

Abstract
The strikingly different reactivity of a series of homo- and heterodinuclear [(MIII)(μ-O)2(MIII)III]2+ (M = Ni; M′ = Fe, Co, Ni and M = M′ = Co) complexes with β–diketiminate ligands in electrophilic and nucleophilic oxidation reactions is reported, and can be correlated to the spectroscopic features of the [(MIII)(μ-O)2(MIII)III]2+ core. In particular, the unprecedented nucleophilic reactivity of the symmetric [NiIII(μ-O)2NiIII]2+ complex and the decay of the asymmetric [NiIII(μ-O)2CoIII]2+ core through aromatic hydroxylation reactions represent a new domain for high-valent bis(μ-oxido)dimetal reactivity.
Keywords: Heterobimetallic Complex, Dioxygen activation, nucleophilic oxidant, Metal-Oxo, NIH-Shift
Unified in differences
The decisive role of the second metal ion (M) in fine tuning the electronic structure and reactivity from nucleophilic to electrophilic of a series of [Ni(μ-O)2M]2+ complexes in oxidation reactions is reported, thereby underlining the importance of subtle electronic changes in the reactivity of biologically relevant metal-dioxygen intermediates.
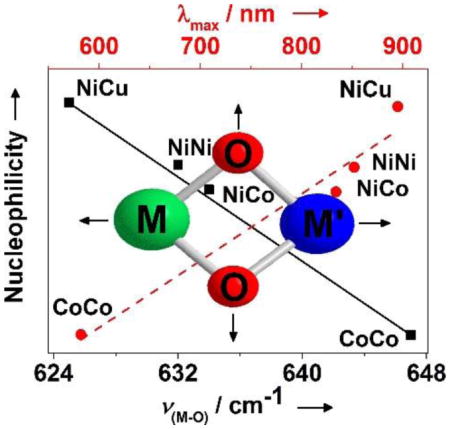
The synthesis and investigation of heterobimetallic bis(μ–oxido) complexes with [M(μ–O)2M′]n+ cores are of particular interest for modeling the structures and reactivities of active sites in chemically related metalloenzymes containing two different metal ions.[1] Moreover, such studies can lead to the discovery of novel species[2] exhibiting reactivity patterns different from their homodinuclear counterparts. The illustrative example (Scheme 1 inset) is the recently reported [L1Ni(μ-O)2Cu(MeAN)]+ (1; L1 = [HC(CMeNC6H3(iPr)2)2]; MeAN = N,N,N′,N′,N″-pentamethyl dipropylenetriamine),[3] complex which, in sharp contrast to the presence of electrophilic oxido groups in symmetric [M2(μ–O)2]n+ (M = Ni, Co, Fe and Mn) analogues,[4] possesses nucleophilic oxido ligands and deformylates aldehydes.
Scheme 1.

Generation and self-decay of the heterodinuclear [NiIII(μ-O)2CoIII]2+ complex 2 and it’s homodinuclear [MIII(μ-O)2MIII]2+ analogues 3 and 4 (M = Ni, Co). Inset: The structure of the previously reported[3] asymmetric bis(μ-oxido)NiIIICuIII compound with nucleophilic oxido ligands.
The unprecedented nucleophilic reactivity of 1 may be attributed to the asymmetric nature of the bis(μ-oxido)dimetal core. Alternatively, the nature of the ancillary ligand may also play a vital role; notably, 1 possesses a dangling amine group, which may induce the change in reactivity, leading to the observed nucleophilicity. Understanding the origin of the nucleophilicity in 1, therefore, warrants a systematic comparison of the reactivity of a series of symmetric and asymmetric dinuclear bis(μ-oxido) complexes involving similar ancillary ligands, which preferably lack any dangling basic/nucleophilic group that could affect the reactivity.
In our continuous effort to uncover structure-reactivity relationships of dinuclear metal-dioxygen intermediates, we now demonstrate spectroscopic trapping of a series of homo- and heterodinuclear [MIII(μ-O)2(MIII)′]2+ (M = Ni; M′ = Co, Ni and M = M′ = Co) complexes of β–diketiminate ligands. A comparison of the reactivity of these complexes in various oxidation reactions reveals a distinct reactivity pattern dependent on the spectroscopic properties of the bis(μ-oxido)dimetal core. Furthermore, the fully symmetric [L1NiIII(μ-O)2NiIIIL1]2+ (3) complex demonstrates a nucleophilic property, which is in contrast to the observed electrophilic behavior of all homodinuclear bis(μ-oxido) cores reported to date.[4] This together with the self-decay of the heterodinuclear [L1Ni(μ-O)2CoL3] species 2 (L3 = [HC(CHNC6H3(iPr)2)2]) by an unprecedented hydroxylation mediated 1,3-migration of an aryl-nitrogen atom, represent a novel domain for high-valent bis(μ-oxido)dimetal reactivity.
Reaction of equimolar amounts of the isolable NiII-superoxo complex [L1NiO2][5] with the CoI precursor [L3Co(C7H8)][6] at −78°C in toluene leads to immediate formation of the intermediate 2 (Fig 1a and Scheme 1) with absorption maxima λmax (ε; M−1cm−1) centered at 576 (1651) and 834 nm (2700). Notably, the near-IR absorption feature of 2 at 834 nm is blue-shifted relative to 1 (Table 1).[3] Moreover, the thermal stability of 2 (t1/2 = 246 s in toluene at −50 °C) is significantly lower than that of 1 (t1/2 = 900 s in CH2Cl2 at −50 °C).[3] The electrospray mass spectrum (ESI-MS; Fig S1) of 2 exhibits the most prominent peak at m/z = 955.4904, with a mass and isotope distribution pattern corresponding to [L1Ni(μ-O)2CoL3]. The rRaman (Fig 1C) spectrum of 2 using 413 nm laser excitation supports the [Ni(μ-O)2Co]2+ assignment of 2. The band at ν = 634 cm−1 of 2-16O16O shifts down to 600 cm−1 [ν(2-18O18O)] in the corresponding 18O2-substituted complex 2. The X-band EPR spectrum (Fig S2) shows a rhombic S=1/2 signal (75 % yield based on spin-quantification) with gx = 1.98, gy = 2.12, gz = 2.76, and observable 59Co hyperfine splitting at gx and gz. The electronic structure of 2 is thus best represented as [L1NiIII(μ-O)2CoIIIL3], where antiferromagnetic coupling between Ni(III) (3d7; S=1/2) and Co(III) (3d6; S=1) ions leads to an S=1/2 ground state.
Figure 1.

A: Absorption spectra of 2–4 at −78 °C in toluene. B–D: rRaman spectra (413 nm excitation) of 2–4 in toluene at −90 °C: B. 3-16O2 in d8-toluene (solid trace) and 3-18O2 in toluene (dash-dotted trace). C: 2-16O2 in d8-toluene (solid trace) and 2-18O2 in toluene (dash-dotted trace). D: 4-16O2 in d8-toluene (solid trace) and 4-18O2 in d8-toluene (dashed trace). See also Fig S22.
Table 1.
Spectroscopic and reactivity properties of 1–4.
X-ray absorption spectroscopy (XAS) studies were performed at the Ni and Co K-edges to directly probe the metal oxidation states in 2. Notably, the Ni K-edge spectrum of 2 is virtually indistinguishable to that reported previously for 1 (where the oxidation state of Ni has been unambiguously determined to be +3; Fig S3A),[3] although the pre-edge is slightly more intense and sharper in 2 (Fig S3A inset). The higher pre-edge intensity (Table S1) of 2 (6.6 units) relative to 1 (5.4 units) at the Ni K-edge may originate from the larger deviation from the ideal square (sq) planar geometry[7] at the Ni center of 2 (dihedral angle ⊗ = 172°; Table S2) relative to 1 (⊗ = 178.1°; Table S2) in the optimized structures derived from Density Functional Theory (DFT) calculations. In addition, a comparison of the Co K-edge spectrum of 2 with that of [L3Co(C7H8)] shows a blue shift in the edge energy (Fig S3B) together with an upshift (from 7709 eV in [L3Co(C7H8)] to 7709.8 eV in 2; Table S1) and intensity increase in the pre-edge peak of 2 (16.9 units) vs [L3Co(C7H8)] (6.2 units). This strongly suggests a change in Co oxidation from +1 to +3 during the conversion of [L3Co(C7H8)] to 2. Thus XAS data also are consistent with a [L1NiIII(μ-O)2CoIIIL3] assignment for 2.
Extended X-ray Absorption Fine Structure (EXAFS) analyses revealed further structural details. For 2 a shell of four N/O scatterers at 1.86 Å is obtained from Ni EXAFS (Table S3-1, Fig S4A), which is in excellent agreement with the DFT calculated average Ni-N/O distance of 1.88 Å for 2. In addition, a peak at 2.78 Å corresponding to the Co scatterer is observed, thereby strongly supporting the presence of a heterodinuclear Ni···Co center in 2. Further support for a Ni···Co separation of ca. 2.78 Å comes from Co EXAFS (Table S3-2, Fig S4B), which detects a Ni scatterer at 2.72 Å. In addition the best fit of the Co EXAFS can be obtained with four N/O scatterers at 1.82 Å. Notably, the DFT calculated average Co-N/O and Co-Ni distances of 1.87 and 2.77 Å, respectively, are again in reasonable agreement with the experiment.
The [L1NiIII(μ-O)2NiIIIL1]2+ (3) and [L3CoIII(μ-O)2CoIIIL3]2+ (4) complexes, which are homometallic (symmetric) analogues of 2, have also been synthesized by reacting [L1NiIIO2] with [(L1Ni)2(C7H8)] and [L3Co(C7H8)] with dioxygen, respectively, in toluene at −78 °C (Scheme 1). Table 1 lists rRaman, absorption spectra and t1/2 data for 1–4. Notably, complex 4 is unique in the series with a significantly blue shifted absorption band (Fig 1a), higher energy metal-oxo (ν(M-O)) stretching vibration frequency and higher stability relative to 1–3. Thermal stability of 4 at room temperature enabled its isolation as dark blue crystals in 80% yields; the X-ray crystal structure (Scheme 1, Fig S5, Table S4) consists of two L3Co-O fragments (Co-O distance of 1.786(1) Å) related by inversion and separated by Co…Co = 2.6715(5) Å. The short metal-metal and metal-oxo distances found in 4 compare favorably to those in related “diamond core” structures.[4] A solution magnetic moment (μeff, C6D6, 298 K) of 2.55 μB for 4 suggests population of a higher spin state at elevated temperatures. The Ni sites in 3 are, however, antiferromagnetically coupled at 298 K stabilizing the singlet (S=0) state. DFT calculations provide some insight into the differences in the magnetic properties of 4 and 3. The parallel spin coupling in 4 can be ascribed to the slightly folded Co2O2 structure, in which two cobalt planes make an angle (∅) of 178° (calculated ∅= 171.7°; Table S2) and initiates ferromagnetic interaction due to loss of orbital overlap. In contrast, a nearly planar Ni2O2 structure (∅ =179.8°; Table S2) is predicted by DFT for 3, which may explain the strong antiferromagnetism leading to an S = 0 ground state. Similar diminution of antiferromagnetic couplings due to a folded Co2O2 core have been observed earlier.4g,h Moreover, the Co-Co separation of 2.6715 Å (DFT: 2.66 Å) in 4 is significantly shorter than the DFT calculated Ni-Ni separation of 2.77 Å (Table S9) for 3. This reduction favors the direct interaction, increasing also the ferromagnetic contribution.
Table 1 (see also Fig S6 and Table S5) also compares the kinetic data for the four complexes with respect to their rates of nucleophilic and electrophilic reactions. In one set of experiments, the nucleophilic oxidation of benzoyl chloride (PhCOCl) by 2–4 was investigated in toluene solutions, resulting in about 80% yield of benzoic acid (Fig 2 and Fig S7). Comparison of the reaction rates with that reported previously for 1[3] reveals a reactivity order of 1>3>2>4. This trend is also found to be valid for reactions with 2-phenylpropionaldehyde (2-PPA), (Fig 2, Fig S8, Table S5) to form acetophenone and formate. Furthermore, when the logarithms of the second order rate constants (log k2) were plotted versus λmax or ν(M-O), an excellent linear correlation was observed (Fig 3), demonstrating that λmax or ν(M-O) reflect the relative nucleophilicities of the bis(μ-oxido)dimetal units in 1–4. The reactivity of 2 and 3 was further investigated using a series of para substituted benzaldehydes (Fig S9). A positive ρ value of 1.33 and 2.26 in the Hammett plots were obtained for 2 and 3, respectively (Fig S10), which, when compared to the ρ value of 2.83 reported previously for 1,[3] again supports the nucleophilicity order of 1>3>2. These results demonstrate that the bis(μ-oxido)dimetal cores of 2 and 3 are active nucleophilic oxidants, similar to that reported previously for 1.
Figure 2.

Reactivity studies of complexes 2–4. Yields are given relative to the metal complexes.
Figure 3.
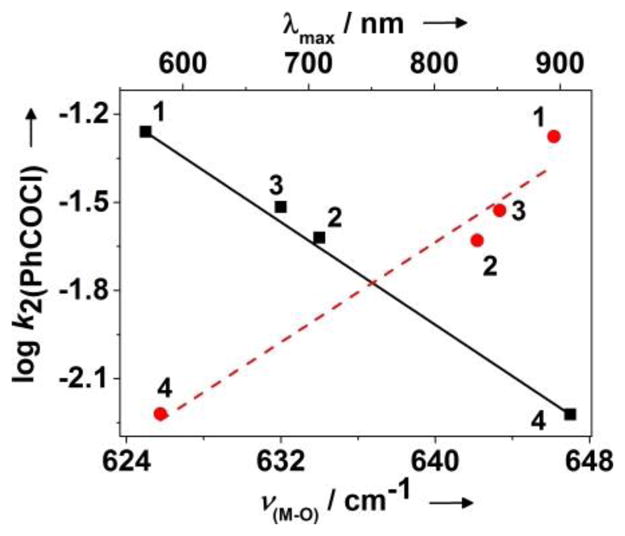
Linear correlation of the logarithms of k2 (PhCOCl) vs λmax (filled circles and dashed line) or ν(M-O) (filled squares and solid line) of the bis(μ-oxido)dimetal units in 1–4.
For comparison, the hydrogen atom abstraction (HAT) rates of 1–4 were also investigated with 1,4-cyclohexadiene (CHD), and substituted phenols. They (Fig S11; Table S5) follow the reactivity order 2>3>4>1. Interestingly, the linear correlation observed for the log k2 values for PhCOCl oxidation versus λmax or ν(M-O) values does not extend to HAT rates of 1–4 (Fig S12 B; Table S5). While the respective rate values associated with 1–3 for the oxidation of CHD follow a linear correlation similar to that observed for PhCOCl, 4 deviate significantly from this pattern and exhibit HAT rates that are few orders of magnitude below what are predicted by these lines. Clearly, the nucleophilic and electrophilic oxidation reactions are not governed by a common set of factors. Based on the observed HAT rates 2 can be considered as the most electrophilic of all the complexes 1–4. Replacement of the Co atom in [L1Ni(μ-O)2CoL3] with Fe further increases the electrophilicity of the bis(μ-oxido)dimetal core to an extent that the [L1Ni(μ-O)2FeL3] (5) complex becomes extremely unstable against self-decay by an intramolecular ligand hydroxylation pathway (Scheme 1). Thus, monitoring of the reaction of [L1NiIIO2] with [L3Fe(C7H8)][8] in toluene by means of UV–Vis spectroscopy (Fig S13) at −90°C did not lead to the observation of any intermediate species. The molecular structure of the resultant product 6 (> 90% yields; Fig S14, Table S7) reveals the presence of β-diketiminato-ligated NiII and FeII centers[9] in sq planar and tetrahedral (Td) coordination environments, respectively. The two metals are bridged by μ-hydroxido (Fig S15) and μ-alkoxido units derived from C-H activation of an isopropyl (iPr) group belonging to the β-diketiminato-ligand bound at the nickel site.
In contrast, decay of a solution of 2 led to the hydroxylation of one of the arene (Scheme 1, Fig S16, Table S7) residues in the major (> 75% yield relative to 2) decay product 7. The X-ray crystal structure analysis of 7 shows a heterodinuclear complex, with β-diketiminato-ligated NiII and CoII centers in sq planar and Td coordination environments, respectively. The oxidation state assignments are based on XANES (Fig S17) studies; Ni K-edge of 7 is near-identical to that of the [L1NiIIO2] precursor (consistent with sq planar NiII) and the Co K-edge of 7 reveals an edge energy intermediate between the CoI precursor, [L3Co(C7H8)], and 2 (consistent with CoII). Notably, the metal atoms are bridged by μ-hydroxido and μ-phenoxido units (Fig S18) derived from hydroxylation induced 1,3- migration of the aryl-nitrogen atom of the β-diketiminato ligand bound at the Ni site. The conversion of 2 to 7 is therefore reminiscent of the NIH shift reaction suggested for enzymatic aromatic hydroxylations.[10] This involves 1,2-migration of an aryl-X (X=H, CH3 or Cl) atom during rearomatization of an oxygenated intermediate (Fig S19A). The cleavage of the aromatic C-N bond during the conversion of 2 to 7 similarly suggests a decay mechanism (Scheme 2; also supported by DFT, Fig S19B) involving intramolecular electrophilic attack of the [NiIII(μ-O)2CoIII]2+ core on an aromatic ring of the Ni-bound β-diketiminato-ligand leading to a cationic intermediate; the rearomatization process now involves cleavage of a C-N bond to form a Ni-nitrene species,[11] which then effects H-atom abstraction from an isopropyl iPr group to yield 7. Thermal decay of a solution of 3 led to the isolation of crystals of the previously reported[5] bis(hydroxido)dinickel(II) species (Fig S20) in 15% yield. The formation of such doubly protonated reduced species has been previously observed in reactions of basic bis(μ-oxido)dimetal cores[12] with protons and is consistent with the presence of nucleophilic oxido ligands in 3. In contrast to the metastable properties of 2, 3 and 5, complex 4 is considerably stable at 25 °C, and no significant decay was observed at this temperature over a period of 24 hours.
Scheme 2.

Self-decay of 2 by an electrophilic aromatic hydroxylation mechanism to form 7.
DFT calculations of 1–5 provided further insight into the different reactivity patterns of the homo and heterodinuclear bis(μ-oxido) cores. The geometry and the ground spin state of the complexes 1–4 are nicely reproduced in the calculation (Tables S8–S9). A quintet (S = 2) ground-state is predicted for 5 akin to 4, which is consistent with the predicted short Ni-Fe distance of 2.69 Å and a folded NiO2Fe core (∅=166.7°; Table S2). Furthermore, the calculated charge densities (QO) on the bridging O-atoms of 1–4 when plotted against the log k2 for the PhCOCl oxidation reactions also fall on a line very similar to the linear correlation obtained from log k2 vs λmax and ν(M-O) plots (Fig S12A). This correlation is however not valid for electrophilic reactions. For example, the room temperature stability of 4 contrasts with the facile decay of 5 by an electrophilic intramolecular ligand hydroxylation step (that prevents its isolation even at low temperatures), although identical QO values (Table S10) have been calculated in both cases. For comparison calculations were also performed on the hypothetical [L1Ni(μ-O)2CuL3] complex (8)[13] that unlike 1 contains a β–diketiminate ligand attached to the Cu center. A trend of increasing QO and decreasing covalency of the [Ni(μ-O)2M] (M= Fe, Co, Ni, Cu) cores with increasing atomic weight of M is now obvious, as also evidenced by the decreasing M-O Mayer Bond Order and increasing Ni-M distances (Tables S9 and S10).
In conclusion, this study presents the first example showing that a fully symmetric bis(μ-oxido)dimetal complex 3 can act as a nucleophilic oxidant in addition to demonstrating the typical HAT reactivities. 3 reveals a characteristic absorption band in the near infrared (IR) region, which acts as a spectroscopic marker for nucleophilic bis(μ-oxido)dimetal cores. Furthermore, the nature of the second metal ion (M) is shown to systematically modulate the electronic structure and reactivity of a series of β–diketiminate [Ni(μ-O)2M]2+ complexes with M = Fe, Co Ni, and Cu. The combined experimental and computational evidences herein indicate that the decreasing nucleophlicity of the oxygen atoms in the order [NiIII(μ-O)2CuIII]2+ > [NiIII(μ-O)2NiIII]2+ > [NiIII(μ-O)2CoIII]2+ > [NiIII(μ-O)2FeIII]2+ correlates to the increasing covalency of the M-O bonds, owing to an increase in the extent of O→M π-backdonation (Fig S21). For M=Fe (d5) with half-filled π*(xz, yz) levels O→M π-backdonation is maximum with the most electrophilic oxygen atoms. The additional electrons in the Co(d6), Ni(d7) and Cu(d8) complexes subsequently fill up the π*(xz, yz) levels and reduce the O→M π-backdonation to an extent that there is essentially no multiple bond character present in the [NiIII(μ-O)2CuIII]2+ complex. Additionally, with increasing oxido nucleophilicity, the energy of the oxygen based highest occupied molecular orbital (HOMO) is found to increase thereby decreasing its energy separation from the metal/β–diketiminate ligand-based lowest unoccupied molecular orbital (LUMO) in the order [NiIII(μ-O)2FeIII]2+ > [NiIII(μ-O)2CoIII]2+ > [NiIII(μ-O)2NiIII]2+ > [NiIII(μ-O)2CuIII]2+ (Fig 4). We tentatively assign the near IR absorption band of the [Ni(μ-O)2M]2+ complexes to originate from this HOMO→LUMO transition, which may explain the red shift of this band with increasing nucleophilicity of the oxido groups. Therefore, the present study highlights the importance of the interplay between metal centers and local environment in governing key physical and chemical properties of biologically relevant dinuclear metal-dioxygen intermediates.
Figure 4.

Nature of the HOMOs and LUMOs and their absolute energies as obtained from the DFT calculations on a series of [L1Ni(μ-O)2ML3] (M= Fe, Co, Ni and Cu) complexes. ΔE represents the HOMO-LUMO energy gap.
Supplementary Material
Acknowledgments
Financial support from the DFG (Cluster of Excellence “Unifying Concepts in Catalysis”; EXC 314-2) is gratefully acknowledged. K.R. also thanks the Heisenberg-Program of DFG for financial support. T.S. is grateful for generous support by The New Széchenyi Plan TAMOP-4.2.2/B-10/1-2010-0009. XAS experiments were conducted at SSRL beamline 2-2 (SLAC National Accelerator Laboratory, USA), with support from the DOE Office of Science (DE-AC02-76SF00515 and DE-SC0012704) and NIH P30-EB-009998.
References
- 1.a) Kaila VR, Verkhovsky MI, Wikström M. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]; b) Klinman JP. Chem Rev. 1996;96:2541–2562. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]; c) Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M. J Biol Chem. 2006;281:8981–8990. doi: 10.1074/jbc.M509785200. [DOI] [PubMed] [Google Scholar]
- 2.a) Garcia-Bosch I, Ribas X, Costas M. Eur J Inorg Chem. 2012:179–187. [Google Scholar]; b) Mankad NP. Chem Eur J. 2016;22:1–9. [Google Scholar]
- 3.Kundu S, Pfaff FF, Miceli E, Zaharieva I, Herwig C, Yao S, Farquhar ER, Kuhlmann U, Bill E, Hildebrandt P. Angew Chem Int Ed. 2013;52:5622–5626. doi: 10.1002/anie.201300861. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:5732–5736. [Google Scholar]
- 4.a) Kieber-Emmons MT, Riordan CG. Acc Chem Res. 2007;40:618–625. doi: 10.1021/ar700043n. [DOI] [PubMed] [Google Scholar]; b) Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hikichi S, Yoshizawa M, Sasakura Y, Komatsuzaki H, Morooka Y, Akita M. Chem Eur J. 2001;7:5011–5028. doi: 10.1002/1521-3765(20011203)7:23<5011::aid-chem5011>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]; d) Tinberg CE, Lippard SJ. Acc Chem Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mandimutsira BS, Yamarik JL, Brunold TC, Gu W, Cramer SP, Riordan CG. J Am Chem Soc. 2001;123:9194–9195. doi: 10.1021/ja016209+. [DOI] [PubMed] [Google Scholar]; f) Citek C, Herres-Pawlis S, Stack TDP. Acc Chem Res. 2015;48:2424–2433. doi: 10.1021/acs.accounts.5b00220. [DOI] [PubMed] [Google Scholar]; g) Larsen PL, Parolin TJ, Powell DR, Hendrich MP, Borovik AS. Angew Chem Int Ed. 2003;42:85–89. doi: 10.1002/anie.200390060. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2003;115:89–93. [Google Scholar]; h) Dai X, Kapoor P, Warren TH. J Am Chem Soc. 2004;126:4798–4799. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]; j) Que L, Jr, Tolman WB. Angew Chem Int Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Yao S, Bill E, Milsmann C, Wieghardt K, Driess M. Angew Chem Int Ed. 2008;47:7110. doi: 10.1002/anie.200802234. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:7218. [Google Scholar]
- 6.Yao S, Lindenmaier N, Xiong Y, Inoue S, Szilvási T, Adelhardt M, Sutter J, Meyer K, Driess M. Angew Chem Int Ed. 2015;54:1250–1254. doi: 10.1002/anie.201409469. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2015;127:1266–1270. [Google Scholar]
- 7.Shadle SE, Penner-Hahn JE, Schugaryl HJ, Hedman B, Hodgson K, Solomon EI. J Am Chem Soc. 1993;115:767–776. [Google Scholar]
- 8.Yao S, Szilvási T, Lindenmaier N, Xiong Y, Inoue S, Adelhardt M, Sutter J, Meyer K, Driess M. Chem Commun. 2015;51:6153–6156. doi: 10.1039/c5cc00147a. [DOI] [PubMed] [Google Scholar]
- 9.Assignment based on a previous study; see Yao S, Herwig C, Xiong Y, Company A, Bill E, Limberg C, Driess M. Angew Chem Int Ed. 2010;49:7054–7058. doi: 10.1002/anie.201001914.Angew Chem. 2010;122:7208–7212.
- 10.a) Guroff G, Daly JW, Jerina DM, Renson J, Witkop B, Udenfriend S. Science. 1967;157:1524–1530. doi: 10.1126/science.157.3796.1524. [DOI] [PubMed] [Google Scholar]; b) Bassan A, Blomberg MRA, Siegbahn PEM. Chem Eur J. 2003;9:4055–4067. doi: 10.1002/chem.200304768. [DOI] [PubMed] [Google Scholar]
- 11.Ray K, Heims F, Pfaff FF. Eur J Inorg Chem. 2013:3784–3807. [Google Scholar]
- 12.a) Aboelella NW, York JT, Reynolds AM, Fujita K, Kinsinger CR, Cramer CJ, Riordan CG, Tolman WB. Chem Commun. 2004:1716. doi: 10.1039/b404640d. [DOI] [PubMed] [Google Scholar]; (b) York JT, Llobet A, Cramer CJ, Tolman WB. J Am Chem Soc. 2007;129:7990. doi: 10.1021/ja071744g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.8 could not be synthesized owing to the lack of [L3CuI] precursor complex (see Shimokawa C, Tachi Y, Nishiwaki N, Ariga M, Itoh S. Bull Chem Soc Jpn. 2006;79:118–125.).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.