

Abstract
One third of type-2 diabetic patients respond poorly to metformin. Despite extensive research, the impact of genetic and non-genetic factors on long-term outcome is unknown. In this study, we combine non-linear mixed effect modeling with computational genetic methodologies to identify predictors of long-term response. 1056 patients contributed their genetic, demographic and long-term HbA1c data. The top 9 variants (of 12,000 variants in 267 candidate genes) accounted for approximately 1/3 of the variability in the disease progression parameter. Average serum creatinine level, age and weight were determinants of symptomatic response, however explaining negligible variability. Two SNPs in CSMD1 gene (rs2617102, rs2954625) and one SNP in pharmacologically relevant SLC22A2 gene (rs316009) influenced disease progression, with minor alleles leading to less and more favorable outcomes respectively. Overall, our study highlights the influence of genetic factors on long-term HbA1c response and provides a computational model, which when validated, may be used to individualize treatment.
Metformin is the first line of therapy for treatment of type 2 diabetes (T2D) and is one of the most frequently prescribed drugs worldwide1–3. Response to the drug is highly variable; greater than 30% of patients taking metformin are considered poor responders and require additional medications such as sulfonylureas and insulin instead of metformin. Metformin lowers both basal and postprandial glucose in patients with T2D and works by inhibiting hepatic glucose production, reducing intestinal glucose absorption, and improving glucose uptake and utilization2,4. Glycosylated hemoglobin (HbA1c) is formed through a non-enzymatic and irreversible reaction between hemoglobin and glucose and is the primary surrogate biomarker for long-term glycemic control and drug response, reflecting the average glucose levels circulating in the blood over previous months5. This biomarker has been shown to be more reliable than fasting plasma glucose in assessing long-term efficacy; several studies have shown that HbA1c levels are strongly linked to adverse T2D-related cardiovascular outcomes and mortality6–8.
Baseline HbA1c levels vary significantly in the T2D population, from 5.5% (37 mmol/mol) to 15% (140 mmol/mol))9,10. Most studies have focused on uncovering the effect of genetic variants in pharmacokinetic (PK) genes on static pharmacological phenotypes of metformin and fail to address the variable nature of metformin response2,10–14. One of the largest studies to date, a genome-wide association study on metformin response in individuals from the United Kingdom, identified variants near the Ataxia Telangiectasia Mutated locus associated with the ability to achieve HbA1c below 7% (53 mmol/mol) in the first 18 months of metformin treatment15. Finally, despite many studies having demonstrated associations between single nucleotide polymorphisms (SNPs) in biologically relevant genes with metformin PK and pharmacodynamics (PD), each variant accounts for only a small fraction of the variation in HbA1c levels.
To date, there have been no studies on the effect of genetic and demographic variables on long-term changes of HbA1c in patients on metformin. These factors may influence the drug’s efficacy or the patient’s underlying disease progression and, once accounted for, may make it easier to detect responders and non-responders to metformin16. The traditional approach considers a glycemic HbA1c change from baseline to evaluate the effectiveness of the drug. This approach however, effectively collapses the time dimension in the data by disregarding the actual trajectory of the biomarker and disease status over time. As a result, this method not only ignores crucial information on disease progression, but also lumps together the short-term effects of a treatment with the long-term effects on the disease.
Longitudinal disease progression analysis allows for a quantitative assessment of drug treatment effect on the time-course of the disease/biomarker. Computational methods use mathematical models to describe or predict changes in the disease status as a function of time16. These methods allow researchers to understand the role of genes as well as any relevant demographic predictors on specific response curve characteristics (such as disease progression and the long-term dynamics of therapeutic effects). Non-linear Mixed Effect Analysis (NLME) is a powerful statistical approach used for this longitudinal analysis that effectively enhances the signal-to-noise ratio and enables the utilization of all data points, irrespective of study design17,18,19.
To date, current mathematical models that capture the time-course of HbA1c in relation to metformin therapy have been limited by small sample sizes and sparse measurements16,20,21. Furthermore, a comprehensive genetic analysis linking genetic variants to long-term HbA1c trajectories has not yet been performed and consequently, there is no current knowledge regarding the influence of genetics on long-term HbA1c dynamics.
The aim of this research is to explain the variance in long-term response, linking genes, demographics, and clinical factors to the upward trajectory of HbA1c levels (a marker of disease progression) using a rich, long-term HbA1c dataset from patients on metformin (Figure 1).
Figure 1.

Workflow of longitudinal modeling, genetic analysis and the potential clinical impact on individualizing metformin therapy. Longitudinal HbA1c modeling is followed by a clinical/demographic analysis of model parameters using a step-wise approach. Once model parameters have been corrected for by clinical and demographic factors, a genetic analysis pipeline was deployed using multiple approaches; Disease-based and pharmacologically relevant genes were selected as part of the candidate gene selection. A hyperlasso regression and a mode-based approach were sequentially used to develop the final HbA1c model. Simulations were then performed using the final HbA1c model in order to determine the clinical impact of identified clinical, demographic and genetic factors. This work sets the stage for future research groups to replicate and validate the clinical impact of identified factors on external datasets.
RESULTS
Summary of data
Baseline characteristics of patients with T2D are summarized in Table 1. A total of 7822 HbA1c measurements from 1056 patients were used to develop a mathematical model of longitudinal HbA1c levels.
Table 1.
Baseline characteristics of patients with Type II Diabetes
| Clinical Site | N (%) |
|---|---|
| Total patients | 1056 |
| Kaiser South East | 154 (15%) |
| Marshfield Clinic | 150 (14%) |
| Vanderbilt | 251 (24%) |
| Kaiser Northern California | 501 (47%) |
| Categorical Variable | N (%) |
| Males | 415 (61%) |
| Females | 641 (39%) |
| European Americans1 | 376 (36%) |
| African Americans | 665 (63%) |
| Asian Americans and Others | 15 (1%) |
| Continuous Variable | Median (range) |
| Age (years) | 55 (23–90) |
| Body weight (kg) | 96 (34–212) |
| Average Serum Creatinine (mg/dL) | 0.91 (0.5–2.0) |
| Baseline HbA1c (%) | 7.6 (5.6–17.9) (60 mmol/mol (38–172 mmol/mol) |
| Metformin daily dose (mg) | 1000 (200–2500) |
| # HbA1c samples/patient | 5 (1–45) |
| Years on study | 1.43 (0.28–13.5) |
Ethnicities reported are all self reported.
This is the average daily dose of metformin calculated from metformin start day up to the day, where minimum HbA1c levels were achieved between 3–18 months (and before other anti-diabetic drug or insulin was added). There was one patient, as noted in the electronic medical record, who had <250 mg average metformin dose due to an early stop of metformin (at 1000 mg) for several months and then restarted the metformin at 500 mg. As a result, the average metformin dose was <250mg.
Of the 7822 total HbA1c measurements, 2928 HbA1c samples (37%) were collected after 2 years following metformin initiation across 344 patients (33%). 1220 HbA1c measurements (15.6%) were collected after 5 years following metformin initiation across 202 patients (19%). 555 HbA1c samples (7%) were collected after 7 years following metformin initiation across 123 patients (12%).
The dataset has a stronger representation of African Americans (63%) compared to European Americans (36%). The average length of time that each patient was under study was 2.78 years (median of 1.43 years, range of 0.28–13.5 years). Mean HbA1c samples provided per patient available for analysis was 7.5 (58 mmol/mol) (median of 5, range of 1–45). Of the 1056 patients, 1220 HbA1c measurements (15.6%) were available for 202 patients (19%) 5 years following metformin initiation. 123 patients (12%) 7 years after metformin initiation, and 28 patients (3%) 10 years after.
Mathematical model development
A turnover HbA1c model with a reversible metformin effect on the synthesis rate of HbA1c best characterized the data. A reversible (symptomatic) metformin effect was implemented because it was assumed that the drug does not directly impact the disease progression and this structure was supported by the data22. The upward trajectory (disease progression) of HbA1c over time was modeled by implementing a separate compartment that represented the HbA1c synthesis rate: KIN(t). Model mechanics and the interplay of disease progression, HbA1c synthesis rate and %HbA1c level over time can be viewed in Supplementary Figure 1. In the model structure, KIN was increasing due to disease progression, which is quantified by the disease progression parameter. The disease progression parameter generates a nonlinear increase of KIN over time, especially when the estimate of disease progression is high. A time dependent increase in the HbA1c synthesis rate captured well the upward HbA1c trajectory observed in the data. In the model, between-subject variability (BSV) was estimated for baseline HbA1c, the magnitude of metformin’s effect (an individual’s specific HbA1c relative change from baseline), and disease progression. The inclusion of a full covariance block for all BSV parameters resulted in a significant improvement in the likelihood ratio. Final selection of the model was based on improvements in the objective function value and visual predictive checks of the longitudinal HbA1c data. Through simulations, the “onset’ of disease progression, which is defined by the time point at which HbA1c levels start to increase (i.e. an upward slope in HbA1c levels), was investigated. The model predicted that the onset of disease progression for a typical patient on metformin is approximately 321 days; at which point, HbA1c levels increased at a rate of 0.1% (1.1 mmol/mol) [0.07%–0.13%] per year through the first three years (Table 2). For patients not on metformin, the model predicts that HbA1c levels would increase at a steady state rate of approximately 0.16% (1.7 mmol/mol) [0.08%–0.22%] per year. Mathematical model parameters along with clinically derived parameters are summarized in Table 2.
Table 2.
Population pharmacodynamic model derived estimates and bootstrap results for model parameters
| Final Model Parameter | Median (%RSE)1 | Median (90% CI)2 |
|---|---|---|
| Baseline HbA1c Level (%) | 7.74 (1) | 7.73 (7.6–7.8) |
| Half Life of Effect (days) | 40.9 (6) | 41.2 (36.8–45.7) |
| Metformin Effect Magnitude EFF | 13.1% (5) | 13.0 (12.1–14.4) |
| Disease Progression Estimate DISPR3 (all patients) | 82.2 (67) | 75.3 (32.6–249) |
| Boxcox transformation parameter on Baseline | 2.38 (9) | 2.41 (1.99–2.78) |
| Boxcox transformation parameter on DISPR | −0.246 (15) | −0.26 (−0.31–−0.20) |
| KLOSS | 0.205 (86%) | 0.266 (0.05–0.657) |
| Between-subject variability (% variance) | ||
| Between-subject variability (Baseline) | 16.9 (3) | 16.6 (15.9–17.8) |
| Between-subject variability (Metformin Effect Magnitude METFEFF) | 76.4 (4) | 75.9 (71.7–81.6) |
| Between-subject variability (Disease Progression DISPR) | 324 (17) | 390 (164–418) |
| Covariance of parameters (%) | ||
| Correlation Baseline-METFEFF | 0.114 (1) | 0.11 (0.101–0.136) |
| Correlation Baseline-DISPR | 0.033 (3.6) | 0.03 (−0.07–0.14) |
| Correlation DISPR-METFEFF | 0.204 (21) | 0.31 (−0.42–0.95) |
| Residual error model | ||
| Proportional error (%) | 0.098 (3) | 0.098 (0.092–0.101) |
| Additive error | 0.1 (FIXED) | 0.1 (NA) |
| Derived Clinical Parameters | Simulated Median (90% CI) | |
| Estimated onset of disease progression4 | 321 (309–332) days | |
| Estimated yearly rate of HbA1c increase on Metformin4 | 0.1 %HbA1c (0.07–0.13) | |
| Estimated yearly rate of HbA1c increase not on Metformin4 | 0.16 %HbA1c (0.08–0.22) | |
Typical value of parameter in final model. RSE= Relative standard error (%), also known as the precision of the parameter estimate.
Confidence interval for the population pharmacodynamic parameter following bootstrap results. Covariance of parameters are shown in untransformed format.
DISPR is the disease progression model parameter that affects the synthesis rate of HbA1c and longitudinal HbA1c levels through the following equations. (1) DADT(A1) = KON*(1+DISPR) - KLOSS*A(1) and (2) DADT(A2) = A(1)*(1-METFEFF) - KOUT*A(2). Where A(1) represents the synthesis rate of HbA1c (KSYN), and A(2) represents HbA1c levels.
Yearly rate of HbA1c increase was based on simulated median yearly increase over the first three years following the onset of disease progression (i.e. 321 days). The median and 90%CI of the onset and yearly rate of HbA1c increase was calculated across simulations. For example, each simulation provided a median, which was then summarized across 1000 simulations.
Final demographic/clinical covariate model
As determined by model diagnostics, the demographic-corrected mathematical model adequately described the data (Figure 2). As expected, average serum creatinine level (a likely surrogate for metformin drug exposure) was a significant predictor on MetfEFFECT, with higher levels leading to improved HbA1c response. Through simulations, a typical patient with a 0.6 mg/dL creatinine level is expected to result in a 0.77% (8.4 mmol/mol) HbA1c improvement from baseline (at 2 years), whereas a patient with a 1.3 mg/dL creatinine level is expected to result in a 0.96% improvement in HbA1c (10.5 mmol/mol) from baseline. This response characteristic is anticipated as pharmacologically, average exposure of metformin is expected to increase by approximately by 20% with a 0.7 mg/dL increase (from 0.6 to 1.3) in serum creatinine level for males and females of age 50.
Figure 2.

Longitudinal HbA1c levels over time and model based visual predictive check. The plot to the left shows raw HbA1c observations over time. On the right plot, a visual predictive check is shown, where the solid black line highlights the median observed profiles. The shaded regions indicate the 95th and 5th percentiles (ends) and the range of median simulated profiles (center) of simulated predictions from the visual predictive check.
Additionally, body weight and clinical site were significant covariates on the MetfEFFECT model parameter. Body weight was inversely related to metformin effect, estimated to result in a 6% decrease in metformin’s effect parameter per 10-kilogram increase in body weight. Clinically, this would result in a 0.99% and 0.80% change in HbA1c (equivalent of 10.8 and 8.7 mmol/mol) from baseline (at 2 years) for patients with body weights of 66 kg and 140 kg (5th and 95th percentile) respectively. For clinical site variable, Vanderbilt and Kaiser Georgia had a 16% and 30% lower estimate on the metformin effect parameter when compared to Kaiser Northern California, respectively.
Age was also a significant covariate on the disease progression model parameter, with a negative correlation observed between age and disease progression. Clinically, this would result in a relative change in HbA1c (at 2 years) to fall between 0.76% and 0.84% improvement for patients between the ages of 49 and 64 years.
Genetic analysis: hyperlasso methodology on model parameters
A total of 267 genes were selected and approximately 12000 variants within a 50-kilobase region around each gene were extracted for analysis. Of the variants investigated, a total of 16 SNPs were linked to the disease progression parameter by hyperLASSO analysis (with a MAF ≥ 5%). Of the remaining 16 variants, 11 were intronic [CSMD1(4), ADCY5(1), PRKAG(1), SLC22A2(1), EMILIN2(1), SULF1(1), FTO(1), WWOX(1)], 1 was missense [SREBF1], and 4 were located within 50 kilobases upstream or downstream of each gene [VPS13C(1), KCNK16(1), PPARG(1), FOXN3(1)].
Genetic analysis: model-based approach for variant selection
Of the prioritized 16 variants from hyperlasso, a model-based methodology was implemented to verify statistical significance and determine effect sizes on the disease progression parameter. SNPs that passed this test were included in the final mathematical model for simulation purposes. From this step, 7 SNPs were removed due to the defined criteria (see Methods). The 9 remaining variants were statistically significant in the model structure and collectively accounted for approximately one-third of the variability in the predicted disease progression model parameter (reduced the BSV of the disease progression model parameter from 324% to 225%). Of the 9 variants, rs12907856 (VPS13C), rs2954625 (CSMD1), and rs3160009 (SLC22A2) individually accounted for approximately 6%, 5%, and 8% of the variability, respectively. The characteristics of each SNP are shown in Table 3.
Table 3.
Summary of Top Genetic Variants Included in Final Population Pharmacodynamic Model of Metformin.
| SNP | Chr | Gene | Minor Allele | Major Allele | Feature | Model-based P-Value | Effect size of minor allele on DP parameter | MAF CEU | MAF YRI | Gene Functions (References cited in this table are in the footnote) |
|---|---|---|---|---|---|---|---|---|---|---|
| rs12907856 | 15 | VPS13C - vacuolar protein sorting-associated 13 gene family | G | A | Proximal | 0.0003 | −0.147 (GA) | 0.30 | 0.30 | This gene encodes a member of a vacuolar protein. SNP in this gene was associated with glucose-stimulated insulin response1–3. |
| rs2815022 | 6 | KCNK16 - Potassium Channel, Two Pore Domain Subfamily K, Member 16 | G | A | Proximal | 0.009 | 0.39 (GA) | 0.48 | 0.26 | This gene encodes the TALK-1 channel, the most abundant potassium channel in human beta-cells and it modulates beta-cells electrical excitability, second-phase insulin secretion and glucose homeostasis4, 5 |
| rs2617102 | 8 | CSMD1 - CUB And Sushi Multiple Domains 1 | C | A | Intron | 0.02 | 0.717 (CA) | 0.19 | 0.20 | This gene encodes an integral membrane protein with unknown molecular function. SNP in this gene was associated with congenital hyperinsulinism of infancy6. |
| rs2954625 | 8 | T | C | Intron | 0.029 | 0.23 (TC) | 0.21 | 0.46 | ||
| rs316009 | 6 | SLC22A2 - Solute Carrier Family 22 (Organic Cation Transporter), Member 2 | T | C | Intron | 0.04 | −0.44 (TC) | 0.10 | 0.08 | This is a transporter in the kidney that secretes metformin into the urine. This SNP is in linkage disequilibrium to a non-synonymous variant, A270S (rs316019), which was associated with metformin disposition7, 8. |
| rs642887 | 18 | EMILIN2 - Elastin Microfibril Interfacer 2 | A | G | Intron | 0.05 | −0.42 (AG) | 0.14 | 0.21 | An extracellular matrix glycoprotein associated with thrombosis. EMILIN2 is involved in regulating platelet activation important for cardiovascular development9, 10, whereas EMILIN1 may be involved in regeneration of islets, which could play a role in blood glucose lowering11. |
| rs6982250 | 8 | SULF1 - Sulfatase 1 | T | C | Intron | 0.0023 | −0.37 (TC) | 0.17 | 0.28 | This gene encodes an enzyme, which is involved in modulating growth factor signaling. Data from sulfatase knockout mice showed that it plays a role in diabetic nephropathy12. |
| rs7159552 | 14 | FOXN3 - Forkhead Box N3 | T | G | Proximal | 0.009 | −0.25 (TG) | 0.27 | 0.35 | This is a transcriptional repressor, which plays an important role in cell cycle arrest. This gene is localized in chromosome 14q24.3-q31, which is a locus associated with insulin-dependent diabetes mellitus susceptibility13. |
| rs7500549 | 16 | WWOX - WW Domain Containing Oxidoreductase | C | T | Intron | 0.029 | −0.16 (CT) | 0.55 | 0.20 | SNP in this locus was associated with reduced insulin secretion14. |
P-value = The significance level that resulted from objective function value changes after SNP addition to the demographic corrected base model. Genetic variant rs3160009 significance threshold was determined through model-based analysis of the SNP effect of carriers of either 1 or 2 alleles; MAF= Minor allele frequency; DP = Disease Progression; CEU= Northern Europeans; YRI=African population.
References for Table 3
Grarup N, Overvad M, Sparso T, Witte DR, Pisinger C, Jorgensen T, Yamauchi T, Hara K, Maeda S, Kadowaki T, Hansen T, Pedersen O. The diabetogenic VPS13C/C2CD4A/C2CD4B rs7172432 variant impairs glucose-stimulated insulin response in 5,722 non-diabetic Danish individuals. Diabetologia. 2011;54(4):789–94. doi: 10.1007/s00125-010-2031-2. PubMed PMID: 21249489.
Holstein JD, Patzer O, Korner A, Stumvoll M, Kovacs P, Holstein A. Genetic variants in GCKR, GIPR, ADCY5 and VPS13C and the risk of severe sulfonylurea-induced hypoglycaemia in patients with type 2 diabetes. Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes Association. 2013;121(1):54–7. doi: 10.1055/s-0032-1321834. PubMed PMID: 22956255.
Windholz J, Kovacs P, Tonjes A, Dittrich K, Bluher S, Kiess W, Stumvoll M, Korner A. Effects of genetic variants in ADCY5, GIPR, GCKR and VPS13C on early impairment of glucose and insulin metabolism in children. PloS one. 2011;6(7):e22101. doi: 10.1371/journal.pone.0022101. PubMed PMID: 21789219; PubMed Central PMCID: PMC3137620.
Sakai K, Imamura M, Tanaka Y, Iwata M, Hirose H, Kaku K, Maegawa H, Watada H, Tobe K, Kashiwagi A, Kawamori R, Maeda S. Replication study for the association of 9 East Asian GWAS-derived loci with susceptibility to type 2 diabetes in a Japanese population. PloS one. 2013;8(9):e76317. doi: 10.1371/journal.pone.0076317. PubMed PMID: 24086726; PubMed Central PMCID: PMC3783369.
Vierra NC, Dadi PK, Jeong I, Dickerson M, Powell DR, Jacobson DA. Type 2 Diabetes-Associated K+ Channel TALK-1 Modulates beta-Cell Electrical Excitability, Second-Phase Insulin Secretion, and Glucose Homeostasis. Diabetes. 2015;64(11):3818–28. doi: 10.2337/db15-0280. PubMed PMID: 26239056; PubMed Central PMCID: PMC4613978.
Proverbio MC, Mangano E, Gessi A, Bordoni R, Spinelli R, Asselta R, Valin PS, Di Candia S, Zamproni I, Diceglie C, Mora S, Caruso-Nicoletti M, Salvatoni A, De Bellis G, Battaglia C. Whole genome SNP genotyping and exome sequencing reveal novel genetic variants and putative causative genes in congenital hyperinsulinism. PloS one. 2013;8(7):e68740. doi: 10.1371/journal.pone.0068740. PubMed PMID: 23869231; PubMed Central PMCID: PMC3711910.
Wang ZJ, Yin OQ, Tomlinson B, Chow MS. OCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenetics and genomics. 2008;18(7):637–45. doi: 10.1097/FPC.0b013e328302cd41. PubMed PMID: 18551044.
Chen Y, Li S, Brown C, Cheatham S, Castro RA, Leabman MK, Urban TJ, Chen L, Yee SW, Choi JH, Huang Y, Brett CM, Burchard EG, Giacomini KM. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenetics and genomics. 2009;19(7):497–504. doi: 10.1097/FPC.0b013e32832cc7e9. PubMed PMID: 19483665; PubMed Central PMCID: PMC3104496.
Huang M, Sannaningaiah D, Zhao N, Gong Y, Grondolsky J, Hoover-Plow J. EMILIN2 regulates platelet activation, thrombus formation, and clot retraction. PloS one. 2015;10(2):e0115284. doi: 10.1371/journal.pone.0115284. PubMed PMID: 25658937; PubMed Central PMCID: PMC4319747.
Bot S, Andreuzzi E, Capuano A, Schiavinato A, Colombatti A, Doliana R. Multiple-interactions among EMILIN1 and EMILIN2 N- and C-terminal domains. Matrix biology : journal of the International Society for Matrix Biology. 2015;41:44–55. doi: 10.1016/j.matbio.2014.10.001. PubMed PMID: 25445627.
Lavoie JR, Creskey MC, Muradia G, Bell GI, Sherman SE, Gao J, Stewart DJ, Cyr TD, Hess DA, Rosu-Myles M. EMILIN-1 and ILK are Novel Markers of Islet Regenerative Function in Human Multipotent Mesenchymal Stromal Cells. Stem cells. 2016. doi: 10.1002/stem.2385. PubMed PMID: 27090767.
Takashima Y, Keino-Masu K, Yashiro H, Hara S, Suzuki T, van Kuppevelt TH, Masu M, Nagata M. Heparan sulfate 6-O-endosulfatases, Sulf1 and Sulf2, regulate glomerular integrity by modulating growth factor signaling. American journal of physiology Renal physiology. 2016;310(5):F395–408. doi: 10.1152/ajprenal.00445.2015. PubMed PMID: 26764203.
Field LL, Tobias R, Thomson G, Plon S. Susceptibility to insulin-dependent diabetes mellitus maps to a locus (IDDM11) on human chromosome 14q24.3-q31. Genomics. 1996;33(1):1–8. doi: 10.1006/geno.1996.0153. PubMed PMID: 8617492.
Chang YC, Chiu YF, Liu PH, Shih KC, Lin MW, Sheu WH, Quertermous T, Curb JD, Hsiung CA, Lee WJ, Lee PC, Chen YT, Chuang LM. Replication of genome-wide association signals of type 2 diabetes in Han Chinese in a prospective cohort. Clinical endocrinology. 2012;76(3):365–72. doi: 10.1111/j.1365-2265.2011.04175.x. PubMed PMID: 21767287.
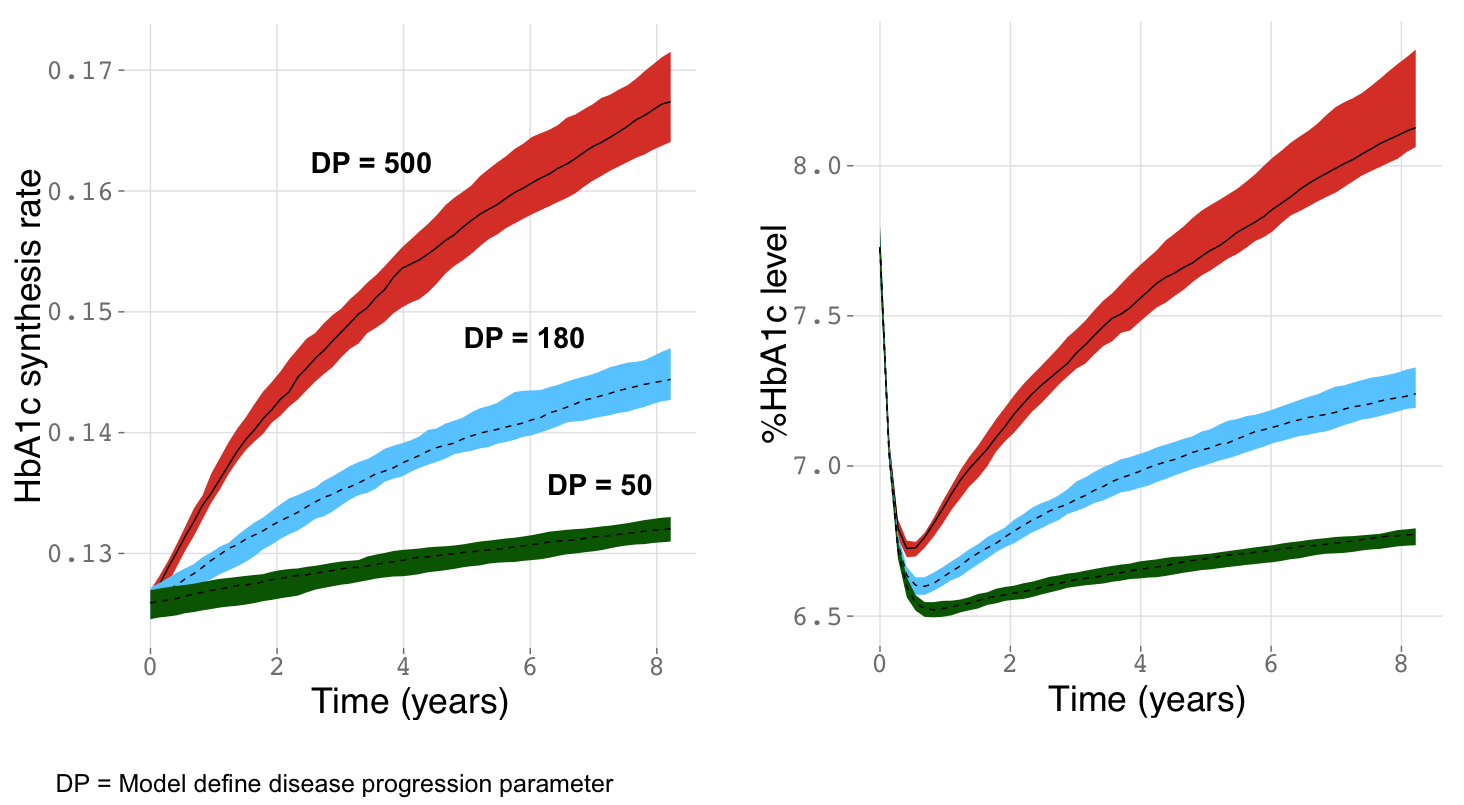
In the final model, several simulations were performed to illustrate the potential clinical impact of each SNP on long-term HbA1c levels. Figure 3 quantitatively summarizes the predicted effects of final model genetic and non-genetic factors on HbA1c levels at the 1-year and 5-year mark. Hypothetical gene/gene interactions were also explored and the combinatorial effects of high risk SNPs in the CSMD1, WWOX, and SLC22A2 genes were also explored in Figure 4.
Figure 3.

Top genetic and demographic covariates on long term HbA1c levels. A. The effect of covariates on the simulated median (bands show 5th and 95th CI of simulated median) of HbA1c levels at the 1-year mark. B. The effect of covariates on the simulated median (5th and 95th CI of simulated median) of HbA1c levels at the 5-year mark. A normal individual here represents a hypothetical patient with no minor alleles of any of the identified variants with median age, body weight, and serum creatinine values.
Figure 4.

Effect of SNP combinations in CSMD1, SLC22A2, and WWOX on the dynamics of HbA1c levels. A: Simulated median HbA1c levels (with 95% CI bands) over 5 years comparing carriers and non-carriers of CSMD1 minor (risk) alleles. B: Simulated median HbA1c levels over 5 years comparing carriers and non-carriers of SLC22A1/WWOX genes minor alleles. Blue shaded region with solid line: Simulated median for patients carrying no minor alleles with 5th and 95th confidence interval. Red/green shade with dashed line: Simulated median for patients carrying minor alleles of labeled gene(s) with 5th and 95th confidence interval of median.
In the exploratory studies, patients carrying one or more minor alleles of the identified variants in the CSMD1 gene (rs2617102 (C), rs2954625 (T)) were predicted to have significantly higher long-term HbA1c levels compared to patients not carrying any CSMD1 minor alleles or patients with homozygous rs3160009 TT (SLC22A2) and/or homozygous rs7500549 CC (WWOX) genotypes.
Functional annotation of top variants
Three out of the nine variants (rs12907856, rs316009, and rs7159552) are located in linkage disequilibrium (LD) to a regulatory region, determined by an algorithmic prediction by RegulomeDB23. In particular, rs316009 and rs7159552 are located in a transcription factor binding motif as identified by the ENCODE project24. The rs316009 variant is in LD to the nonsynonymous variant of SLC22A2 - rs316019 - which is known to play a role in metformin pharmacokinetics,25,26,27. Another variant, rs6982250, is in an intronic region of SULF1. Several SNPs in SULF1 have been associated with many phenotypes28 with one such variant associated with fasting insulin-related traits29.
DISCUSSION
Previous pharmacogenetic studies of metformin response have focused on the effect of selected variants in relevant pharmacogenes on single-time point outcomes of metformin (i.e. HbA1c levels after 90 days, FPG levels, etc.)15,30,31,32. Long-term, time-dependent changes of HbA1c have been previously overlooked, resulting in a collapse of valuable biomarker information that may inform disease progression as well as temporal response patterns.
Here, we developed a longitudinal HbA1c model by leveraging a large T2D dataset and subsequently investigated the role of genetic and non-genetic factors on long-term dynamics of HbA1c following metformin initiation. Special focus was given to identifying factors that are responsible for the long-term variance in HbA1c levels.
Three important findings emerged from this analysis: (i) a mathematical model incorporating disease progression and a reversible metformin effect best characterized the long-term HbA1c data in T2D Patients. (ii) The model presented herein predicted that the onset of disease progression for patients on metformin is approximately 321 days, at which point, levels increase, on average, at a rate of 0.1% (1.1 mmol/mol) [0.04%–0.16%] HbA1c per year; HbA1c levels are expected to increase at a steady state rate of approximately 0.16% (1.76 mmol/mol) [0.08%–0.22%] per year in patients not treated with metformin. (iii) Nine variants in 8 genes (of 267 genes interrogated) accounted for approximately one-third of the total estimated variability in the disease progression model parameter. Variants in three of these genes (CSMD1, WWOX, and SLC22A2) were identified as significant influencers of disease progression on metformin therapy.
The development of the final mathematical model resulted from the exploration of several approaches with various empirical and semi-mechanistic considerations. The structural parameters from the model were estimated with high precision. The between-subject variability estimates of baseline, metformin effect, and disease progression were also estimated with relatively high precision (3%, 4%, and 17% relative standard error (RSE), respectively). The high degree of parameter confidence was due to the abundance of available HbA1c data, allowing for the reliable assessment of clinical, demographic, and genetic covariates on disease progression. Disease progression (upward trajectory of HbA1c levels) is a function of both the patient’s underlying disease as well as the build up of metformin resistance. In order to differentiate between the effects of a patient’s biology and a reduction of metformin’s reversible effect, it is necessary to model longitudinal HbA1c data prior to the administration of treatment; unfortunately, this was not possible in our analysis as this would require patients to be off treatment during the duration of the disease. The HbA1c model was however able to adequately predict the dynamics of HbA1c levels, capturing the long-term upward trend observed in this population. The ability to predict long-term HbA1c changes is especially valuable: the onset of disease progression and the rate of HbA1c increase were quantified for patients on metformin therapy (~0.1% increase per year for the first three years after 321 days, which is the estimated onset of disease progression) leveraging the richness of HbA1c data available. This finding was particularly interesting in relation to the study by Winter et al. where the authors noted a slight rise in patients’ HbA1c levels, between 200 and 400 days after metformin initiation; however, they were unable to quantify this upward trend through their simulations – a limitation which resulted from the lack of longitudinal data points available after 400 days16. In our analysis, the average length of time in the study was 1014 days and up to 10 years worth of HbA1c measurements were available to inform disease progression – allowing the characterization and quantification of this upward trend with high precision. The robustness in the model enabled the simulation of patient-specific disease progression with an underlying assumption of no metformin administration (approximate increase of 0.16% (1.7 mmol/mol) in HbA1c per year). The ability to separate disease progression and metformin effect is based on early HbA1c data (up to 1 year following metformin initiation). Simulations of disease progression assuming no metformin administration were explored by removing metformin’s estimated effect on the HbA1c synthesis rate within the model structure. The simulations demonstrate that on average, disease progression in patients who are metformin-naive will occur faster than in patients taking metformin for several months. Comparing this estimate to existing literature is problematic since T2D progression is a gradual process that typically takes place over several years and thus allows only a small trajectory of change within the limited time frame available for most studies. In the few studies reported, the rate of HbA1c increase was estimated to be approximately 0.2% (2.2 mmol/mol) per year, a value consistent with our observations33.
A stepwise multivariate analysis was performed to identify statistically significant demographic and clinical covariates on model parameters. Average serum creatinine level surfaced as a significant factor that influenced the magnitude of metformin’s effect. This finding was expected since serum creatinine is considered a likely surrogate for metformin exposure. Serum creatinine directly influences a patient’s creatinine clearance, which ultimately influences a patient’s systemic exposure to metformin by affecting the apparent clearance pharmacokinetic parameter. The effect of age was also noted - an inverse relationship was observed between age and the magnitude of disease progression. It is important to note that although age was statistically significant through a stepwise analysis, the effect size was quite small and a reproduction of these results is required to inspire greater conviction of this correlation. Previously, in a study by Williams et al., lower HbA1c levels were reported in African Americans compared to European American individuals34. In our analysis, however, there was no significant effect of self-reported ethnicity on any of the model parameters, including disease progression.
We used multiple genetic methods to prioritize influential variants on disease progression. Hyperlasso methodology was selected over a stepwise procedure, as well as several other algorithms. This is because the hyperlasso approach has been shown to be robust when investigated covariates are correlated, which is the case here with strong LD patterns in the genotype data. The final selection of variants was based on the performance of individual variants within the demographic-corrected model so that the correlation across various model parameters may also be taken into consideration.
Nine variants emerged were linked to the progression of HbA1c levels on metformin. Collectively, the variants accounted for approximately one-third of the variance in the disease progression model parameter. It was also observed that these genetic variants had larger effects on HbA1c levels than the demographic and clinical covariates identified from the stepwise analysis.
Of the top genes, minor alleles of two SNPs (rs2617102, rs2954625) in the CSMD1 (CUB and Sushi multiple domains 1) gene had the strongest impact on disease progression. Although the pharmacological and biological mechanism remains unclear, CSMD1 has been previously linked to insulin sensitivity and lipid levels35,36. CSMD1 variants may have a significant impact on longitudinal HbA1c levels, especially at the five-year mark when the simulated HbA1c improvement from baseline becomes nominal – especially for homozygous carriers (TT) of rs2617102. The simulated 5-year HbA1c level was very similar to baseline levels (Figure 4) – which means that HbA1c levels rebounded back to its baseline state. Furthermore, the effect on HbA1c levels at the 5-year mark was higher for hypothetical homozygous carriers of both CSMD1 SNPs (rs2617102, rs2954625) – where HbA1c levels were predicted to be significantly higher than baseline levels.
Minor alleles of SNPs in genes SLC22A2, WWOX, EMILIN2, and FOXN3 were associated with more favorable trajectories (lower disease progression) of HbA1c levels compared to major allele carriers. Of these genes, SLC22A2 (rs316009 (T)) and WWOX (rs7500549 (C)) showed the strongest effect. In contrast to homozygous carriers of CSMD1 risk alleles, homozygous carriers of both SLC22A2 and WWOX SNPs were predicted to have a favorable clinical outcome - maintaining their peak HbA1c level improvement from baseline through 5 years of metformin therapy. The rs316009 variant is in LD to a nonsynonymous variant of SLC22A2 (rs316019), a SNP that has been previously shown to alter transporter function as well as modulate metformin pharmacokinetics.26,27 Therefore, the clinical expectation that the reduced function rs316009 (T) allele would lead to a more favorable outcome is pharmacologically sound. OCT2 (SLC22A2) is predominantly expressed at the basolateral membrane in distal renal tubules and is responsible for the uptake of metformin from circulation into renal epithelial cells, working in concert with other renal transporters to excrete metformin. Though functional studies have been controversial25, loss of transporter function is expected to increase plasma levels of metformin, potentially leading to a more favorable pharmacodynamic outcome with relatively low HbA1c levels.
Also of clinical interest, the gene WWOX has been previously associated with several T2D traits including body weight, C-reactive protein, insulin, obesity, and lipid levels37. WWOX encodes for an enzyme that is found in all eukaryotes and has been biologically shown to play an important role in the regulation of a wide variety of cellular functions such as protein degradation, transcription, and RNA splicing. Unlike SLC22A2, a pharmacological mechanism for WWOX is not clear. However, the clinical impact (if replicated) would mean that carriers of the rs7500549 (C) allele would respond favorably to metformin therapy. Future studies should focus on elucidating the biology of WWOX and replicating the genetic findings on disease progression.
Although this computational approach represents a novel way to uncover factors that influence long-term drug response, several important limitations must be highlighted. First, the demographic distribution used for this analysis does not appropriately reflect the national population distribution due to the disproportionate representation of African Americans in this cohort. As a result, it will be critical to replicate both genetic and non-genetic findings in separate cohorts for validation purposes. Furthermore, the retrospective data set lacks a control group and is reflecting multiple studies across multiple sites. As such, validation of the model-based simulations, which quantify metformin’s effect on long term HbA1c dynamics with consideration of impactful covariates, is required.
Overall, our study has successfully integrated robust model-based approaches with genetic analyses methods to uncover genes linked to the progression of HbA1c on metformin therapy in a large T2D cohort. If replicated, these genetic findings may have a significant influence on T2D treatment strategy. Ultimately, the long-term goal of this research is to translate this computational model into clinical practice and enable clinicians to provide data-driven, personalized treatment advice to T2D patients based on individual patient characteristics.
METHODS
Patients with type 2 diabetes
Diabetic patients of European American, African American, and Asian American ancestry were recruited into a multicenter retrospective study as described previously13,32. All patients were metformin-naive, had HbA1c levels measured before and after initiation of metformin therapy (between 3 and 18 months), and had a medication possession ratio greater than 80%. The institutional review boards (IRBs) of Marshfield Clinic Research Foundation, Kaiser Permanente Northern California, Kaiser Permanente South East, Georgia, approved this study and informed consent was obtained. At Vanderbilt, an opt-out consent model was used. In diabetic patients, metformin was administered for at least three months, so steady state drug concentration levels were achieved, since the half-life of metformin is roughly 5 hours. Patients were in the study for an average of 2.8 years (median = 1.43 years) with on average 7.4 (median = 5) HbA1c measurements. HbA1c results were reported in the NGSP format (National Glycohemoglobin standardization program). The median metformin dose across the patient population was 1000 mg (Table 1). Patients were genotyped using an Illumina OmniExpress genotype array (see supplementary methods section for further details).
Development of mathematical model
Patient data were analyzed using non-linear mixed effect modeling (NONMEM 7) with first order conditional estimation method with interaction (FOCE-I). Several semi-mechanistic approaches were explored to best describe the longitudinal HbA1c versus time profiles. Model selection was determined using the objective function value (OFV, −2 times the log of the likelihood) and visual inspection of diagnostic plots. The selected longitudinal HbA1c profiles were described by the following equations:
| (1) |
| (2) |
Equation 1 defines the synthesis rate of HbA1c, which includes a non-linear time sensitive parameter dependent on the baseline synthesis rate and the extent of patient-specific disease progression. In Equation 1, DisprEFFECT, KLOSS and KSYN represent the disease progression effect parameter, loss rate of KIN and synthesis rate of the KIN parameter, respectively. Equation 2 defines the dynamics of HbA1c, parameterized by the synthesis rate of HbA1c (KIN(t)), metformin’s effect from baseline (MetfEFFECT), and the loss rate of HbA1c (KOUT). A more detailed explanation and the model source code can be found in the supplementary methods section. A simulated demonstration of the dynamics of this model can viewed in supplementary figure 1. A patient’s individual administered doses were taken into consideration by examining the effect of the average daily dose. The average daily dose of metformin was calculated from metformin start day up to the day, where minimum HbA1c levels were achieved between 3–18 months (and before other anti-diabetic drug or insulin was added). Although no drug concentration was directly used for this analysis, surrogate PK information was taken into account in the model structure by investigating the effect of average serum creatinine level (a major predictor on metformin individual clearance) or imputed exposure (based on estimated individual clearance of metformin and average daily dose). Individual clearance was estimated based on the clearance equation previously described10. Both average dose and metformin exposure were tested on the MetfEFFECT parameter.
Demographic analysis
Using the mathematical model described above, agnostic stepwise forward selection (P < 0.05) and backward elimination (P < 0.01) were applied to identify statistically significant demographic and clinical covariates on model parameter estimates, which helped guide the selection of the demographic-corrected final model. The effect of concomitant medications was taken into account by investigating the effect of added drug on model parameters. The subsequent demographic-corrected mathematical model served as a basis to investigate the effect of genetic variants on the variance of long-term response.
Genetic analyses of model parameters
A comprehensive list of candidate genes was selected using the GWAS Integrator tool on the HuGE Navigator38 (details found in supplementary methods section). A penalized regression-based approach called hyperlasso was implemented to statistically prioritize the variants associated with phenotypes outputted from the mathematical model (e.g. disease progression, metformin effect, and baseline). This methodology was originally proposed by Hoggart et al., and is a generalization of Lasso39,40. Further information about the hyperlasso method can be found in supplementary methods section.
Model based genetic analysis of identified variants
The top SNPs from hyperlasso were subsequently investigated in the developed demographic-corrected mathematical model described above. Model based analyses are advantageous because they account for correlations across various model parameters as well as potential SNP/SNP interactions. Two key steps were taken to select the final mathematical model; (1) removal of non-significant SNPs, which resulted from a univariate analysis of each variant in the demographic-adjusted mathematical model, and (2) removal of variants from the full genetic model that had very low, clinically irrelevant effect sizes. Details of this step may be found in the supplemental section.
Supplementary Material
{kind=link}
RESEARCH HIGHLIGHTS.
What is the current knowledge on the topic?
Previous studies have focused on the effect of genetic polymorphisms in candidate genes on short-term changes in metformin response. Additionally, studies have developed computational models to capture short-term pharmacodynamic changes without consideration of long-term disease progression.
What question did this study address?
In this study, we combined quantitative pharmacology with computational genetic analysis techniques to investigate the effect of genetic variants in biologically and pharmacologically meaningful genes on long-term disease progression of patients with type 2 diabetes on metformin therapy.
What this study adds to our knowledge
This study provides evidence that genetic polymorphisms in CSMD1 and membrane transporter gene SLC22A2 are significant influencers of disease progression, affecting the long-term trajectory of HbA1c levels. This study also adds a robust quantitative pharmacology model that predicts long-term changes in HbA1c levels, which if validated, may be used as a valuable tool to predict long-term outcomes for patients.
How this might change clinical pharmacology or translational science
To date, this is the first study to explore the effect of biologically and pharmacologically relevant genes on long-term disease progression of patients taking metformin. This is also the first study to investigate long-term HbA1c disease trajectories. In the future, combining genotyping of biologically and pharmacologically relevant genes with proper consideration of demographic and clinical predictors may be used to inform metformin therapy in T2D patients.
Acknowledgments
This research was supported by grants from the National Institutes of Health (NIH) National Institute of General Medical Sciences (GM61390 and GM117163), NIH P30 DK063720, and the NIH Pharmacogenomics Research Network (PGRN)-RIKEN Strategic Alliance.
Footnotes
AUTHOR CONTRIBUTIONS
R.S., S.G., and K.M.G. wrote the manuscript; R.S., S.G., S.W.Y., J.D.M., and K.M.G. designed the research; R.S., S.G., S.W.Y., F.X., S.B.S., M.K., R.D., and K.M.G. performed the research; R.S., S.G., S.W.Y., A.T., S.M., D.R., M.M.H., and K.M.G. analyzed the data.
Contributor Information
Srijib Goswami, University of California, San Francisco.
Sook Wah Yee, University of California, San Francisco.
Fei Xu, Kaiser Permanente Northern California.
Sneha B. Sridhar, Kaiser Permanente Northern California
Jonathan JD Mosley, Vanderbilt University.
Atsushi Takahashi, RIKEN Institute, Center for Genomic Medicine.
Michiaki Kubo, RIKEN Institute, Center for Genomic Medicine.
Shiro Maeda, RIKEN Institute, Center for Genomic Medicine.
Robert L. Davis, Kaiser Permanente Georgia
Dan M. Roden, Vanderbilt University
Monique M. Hedderson, Kaiser Permanente Northern California
Kathleen M. Giacomini, University of California, San Francisco
Radojka M. Savic, University of California, San Francisco
References
- 1.Scarpello JH, Howlett HC. Metformin therapy and clinical uses. Diab Vasc Dis Res. 2008;5:157–67. doi: 10.3132/dvdr.2008.027. [DOI] [PubMed] [Google Scholar]
- 2.Gong L, Goswami S, Giacomini KM, Altman RB, Klein T. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2013;22:820–27. doi: 10.1097/FPC.0b013e3283559b22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–70. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick CM, Ray JE, Timmins P, Williams KM. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50:81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Hamrén B, Björk E, Sunzel M, Karlsson M. Models for plasma glucose, HbA1c, and hemoglobin interrelationships in patients with type 2 diabetes following tesaglitazar treatment. Clin Pharmacol Ther. 2008;84:228–35. doi: 10.1038/clpt.2008.2. [DOI] [PubMed] [Google Scholar]
- 6.Menon V, Greene T, Pereira AA, Wang X, Beck GJ, Kusek JW, Collins AJ, Levey AS, Sarnak MJ. Glycosylated hemoglobin and mortality in patients with nondiabetic chronic kidney disease. J Am Soc Nephrol. 2005;16:3411–27. doi: 10.1681/ASN.2005050552. [DOI] [PubMed] [Google Scholar]
- 7.Zoungas S, Chalmers J, Ninomiya T, Li Q, Cooper ME, Colagiuri S, Fulcher G, de Galan BE, Harrap S, Hamet P, Heller S, MacMahon S, Marre M, Poulter N, Travert F, Patel A, Neal B, Woodward M. Association of HbA1c levels with vascular complications and death in patients with type 2 diabetes: evidence of glycaemic thresholds. Diabetologia. 2012;55:636–43. doi: 10.1007/s00125-011-2404-1. [DOI] [PubMed] [Google Scholar]
- 8.Andersson C, van Gaal L, Caterson ID, Weeke P, James WPT, Coutinho W, Couthino W, Finer N, Sharma AM, Maggioni AP, Torp-Pedersen C. Relationship between HbA1c levels and risk of cardiovascular adverse outcomes and all-cause mortality in overweight and obese cardiovascular high-risk women and men with type 2 diabetes. Diabetologia. 2012;55:2348–55. doi: 10.1007/s00125-012-2584-3. [DOI] [PubMed] [Google Scholar]
- 9.Cook MN, Girman CJ, Stein PP, Alexander CM. Initial monotherapy with either metformin or sulphonylureas often fails to achieve or maintain current glycaemic goals in patients with type 2 diabetes in UK primary care. Diabet Med. 2007;24:350–58. doi: 10.1111/j.1464-5491.2007.02078.x. [DOI] [PubMed] [Google Scholar]
- 10.Goswami S, Yee SW, Stocker S, Mosley JD, Kubo M, Castro R, Mefford J, Wen C, Liang X, Witte J, Brett C, Maeda S, Simpson MD, Hedderson MM, Davis RL, Roden DM, Giacomini KM, Savic RM. Genetic variants in transcription factors are associated with the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2014;96:370–90. doi: 10.1038/clpt.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shu Y, Leabman MK, Feng B, Mangravite LM, Huang CC, Stryke D, Kawamoto M, Johns SJ, DeYoung J, Carlson E, Ferrin TE, Herskowitz I, Giacomini KM. Evolutionary conservation predicts function of variants of the human organic cation transporter, OCT1. Proc Natl Acad Sci U S A. 2003;100:5902–7. doi: 10.1073/pnas.0730858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1 ( OCT1 ) on metformin action. J Clin Invest. 2007;117:1422–31. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stocker SL, Morrissey KM, Yee SW, Castro R, Xu L, Dahlin A, Ramirez H, Roden DM, Wilke R, McCarty C, Davis RL, Brett CM, Giacomini KM. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2013;93:186–94. doi: 10.1038/clpt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christensen MMH, Brasch-Andersen C, Green H, Nielsen F, Damkier P, Beck-Nielsen H, Brosen K. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. 2011;21:837–50. doi: 10.1097/FPC.0b013e32834c0010. [DOI] [PubMed] [Google Scholar]
- 15.Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley S, Donnelly L, Schofield C, Groves CJ, Burch L, Carr F, Strange A, Freeman C, Blackwell JM, Bramon E, Brown M, Casas JP, Corvin A, Craddock N, Deloukas P, Dronov S, Duncanson A, Edkins S, Gray E, Hunt S, Jankowski J, Langford C, Markus HS, Mathew CG, Plomin R, Rautanen A, Sawcer SJ, Samani NJ, Trembath R, Viswanathan AC, Wood NW, Harries LW, Hattersley AT, Doney ASF, Colhoun H, Morris AD, Sutherland C, Hardie DG, Peltonen L, McCarthy MI, Holman RR, Palmer CN, Donnelly P, Pearson ER. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43:117–20. doi: 10.1038/ng.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Winter W, DeJongh J, Post T, Ploeger B, Urquhart R, Moules I, Eckland D, Danhof M. A mechanism-based disease progression model for comparison of long-term effects of pioglitazone, metformin and gliclazide on disease processes underlying type 2 diabetes mellitus. J Pharmacokinet Pharmacodyn. 2006;33:313–43. doi: 10.1007/s10928-006-9008-2. [DOI] [PubMed] [Google Scholar]
- 17.Ette EI, Williams PJ. Population pharmacokinetics I: background, concepts, and models. Ann Pharmacother. 2004;38:1702–16. doi: 10.1345/aph.1D374. [DOI] [PubMed] [Google Scholar]
- 18.Ette EI, Williams PJ, Lane JR. Population pharmacokinetics III: design, analysis, and application of population pharmacokinetic studies. Ann Pharmacother. 2004;38:2136–44. doi: 10.1345/aph.1E260. [DOI] [PubMed] [Google Scholar]
- 19.Pillai GC, Mentré F, Steimer JL. Non-linear mixed effects modeling - from methodology and software development to driving implementation in drug development science. J Pharmacokinet Pharmacodyn. 2005;32:161–83. doi: 10.1007/s10928-005-0062-y. [DOI] [PubMed] [Google Scholar]
- 20.Hong Y, Rohatagi S, Habtemariam B, Walker JR, Schwartz SL, Mager DE. Population exposure-response modeling of metformin in patients with type 2 diabetes mellitus. J Clin Pharmacol. 2008;48:696–707. doi: 10.1177/0091270008316884. [DOI] [PubMed] [Google Scholar]
- 21.Møller JB, Kristensen NR, Klim S, Karlsson MO, Ingwersen SH, Kjellsson MC. Methods for predicting diabetes phase III efficacy outcome from early data: superior performance obtained using longitudinal approaches. CPT pharmacometrics Syst Pharmacol. 2014;3:122–30. doi: 10.1038/psp.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan PL, Holford NH. Drug treatment effects on disease progression. Annu Rev Pharmacol Toxicol. 2001;41:625–59. doi: 10.1146/annurev.pharmtox.41.1.625. [DOI] [PubMed] [Google Scholar]
- 23.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub Ma, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–97. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myers RM, Stamatoyannopoulos J, Snyder M, Dunham I, Hardison RC, Bernstein BE, Gingeras TR, Good PJ, et al. A user’s guide to the Encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:245–99. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Li S, Brown C, Cheatham S, Castro RA, Leabman MK, Urban TJ, Chen L, Yee SW, Choi JH, Huang Y, Brett CM, Burchard EG, Giacomini KM. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics. 2009;19:497–504. doi: 10.1097/FPC.0b013e32832cc7e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang ZJ, Yin OQP, Tomlinson B, Chow MSS. OCT2 polymorphisms and in-vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenet Genomics. 2008;18:637–45. doi: 10.1097/FPC.0b013e328302cd41. [DOI] [PubMed] [Google Scholar]
- 27.Christensen MMH, Pedersen RS, Stage TB, Brasch-andersen C, Nielsen F, Damkier P, Beck-nielsen H, Brøsen K. A gene – gene interaction between polymorphisms in the OCT2 and MATE1 genes influences the renal clearance of metformin. Pharmacogenet Genomics. 2013;23:526–34. doi: 10.1097/FPC.0b013e328364a57d. [DOI] [PubMed] [Google Scholar]
- 28.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Langenberg C, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–69. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Florez JC, Barrett-Connor E, Jablonski K, Knowler WC, Taylor A, Shuldiner AR, Mather K, Pollin TI, Horton E, White NH. The C allele of ATM rs11212617 does not associate with metformin response in the diabetes prevention program. Diabetes Care. 2012;35:1864–67. doi: 10.2337/dc11-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tzvetkov MV, Saadatmand AR, Bokelmann K, Meineke I, Kaiser R, Brockmöller J. Effects of OCT1 polymorphisms on the cellular uptake, plasma concentrations and efficacy of the 5-HT3 antagonists tropisetron and ondansetron. Pharmacogenomics J. 2012;12:22–29. doi: 10.1038/tpj.2010.75. [DOI] [PubMed] [Google Scholar]
- 32.Choi JH, Yee SW, Ramirez H, Morrissey KM, Jang GH, Joski PJ, Mefford J, Hesselson SE, Schlessinger A, Jenkins G, Castro R, Johns SJ, Stryke D, Sali A, Ferrin TE, Witte JS, Kwok PY, Roden DM, Wilke R, McCarty C, Davis RL, Giacomini KM. A common 5′-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. 2011;90:674–84. doi: 10.1038/clpt.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner R. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–65. [PubMed] [Google Scholar]
- 34.Keoki Williams L, Padhukasahasram B, Ahmedani BK, Peterson EL, Wells KE, González Burchard E, Lanfear DE. Differing effects of metformin on glycemic control by race-ethnicity. J Clin Endocrinol Metab. 2014:39–55. doi: 10.1210/jc.2014-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irvin MR, Wineinger NE, Rice TK, Pajewski NM, Kabagambe EK, Gu CC, Pankow J, North KE, Wilk JB, Freedman BI, Franceschini N, Broeckel U, Tiwari HK, Arnett DK. Genome-wide detection of allele specific copy number variation associated with insulin resistance in african americans from the hyperGEN study. PLoS One. 2011;6:9–11. doi: 10.1371/journal.pone.0024052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong KW, Go MJ, Jin HS, Lim JE, Lee JY, Han BG, Hwang SY, Lee SH, Park HK, Cho YS, Oh B. Genetic variations in ATP2B1, CSK, ARSG and CSMD1 loci are related to blood pressure and/or hypertension in two Korean cohorts. J Hum Hypertens. 2010;24:367–72. doi: 10.1038/jhh.2009.86. [DOI] [PubMed] [Google Scholar]
- 37.Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta. 2014;1846:188–200. doi: 10.1016/j.bbcan.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu W, Clyne M, Khoury MJ, Gwinn M. Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics. 2010;26:145–56. doi: 10.1093/bioinformatics/btp618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoggart CJ, Whittaker JC, De Iorio M, Balding DJ. Simultaneous analysis of all SNPs in genome-wide and re-sequencing association studies. PLoS Genet. 2008;4:1–8. doi: 10.1371/journal.pgen.1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertrand J, Balding DJ. Multiple single nucleotide polymorphism analysis using penalized regression in nonlinear mixed-effect pharmacokinetic models. Pharmacogenet Genomics. 2013;23:167–74. doi: 10.1097/FPC.0b013e32835dd22c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.