); thus, we cannot establish the baseline (prior to infection) microbiota of subjects. Although the Procrustes plot and the paired samples for FT babies are less different, we cannot conclude if this change is only due to prematurity or if it is because these communities are more resilient due to other factors such as the timing of the second sample (eg, ~1 month). Also, we could argue that FT babies may be more prone to RV since the microbiota has not changed much during and postinfection. Another limitation of our study is the small number of sample size; the microbiota changes described here could vary when more samples are analyzed.
Longitudinal studies are critically needed to further investigate the interplay between PM birth, NP microbiota and the development of airway immune responses against respiratory viruses in early life. Given that viral respiratory infections are the most common cause of hospitalization and death in PM infants,35,36 these studies may have a significant impact leading to novel diagnostic and therapeutic strategies to improve respiratory outcomes in this vulnerable population.
Significance of this study.
What is already known about this subject?
-
▸
Nasopharyngeal (NP) microbial composition correlates with individual nasal cytokine signatures, lower respiratory tract infection risk and asthma.
-
▸
Severe preterm infants have persistent abnormal nasal airway immune responses against respiratory viruses during the first 2 years of life.
-
▸
Viral infections alter the nasal airway microbiome.
What are the new findings?
-
▸
Infants born prematurely have distinct NP microbiota signatures.
-
▸
Premature infants have an NP microbiota with high within-group dissimilarity (heterogeneity) relative to full-term age-matched controls.
-
▸
Prematurity-related microbiota features persisted during rhinovirus infection.
How might these results change the focus of research or clinical practice?
-
▸
The NP microbiota of newborns and infants plays a key role in modulating airway inflammation and respiratory symptoms during viral infections. Given that viral respiratory infections are the most common cause of hospitalization and death in premature infants, identifying the microbiome changes in this population could lead to therapeutic strategies to improve respiratory outcomes in this vulnerable population.
Acknowledgments
Work partially supported by Parsons Foundation Grant; grant numbers NHLBI/HL090020 (K12 Genomics of Lung), NICHC/HD001399 (K12 Child Health Research Career Development Award), UL1TR000075 KL2TR000076. Awards from the NIH National Center for Advancing Translational Sciences and K12 Career Development Program K12HL119994, the NIH National Center for Advancing Translational Sciences (award numbers UL1TR001876 and KL2TR001877).
Funding: Parsons Foundation Grant, K12 Genomics of Lung, NHLBI/HL090020, K12 Child Health Research Career Development Award, NICHC/HD001399.
Footnotes
MCR deceased.
Competing interests: None declared.
Ethics approval: Institutional IRB.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Moss ML. The veloepiglottic sphincter and obligate nose breathing in the neonate. J Pediatrics. 1965;67:330–5. [Google Scholar]
- 2.Stocks J. The functional growth and development of the lung during the first year of life. Early Hum Dev. 1977;1:285–309. doi: 10.1016/0378-3782(77)90041-x. [DOI] [PubMed] [Google Scholar]
- 3.Følsgaard NV, Schjørring S, Chawes BL, et al. Pathogenic bacteria colonizing the airways in asymptomatic neonates stimulates topical inflammatory mediator release. Am J Respir Crit Care Med. 2013;187:589–95. doi: 10.1164/rccm.201207-1297OC. [DOI] [PubMed] [Google Scholar]
- 4.Vissing NH, Chawes BL, Bisgaard H. Increased risk of pneumonia and bronchiolitis after bacterial colonization of the airways as neonates. Am J Respir Crit Care Med. 2013;188:1246–52. doi: 10.1164/rccm.201302-0215OC. [DOI] [PubMed] [Google Scholar]
- 5.Bisgaard H, Hermansen MN, Buchvald F, et al. Childhood asthma after bacterial colonization of the airway in neonates. N Engl J Med. 2007;357:1487–95. doi: 10.1056/NEJMoa052632. [DOI] [PubMed] [Google Scholar]
- 6.Mika M, Mack I, Korten I, et al. Dynamics of the nasal microbiota in infancy: a prospective cohort study. J Allergy Clin Immunol. 2015;135:905–12.e11. doi: 10.1016/j.jaci.2014.12.1909. [DOI] [PubMed] [Google Scholar]
- 7.Biesbroek G, Tsivtsivadze E, Sanders EA, et al. Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am J Respir Crit Care Med. 2014;190:1283–92. doi: 10.1164/rccm.201407-1240OC. [DOI] [PubMed] [Google Scholar]
- 8.Teo SM, Mok D, Pham K, et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 2015;17:704–15. doi: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perez GF, Pancham K, Huseni S, et al. Rhinovirus-induced airway cytokines and respiratory morbidity in severely premature children. Pediatr Allergy Immunol. 2015;26:145–52. doi: 10.1111/pai.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pancham K, Perez GF, Huseni S, et al. Premature infants have impaired airway antiviral IFNγ responses to human metapneumovirus compared to respiratory syncytial virus. Pediatr Res. 2015;78:389–94. doi: 10.1038/pr.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez GF, Pancham K, Huseni S, et al. Rhinovirus infection in young children is associated with elevated airway TSLP levels. Eur Respir J. 2014;44:1075–8. doi: 10.1183/09031936.00049214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nino G, Huseni S, Perez GF, et al. Directional secretory response of double stranded RNA-induced thymic stromal lymphopoetin (TSLP) and CCL11/eotaxin-1 in human asthmatic airways. PLoS ONE. 2014;9:e115398. doi: 10.1371/journal.pone.0115398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toma Toma I, Siegel MO, Keiser J, et al. Single-molecule long-read 16S sequencing to characterize the lung microbiome from mechanically ventilated patients with suspected pneumonia. J Clin Microbiol. 2014;52:3913–21. doi: 10.1128/JCM.01678-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE. 2011;6:e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pérez-Losada M, Alamri L, Crandall KA, et al. Nasopharyngeal microbiome diversity changes over time in children with asthma. PLoS ONE. 2017;12:e0170543. doi: 10.1371/journal.pone.0170543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Q, Garrity GM, Tiedje JM, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paulson JN, Stine OC, Bravo HC, et al. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–2. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh B, Amado-Sierra Mdel R, Holmes D, et al. A One-pot allylation-hydrostannation sequence with recycling of the intermediate tin waste. Org Lett. 2014;16:2318–21. doi: 10.1021/ol500460u. [DOI] [PubMed] [Google Scholar]
- 21.Price MN, Dehal PS, Arkin AP. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10. [Google Scholar]
- 23.Gower JC. Generalized Procrustes analysis. Psychometrika. 1975;40:33–51. [Google Scholar]
- 24.Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30. [Google Scholar]
- 25.Parks DH, Tyson GW, Hugenholtz P, et al. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–4. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.RStudioTeam. RStudio: integrated development for R. Boston, MA: RStudio Inc; 2015. http://www.rstudio.com/ [Google Scholar]
- 27.Melville JM, Moss TJ. The immune consequences of preterm birth. Front Neurosci. 2013;7:79. doi: 10.3389/fnins.2013.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadeghi K, Berger A, Langgartner M, et al. Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. J Infect Dis. 2007;195:296–302. doi: 10.1086/509892. [DOI] [PubMed] [Google Scholar]
- 29.Hofer N, Kothari R, Morris N, et al. The fetal inflammatory response syndrome is a risk factor for morbidity in preterm neonates. Am J Obstet Gynecol. 2013;209:542.e1–542.e11. doi: 10.1016/j.ajog.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 30.Segura-Cervantes E, Mancilla-Ramírez J, González-Canudas J, et al. Inflammatory response in preterm and very preterm newborns with sepsis. Mediators Inflamm. 2016;2016:6740827. doi: 10.1155/2016/6740827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lal CV, Travers C, Aghai ZH, et al. The airway microbiome at birth. Sci Rep. 2016;6:31023. doi: 10.1038/srep31023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel RM, Denning PW. Intestinal microbiota and its relationship with necrotizing enterocolitis. Pediatr Res. 2015;78:232–8. doi: 10.1038/pr.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Steenhuijsen Piters WA, Heinonen S, Hasrat R, et al. Nasopharyngeal microbiota, host transcriptome and disease severity in children with respiratory syncytial virus infection. Am J Respir Crit Care Med. 2016;194:1104–15. doi: 10.1164/rccm.201602-0220OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellinghausen C, Gulraiz F, Heinzmann AC, et al. Exposure to common respiratory bacteria alters the airway epithelial response to subsequent viral infection. Respir Res. 2016;17:68. doi: 10.1186/s12931-016-0382-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meissner C. Viral bronchiolitis in children. N Engl J Med. 2016;374:62–72. doi: 10.1056/NEJMra1413456. [DOI] [PubMed] [Google Scholar]
- 36.Jain S, Williams DJ, Arnold SR, et al. Community-acquired pneumonia requiring hospitalization among U.S. children N Engl J Med. 2015;372:835–45. doi: 10.1056/NEJMoa1405870. [DOI] [PMC free article] [PubMed] [Google Scholar]