

Abstract
The eupolypods II ferns represent a classic case of evolutionary radiation and, simultaneously, exhibit high substitution rate heterogeneity. These factors have been proposed to contribute to the contentious resolutions among clades within this fern group in multilocus phylogenetic studies. We investigated the deep phylogenetic relationships of eupolypod II ferns by sampling all major families and using 40 plastid genomes, or plastomes, of which 33 were newly sequenced with next-generation sequencing technology. We performed model-based analyses to evaluate the diversity of molecular evolutionary rates for these ferns. Our plastome data, with more than 26,000 informative characters, yielded good resolution for deep relationships within eupolypods II and unambiguously clarified the position of Rhachidosoraceae and the monophyly of Athyriaceae. Results of rate heterogeneity analysis revealed approximately 33 significant rate shifts in eupolypod II ferns, with the most heterogeneous rates (both accelerations and decelerations) occurring in two phylogenetically difficult lineages, that is, the Rhachidosoraceae–Aspleniaceae and Athyriaceae clades. These observations support the hypothesis that rate heterogeneity has previously constrained the deep phylogenetic resolution in eupolypods II. According to the plastome data, we propose that 14 chloroplast markers are particularly phylogenetically informative for eupolypods II both at the familial and generic levels. Our study demonstrates the power of a character-rich plastome data set and high-throughput sequencing for resolving the recalcitrant lineages, which have undergone rapid evolutionary radiation and dramatic changes in substitution rates.
Keywords: Aspleniaceae, Athyriaceae, backbone phylogeny, plastome, substitution rate, Rhachidosoraceae
Introduction
With the development of next-generation sequencing (NGS) technology, genomic data sets containing unprecedented numbers of informative sites and loci are now available to explore the deepest relationships in the plant tree of life (Xi et al. 2012; Zhang et al. 2012; Zeng et al. 2014; Rothfels et al. 2015). Character-rich data sets, often of genomic-scale, can provide phylogenetic resolution even for particularly recalcitrant lineages, such as those that have undergone rapid evolutionary radiations or have considerable substitution rate heterogeneity (Wu and Ge 2012; Lu et al. 2015; Barrett et al. 2016; Zhang et al. 2016). Furthermore, NGS continues to provide important, new opportunities to explore the relationships among lineages that have contentious or unresolved phylogenies.
The eupolypods II (Aspleniineae in PPG I 2016) is an unranked, highly supported clade (Schneider et al. 2004a; Schuettpelz and Pryer 2007; Rothfels et al. 2012a) comprising approximately one-third the world’s fern diversity and delimited by linear sori and two vascular bundles in the pedicels/stipes (Smith et al. 2006; Rothfels et al. 2012a; Sundue and Rothfels 2014). This group of ferns is remarkable for its great morphological diversity, especially within several large genera including Asplenium (c. 700 spp.) in Aspleniaceae, Athyrium (c. 230 spp.) and Diplazium (c. 350 spp.) in Athyriaceae, Goniopteris (c. 120 spp.), and Sphaerostephanos (c. 185 spp.) in Thelypteridaceae (Ching 1964; Holttum 1982; Tryon and Tryon 1982; Smith 1990; Schneider et al. 2004b; He and Zhang 2012; Wei et al. 2013; PPG I 2016). They have a world-wide distribution, with species specialized to various environmental conditions, from epiphytes or lithophytes (e.g., species of Asplenium, Blechnidium, and Woodsia) to terrestrial members (e.g., Rhachidosorus, Athyrium and Diplazium) (Rothfels et al. 2012a, Sundue and Rothfels 2014).
Phylogenetic relationships within eupolypods II have been difficult to resolve due to a rapid radiation in their history and high substitution rate heterogeneity (Rothfels et al. 2012b). Rapid radiations yield few synapomorphies among clades, and, therefore, may confound phylogenetic analyses (Jian et al. 2008). In contrast, rate heterogeneity can cause convergence among distantly related groups and, therefore, lead to erroneous inferences of close relationships among groups with similar substitution rates (Ho and Jermiin 2004; Soubrier et al. 2012). Although efforts have been made to resolve relationships in eupolypods II in the past decade, notable uncertainties persist. In particular, deep relationships among eupolypod II lineages remain contentious, probably as a predictable consequence of early rapid radiation of the clade (Rothfels et al. 2012b), and several alternative phylogenetic hypotheses have been proposed (Sano et al. 2000; Wang et al. 2003; Smith et al. 2006; Schuettpelz and Pryer 2007; Wei et al. 2010, 2013; Kuo et al. 2011; Li et al. 2011; Rothfels et al. 2012a, 2015; Shao et al. 2015; Mynssen et al. 2016; PPG I 2016). Taken together, these studies highlight that the placement of Rhachidosoraceae and the circumscription of Athyriaceae have been particularly problematic. For example, Kuo et al. (2011) find that Rhachidosoraceae is closely related to the Thelypteridaceae-Blechnaceae clade (fig. 1a) based on sequences of the chloroplast matK gene. However, Rothfels et al. (2012b) obtain a sister relationship between Rhachidosoraceae and the Aspleniaceae-Diplaziopsidaceae clade (fig. 1b) based on five chloroplast loci. With respect to Athyriaceae, its monophyly has not been strongly supported in previous phylogenetic studies because of the ambiguous relationships among several genera, that is, Deparia and Athyrium-Diplazium, which are traditionally circumscribed in the family (Kuo et al. 2011; Rothfels et al. 2012b; Wei et al. 2013; also see Schuettpelz and Pryer 2007) (fig. 1c). Most recently, Rothfels et al. (2015) report a phylogenetic framework for leptosporangiate ferns based on 25 low-copy nuclear genes and a comprehensive taxonomic sampling. However, deep relationships among eupolypod II genera represent one of few areas of low support within the resulting topology (fig. 1d), and some important taxa, such as Rhachidosorus (Rhachidosoraceae) and Diplaziopsis (Diplaziopsidaceae), are not included. Additional phylogenetic studies using more character-rich data sets are needed to address these fundamental and persistent uncertainties within the eupolypod II clade.
Fig. 1.—

Hypotheses on the backbone relationship of eupolypod II ferns with special reference to the positions of Rhachidosoraceae and Athyriaceae. (a) Topology resolving Rhachidosoraceae as sister to the Thelypteridaceae-Blechnaceae clade in Kuo et al. (2011); (b) topology resolving Rhachidosoraceae as sister to the Aspleniaceae-Diplaziopsidaceae clade in Rothfels et al. (2012b); (c) topology showing Athyriaceae as nonmonophyletic in Kuo et al. (2011), Rothfels et al. (2012b) and Wei et al. (2013); and (d) topology showing unresolved relationships within eupolypod II ferns in Rothfels et al. (2015) based on 25 low-copy nuclear genes.
The utilities of whole chloroplast genomes, or plastomes, in fern phylogenetics have been documented in earlier studies (Wolf et al. 2003, 2011; Der 2010; Lu et al. 2015). Our primary objective of this study is to resolve relationships within eupolypods II using a character-rich plastome data set. The specific aims are (1) to explore the position of Rhachidosoraceae and the monophyly of Athyriaceae sensuPPG I (2016) within this group of ferns, and (2) to investigate substitution rate heterogeneity across eupolypods II based on our well-established plastome phylogeny. We believe that our results will provide new insights into the value of plastome data for resolving phylogenetic relationships within fern lineages that have experienced rapid evolutionary radiations.
Materials and Methods
Taxon Sampling
Our sampling strategy followed the classification proposed by PPG I (2016) and comprised 40 samples representing 32 ingroup species and 8 outgroup species (table 1). The outgroup included two species from Pteridaceae (Adiantum and Myriopteris) and one was from Dennstaedtiaceae (Pteridium). The outgroups also included five species representing eupolypods I: one species of Hypodematiaceae (Hypodematium), two species from Dryopteridaceae (Cyrtomium and Dryopteris), and two species from Polypodiaceae (Lepisorus and Polypodium). Among the ingroup, we included two species of Cystopteris from Cystopteridaceae, one species of Rhachidosorus from Rhachidosoraceae, three species of Diplaziopsis and Homalosorus representing Diplaziopsidaceae, three species of Asplenium and Hymenasplenium to represent Aspleniaceae, one species each of Ampelopteris, Christella, Macrothelypteris, Pseudophegopteris, and Stegnogramma from Thelypteridaceae two species of Woodsia from Woodsiaceae, one species each from Austroblechnum and Woodwardia to represent Blechnaceae, one representative species each of Matteuccia and Onoclea from Onocleaceae, and 12 species from Athyrium, Deparia and Diplazium representing Athyriaceae. Our sampling covered almost all major lineages of eupolypods II except Hemidictyaceae and Desmophlebiaceae, which were strongly supported as sisters to Aspleniaceae in prior studies (Schuettpelz and Pryer 2007; Rothfels et al. 2012b; Mynssen et al. 2016). We generated new plastome sequences for 33 species that had no data in GenBank, including 30 from eupolypods II and three from eupolypods I. Finally, we included seven additional plastomes from GenBank. All voucher information is listed in table 1.
Table 1.
Data of the Sampled Species in This Study and Information on the Plastome Assembly
| Species | Family | No. Voucher/References | No. Accession | No. Reads (trimmed) | Coverage of Plastome | Plastome Size (bp) | GC % |
|---|---|---|---|---|---|---|---|
| Ingroup taxa | |||||||
| Ampelopteris prolifera (Retz.) Copel. | Thelypteridaceae | China, Yunnan, WR0326 | KY427329 | 16,252,690 | 606 | 151,772 | 42.4 |
| Anisocampium sheareri (Baker) Ching | Athyriaceae | China, Guizhou, 7194 | KY427330 | 16,594,594 | 239 | 151,068 | 43.5 |
| Asplenium pekinense Hance | Aspleniaceae | China, Beijing, WR0321 | KY427331 | 15,770,830 | 412 | 152,479 | 41.2 |
| Asplenium prolongatum Hook. | Aspleniaceae | China, Guizhou, WR0323 | KY427332 | 15,399,230 | 345 | 151,115 | 40.8 |
| Athyrium devolii Ching | Athyriaceae | China, cultivated, 7057 | KY419703 | 15,573,042 | 411 | 151,284 | 44.0 |
| Athyrium sinense Rupr. | Athyriaceae | China, Hebei, WR0324 | KY427333 | 7,931,306 | 152 | 151,319a | 43.8 |
| Blechnum melanocaulon (Brack.) T.C. Chambers & P.A. Farrant | Blechnaceae | Indonesia, Java, WR0329 | KY427334 | 6,774,584 | 210 | 150,202 | 43.7 |
| Cornopteris opaca (D. Don) Tagawa | Athyriaceae | China, Guizhou, 7148 | KY427335 | 15,459,732 | 337 | 150,979 | 43.7 |
| Cyclosorus procerus S. Lindsay & D.J. Middleton | Thelypteridaceae | China, Yunnan, WR0325 | KY427336 | 14,590,872 | 444 | 151,517 | 42.5 |
| Cystopteris chinensis (Ching) X.C. Zhang & R. Wei | Cystopteridaceae | China, Sichuan, WR0319 | KY427337 | 15,426,778 | 160 | 151,269a | 42.9 |
| Cystopteris protrusa (Weath.) Blasdell | Cystopteridaceae | Marchant et al. unpublished | KP136830 | N/A | N/A | 131,837b | 42.7 |
| Deparia lancea (Thunb.) Fraser-Jenk. | Athyriaceae | China, cultivated, Sh002 | KY427338 | 14,456,502 | 255 | 151,011 | 43.9 |
| Deparia pycnosora (Christ) M. Kato | Athyriaceae | China, Beijing, WR0322 | KY427339 | 28,120,814 | 415 | 151,126 | 44.0 |
| Deparia viridifrons (Makino) M. Kato | Athyriaceae | China, Guizhou, 7434 | KY427340 | 15,633,288 | 171 | 150,939 | 43.8 |
| Diplaziopsis cavaleriana (Christ) C. Chr. | Diplaziopsidaceae | China, Guizhou, 7154 | KY427341 | 18,796,214 | 341 | 151,934 | 43.1 |
| Diplaziopsis javanica (Blume) C. Chr. | Diplaziopsidaceae | Indonesia, Sumatra, 2895 | KY427342 | 17,699,800 | 350 | 151,496 | 42.6 |
| Diplazium bellum (C.B. Clarke) Bir | Athyriaceae | China, Yunnan, WR0206 | KY427343 | 15,094,718 | 255 | 151,601 | 42.6 |
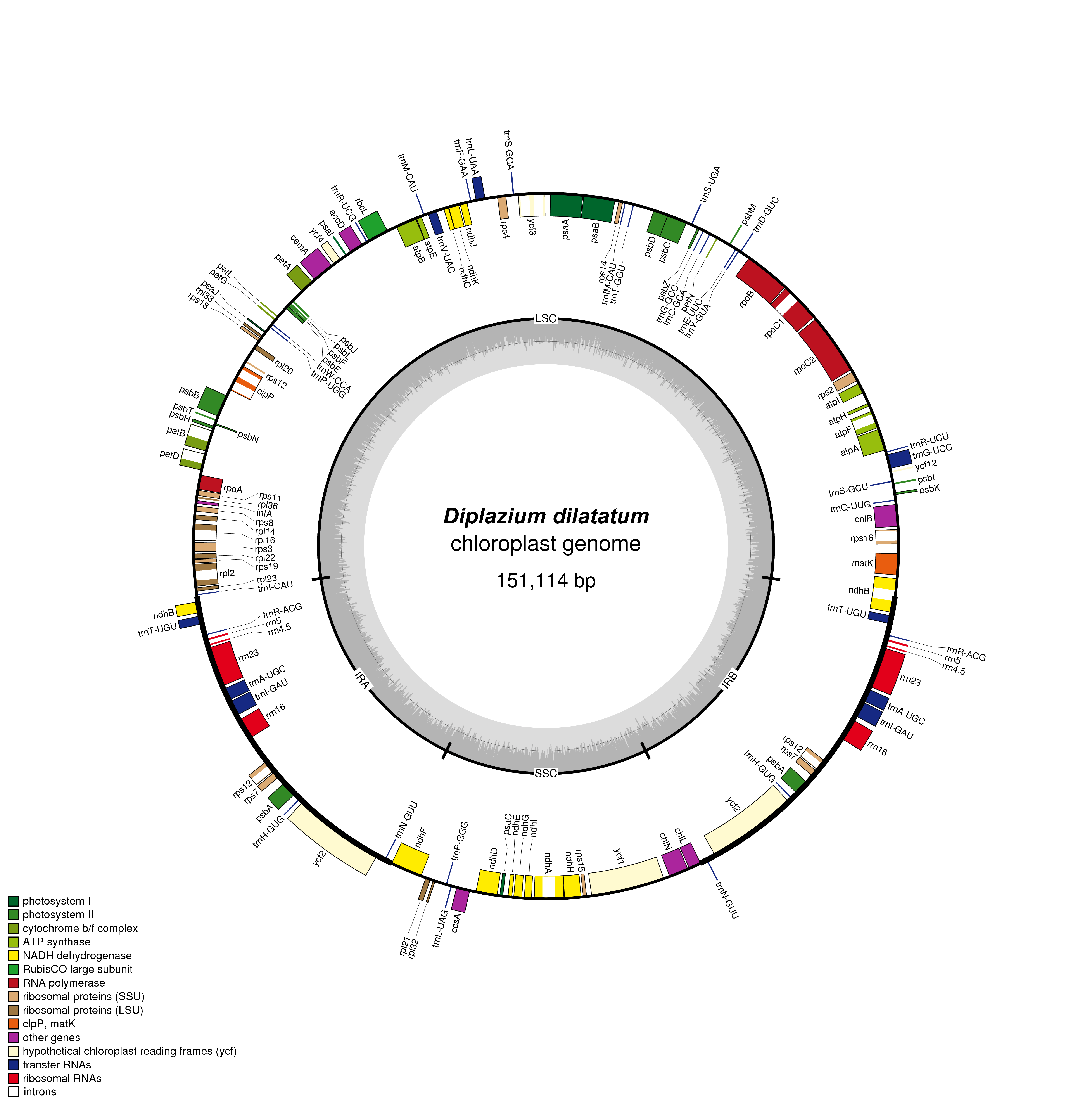
| Diplazium dilatatum Blume | Athyriaceae | China, Guangdong, WR0183 | KY427344 | 16,471,548 | 244 | 151,114 | 43.9 |
| Diplazium dushanense (Ching ex W.M. Chu & Z.R. He) R. Wei & X.C. Zhang | Athyriaceae | China, Guizhou, WR0320 | KY427345 | 15,433,246 | 362 | 150,179 | 43.2 |
| Diplazium striatum (L.) C. Presl | Athyriaceae | Cuba, Santiago, JL455 | KY427346 | 13,811,138 | 489 | 150,779 | 43.8 |
| Diplazium unilobum Hieron. | Athyriaceae | Cuba, Santiago, JL452 | KY427347 | 13,964,600 | 852 | 127,840b | 43.6 |
| Homalosorus pycnocarpos (Spreng.) Pic. Serm. | Diplaziopsidaceae | USA, North Carolina, X004 | KY427349 | 16,754,236 | 1536 | 152,159 | 43.2 |
| Hymenasplenium unilaterale (Lam.) Hayata | Aspleniaceae | China, Guizhou, 7191 | KY427350 | 14,595,828 | 213 | 151,723 | 42.0 |
| Macrothelypteris torresiana (Gaudich.) Ching | Thelypteridaceae | China, Guizhou, 7471 | KY427352 | 15,718,050 | 450 | 151,130 | 43.1 |
| Matteuccia struthiopteris (L.) Tod. | Onocleaceae | China, Beijing, WR0331 | KY427353 | 16,356,718 | 556 | 151,003 | 44.3 |
| Onoclea sensibilis L. | Onocleaceae | China, Beijing, WR0327 | KY427354 | 15,633,816 | 377 | 148,395 | 44.4 |
| Pseudophegopteris aurita (Hook.) Ching | Thelypteridaceae | China, Jiangxi, WR0326 | KY427355 | 8,070,614 | 712 | 149,917a | 43.1 |
| Rhachidosorus consimilis Ching | Rhachidosoraceae | China, Guizhou, 7449 | KY427356 | 18,855,194 | 764 | 153,190a | 43.5 |
| Stegnogramma sagittifolia (Ching) L.J. He & X.C. Zhang | Thelypteridaceae | China, Guizhou, 7486 | KY427357 | 14,368,186 | 225 | 151,132 | 43.0 |
| Woodsia macrochlaena Mett. ex Kuhn | Woodsiaceae | China, Heilongjiang, Wu126 | KY427358 | 13,930,412 | 348 | 150,987 | 42.7 |
| Woodsia polystichoides D.C. Eaton | Woodsiaceae | China, Heilongjiang, Wu48 | KY427359 | 15,593,392 | 613 | 150,685 | 42.6 |
| Woodwardia unigemmata (Makino) Nakai | Blechnaceae | Lu et al. 2015 | NC_028543 | N/A | N/A | 153,717 | 43.2 |
| Outgroup taxa | |||||||
| Adiantum capillus-veneris L. | Pteridaceae | Wolf et al. 2003 | NC_004766 | N/A | N/A | 150,568 | 42.0 |
| Cheilanthes lindheimeri Hook. | Pteridaceae | Wolf et al. 2011 | NC_014592 | N/A | N/A | 155,770 | 42.7 |
| Cyrtomium devexiscapulae (Koidz.) Koidz. & Ching | Dryopteridaceae | Lu et al. 2015 | NC_028542 | N/A | N/A | 151,628 | 42.3 |
| Dryopteris decipiens (Hook.) Kuntze | Dryopteridaceae | China, Guizhou, 7333 | KY427348 | 12,461,232 | 134 | 150,987 | 42.7 |
| Hypodematium crenatum (Forssk.) Kuhn & Decken | Hypodematiaceae | China, Guizhou, 7078 | KY427351 | 12,451,746 | 287 | 149,794a | 41.1 |
| Lepisorus clathratus (C.B. Clarke) Ching | Polypodiaceae | China, Beijing, jingB-1 | KY419704 | 18,095,858 | 540 | 156,998 | 41.8 |
| Polypodium glycyrrhiza D.C. Eaton | Polypodiaceae | Marchant et al. unpublished | KP136832 | N/A | N/A | 129,223b | 40.1 |
| Pteridium aquilinum var. aquilinum (L.) Kuhn | Dennstaedtiaceae | Der, 2010 | NC_014348 | N/A | N/A | 152,362 | 41.5 |
Note.—Vouchers of the specimens are deposited at the Chinese National Herbarium (PE) in Institute of Botany, Chinese Academy of Sciences. Accession numbers in bold indicate newly generated data in this study. N/A, data not available.
Plastid genome with a few small gaps to be bridged.
Partial genome.
Data Extraction and Plastome Assembly
We extracted total genomic DNA from silica-dried material using a modified CTAB method (Doyle and Doyle 1987). We sent the DNA extracts to the Novogene Corporation (Beijing, China), which constructed sequencing libraries using a TruSeq Nano DNA HT Sample Preparation Kit (Illumina, San Diego, CA) following the manufacturer’s recommendations. Specifically, each DNA sample was indexed with a unique marker for downstream identification of its sequences. The samples were sheared into 350-bp fragments, which were end polished, A-tailed, and ligated with the full-length adapter for Illumina sequencing with PCR amplification. The libraries were sequenced on an Illumina HiSeq X Ten platform (Illumina, San Diego, CA), and 150-bp paired-end reads were generated with insert size ∼350 bp. Following enrichment, we obtained the NGS data for 33 species, ranging from c. 4–6 Gb.
We filtered the raw reads using default settings (-L:5 -p:0.5 -N:0.1) in ng_QC v2.0, which was developed by Novogene Corporation. To assemble the reads, we used both de novo and reference-guided methods. Frist, we performed de novo assemblies in VELVET (Zerbino and Birney 2008) using the k-mer size ranging from 57 to 87 bp. We merged contigs in Geneious R9.1.2 (Kearse et al. 2012) and then mapped them to plastomes of Woodwardia unigemmata (NC_028543) and Cyrtomium devexiscapulae (NC_028542) (Lu et al. 2015), which we downloaded from GenBank to use as references. To correct errors and assembly ambiguities mainly resulting from the reference-guided assembly method, we extracted the consensus sequence obtained from reference-guided assembly in Geneious, and used it as the reference sequence to remap the contigs produced by de novo assembly, and we manually curated the remapped contigs. To improve curation of the assemblies, we detected the boundaries of large single copy (LSC), small single copy (SSC) and inverted repeat (IR) regions using BLAST via the NCBI website (http://blast.ncbi.nlm.nih.gov/Blast.cgi; last accessed June 21, 2017). We annotated the final plastome assemblies in DOGMA with an E-value of 5 and a 60% or 80% percent identity cutoff for protein coding genes and tRNAs (Wyman et al. 2004) or manually in Geneious. We deposited all 33 annotated plastomes in GenBank (accession numbers: KY419703, KY419704, KY427329 − KY427359).
Phylogenetic Analyses
We generated three data matrices from 40 plastomes: (1) the complete plastid genome, (2) 88 genes with complete introns included, (3) 83 protein-coding (CDS) genes excluding introns, four ribosomal RNA genes (4.5S, 5S, 16S, and 23S) and one NAD(P)H oxidoreductase gene ndhB (supplementary table S1, Supplementary Material online). Only one copy of the IR region was included in the data matrices. For the complete plastomes, sequences were aligned using MAFFT v7 (Katoh and Standley 2013) implemented in Geneious. For the data sets of 88 genes and 83 CDS genes, we first aligned the genes independently in MAFFT and then concatenated them in BioEdit v7.1.1 (Hall 1999). We excluded poorly aligned regions from the matrices with GBLOCKS (Talavera and Castresana 2007) using codons as the type of sequences (-t), up to half gap positions allowed (b5: half), the block lengths of 2 at minimum (b4: 2), and no more than 13 contiguous nonconserved positions (b3:13).
We performed maximum likelihood (ML) analyses in RAxML v8 (Stamatakis 2006) on the CIPRES Science Gateway (http://www.phylo.org(last accessed June 21, 2017); Miller et al. 2010). For the complete plastome alignment, we conducted the ML analysis without partitioning, because spacers can be difficult to assign into partitions. In order to obtain a reliable phylogenetic reconstruction, we applied three partitioning strategies to the 88- and 83-gene data sets for comparison: (1) no partitioning, (2) partitioning by gene, and (3) partitioning according to results from PartitionFinder v1.1.1 (Lanfear et al. 2012) with pre-defined partitioning by genes. For PartitionFinder and the ML analyses, we specified the GTR + G + I evolutionary model based on results from jModelTest2 (Darriba et al. 2012) for each partition according to the corrected Akaike information criterion (AICc, Burnhan and Anderson 2002). We performed 1,000 rapid bootstrap replications, and we visualized the ML trees and bootstrap values (BS) using FigTree v1.4 (Rambaut 2009). We compared ML outcomes for the three data sets and different partitioning strategies using the AICc criterion. The AICc was calculated as 2(−logeL) + 2K(n/(n − K − 1)) (Akaike 1974; Hurvich and Tsai 1993), where −logeL is the likelihood obtained in the ML analysis, n is the number of sites in the alignment, and K is the number of free model parameters.
We performed Bayesian inference (BI) analyses in MrBayes v3.2.1 (Ronquist et al. 2012). To shorten the calculation time, we analyzed the complete plastome, 88- and 83-gene matrices without partitioning. We applied the GTR + G + I determined in jModelTest2 to each data set. Each Bayesian analysis comprised two independent runs of 100 million generations from a random starting tree with one cold chain and three hot chains, and we sampled the cold chain every 1,000 generations. We observed statistical output from the BI runs in Tracer v1.6 (Rambaut and Drummond 2009) to check for convergence between simultaneous runs, determine appropriate burn-ins, and ensure effective sampling sizes (ESS) of all parameters > 200. A burn-in of 25% was appropriate for all runs. We combined simultaneous, independent runs to obtain majority rule consensus trees and to calculate posterior probabilities (PP).
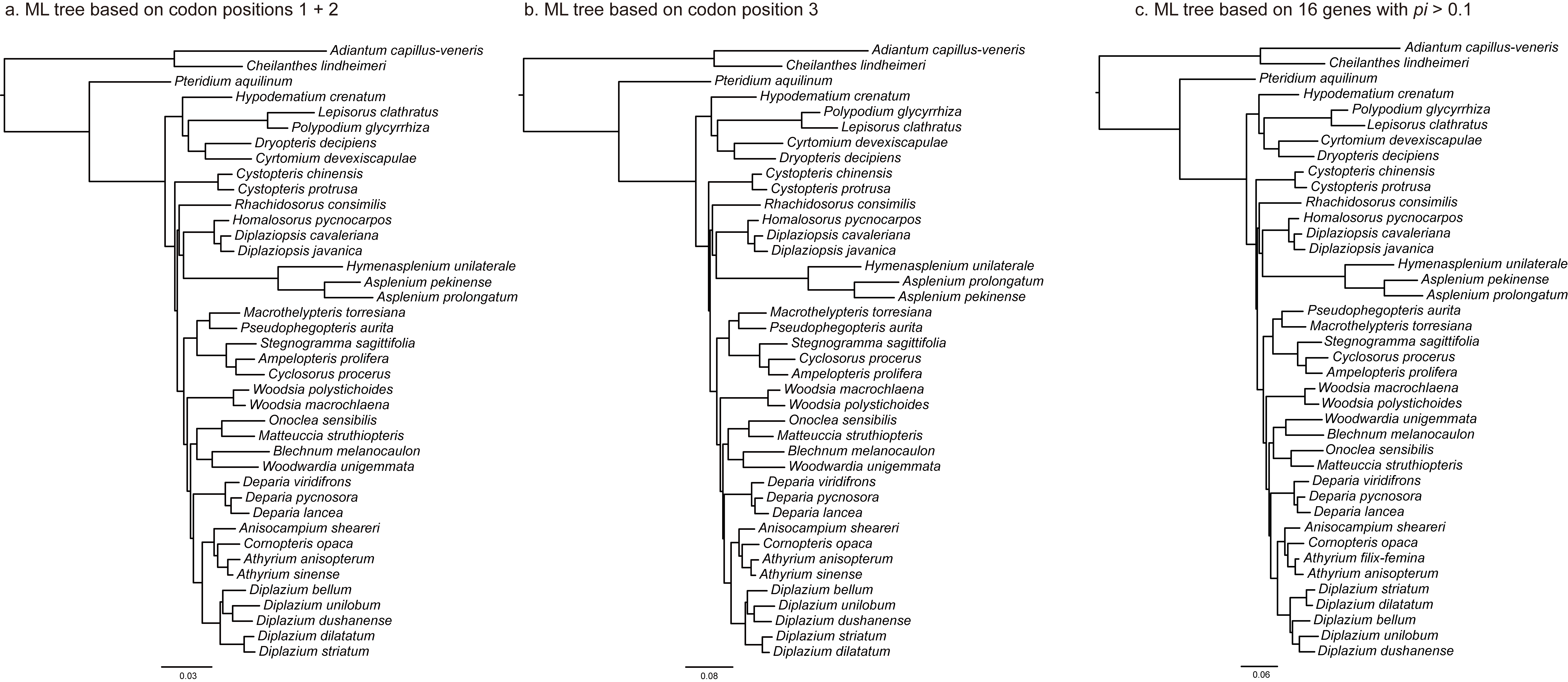
We assessed the effects on tree topology of possible saturation at the third codon position (fast evolving) by performing independent ML analyses of the first and second positions (slowly evolving) combined and the third position. For these additional analyses, we used the same procedure ML described earlier with no partitioning and the GTR + G + I model. We compared tree topologies obtained from the complete plastomes, 88-gene matrix, 83-gene matrix, codon positions 1 + 2, and position 3 using a patristic distance correlation analysis in Mesquite v3.10 (Maddison and Maddison 2015).
Rate heterogeneity, such as in eupolypods II, may sometimes co-occur with other confounding evolutionary signals, such as base compositional heterogeneity (Ho and Jermiin 2004). Base compositional heterogeneity violates the assumptions of phylogenetic inferences methods and can cause misleading results (Sheffield 2013). Thus, we carried out a χ2 test of base-frequency homogeneity for each gene in PAUP* 4.0b10 (Swofford 2002). We detected significant compositional heterogeneity in the ndhF and ycf1 genes, but found that excluding these genes from our data sets yielded results (not shown) highly congruent with those from analyses in which they were not excluded. Thus, we included them in all analyses presented here.
Divergence Time Estimation
To infer a time-scale of rapid radiation within eupolypod II ferns, we carried out a Bayesian divergence time estimation using a relaxed clock model in BEAST v1.7.2 (Drummond and Suchard 2010; Drummond et al. 2012). For this analysis, we used the unpartitioned 83-gene data set, to minimize computational complexity and achieve statistical convergence. We employed two secondary nodal calibrations (Schuettpelz and Pryer 2009; Rothfels et al. 2015). First, we constrained the root age of Pteridaceae and other polypod ferns (Node 1) using a normal prior distribution with a mean of 165.44 and an SD of 31.4 to cover the range of estimated split times (114–194 Ma reported in Schneider et al. 2004a; Schuettpelz and Pryer 2009; Rothfels et al. 2015). For the second calibration node (Node 2), we used a normal prior distribution with a mean of 103.1 and an SD of 10.3 to constrain the crown age of eupolypods II according to the results of Rothfels et al. (2015). As extinction may be an important process in the biogeographical history of this group of ferns, the data set was run using a birth–death speciation prior. For MCMC analyses, we performed three independent runs of 10 million generations with sampling every 1000 generation on the CIPRES Science Gateway. The resulting log files were combined in LOGCOMBINER after discarding the first 10% of generations as burning. We checked the combined logs in TRACER ensure that effective sample sizes (ESS) for the relevant estimated parameters were well >200. We resampled the tree files in LOGCOMBINER using the same burn-in strategy. We obtained the time-calibrated maximum clade credibility (MCC) topology with a posterior probability limit of 0.5 and mean branch lengths using TREEANNOTATOR. We visualized our results in FigTree to check high probability densities (HPDs) of the chronogram.
Plastid Substitution Rate Analyses
To explore the variation in substitution rates across the eupolypod II ferns and the degree to which it influences phylogenetic reconstruction, we used a random local clock (RLC) model within a Bayesian framework as implemented in BEAST. We applied the model to our 83-gene data set and ran the analyses with unpartitioned strategy using the GTR + G + I model with four rate categories, birth–death speciation prior, Poisson rate change prior, and CTMC rate reference for the clock rate with initial value of 1.0 (Ferreira and Suchard 2008; Barrett et al. 2016). Moreover, we performed an independent run using the same data set but with priors only for comparison to ensure that the rate variations that we detected resulted from our sequence data and were not simply constructs of our prior settings. We ran three independent MCMC runs of 10 million generations each from a random starting tree. We used Tracer to confirm stationarity of runs and to verify an ESS >200 for relevant estimated parameters. We discarded the first 10% of generations as burn-in using LOGCOMBINER and generated a MCC tree with relative rates in TREEANNOTATOR.
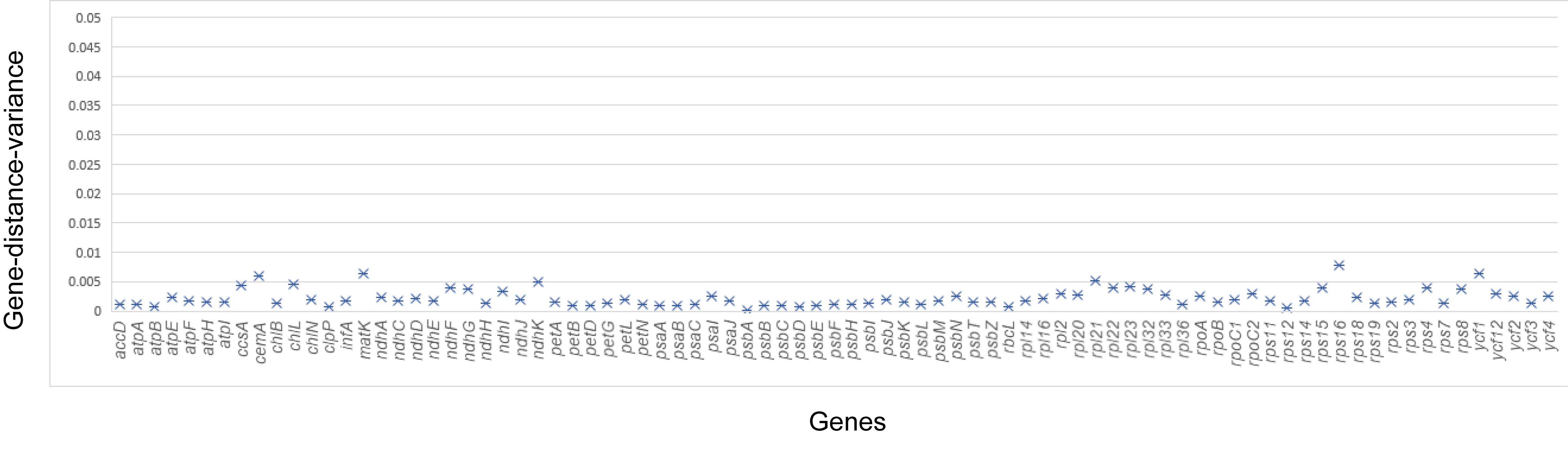
In addition, we tested whether heterogeneous substitution rates among taxa were plastome-wide (global heterogeneity) or primarily restricted to a few genes (local heterogeneity). To accomplish this, we compared within-gene variance of pairwise distances across genes. We performed this analysis using a custom python script (see Supplementary Material online).
Detection of Informative Plastid Markers for Deep Phylogeny of Eupolypod II Ferns
We sought to detect the most variable plastid genes in eupolypod II ferns and determine their potential for resolving recalcitrant nodes, especially due to the rapid radiation. To accomplish this, we measured parsimony-informative characters (PICs) per site (Pi values) with DnaSP v5.1 (Librado and Rozas 2009) for ingroup taxa. We applied a sliding scale window with nonoverlapping 1,000-bp segments in order to take into account both long and short loci. We tested the phylogenetic utility of plastid markers with Pi > 0.1 by using them exclusively to perform an ML analysis. We examined the correlations between the topology based on these genes to the complete-plastome matrix, the 88-gene matrix, and the 83-gene matrix in Mesquite.
Results
We obtained new plastome sequences for 33 species, and the coverage of the plastid genomes ranged from 134× (Dryopteris decipiens) to 1,536× (Homalosorus pycnocarpos) (table 1). We found that plastome structure was conserved across the species sampled and that the complete length of the plastomes without gaps ranged from 148,359 to 156,998 bp (table 1 andsupplementary fig. S1, Supplementary Material online). The GC content of the plastomes ranged from 40.8% to 44.4% (table 1).
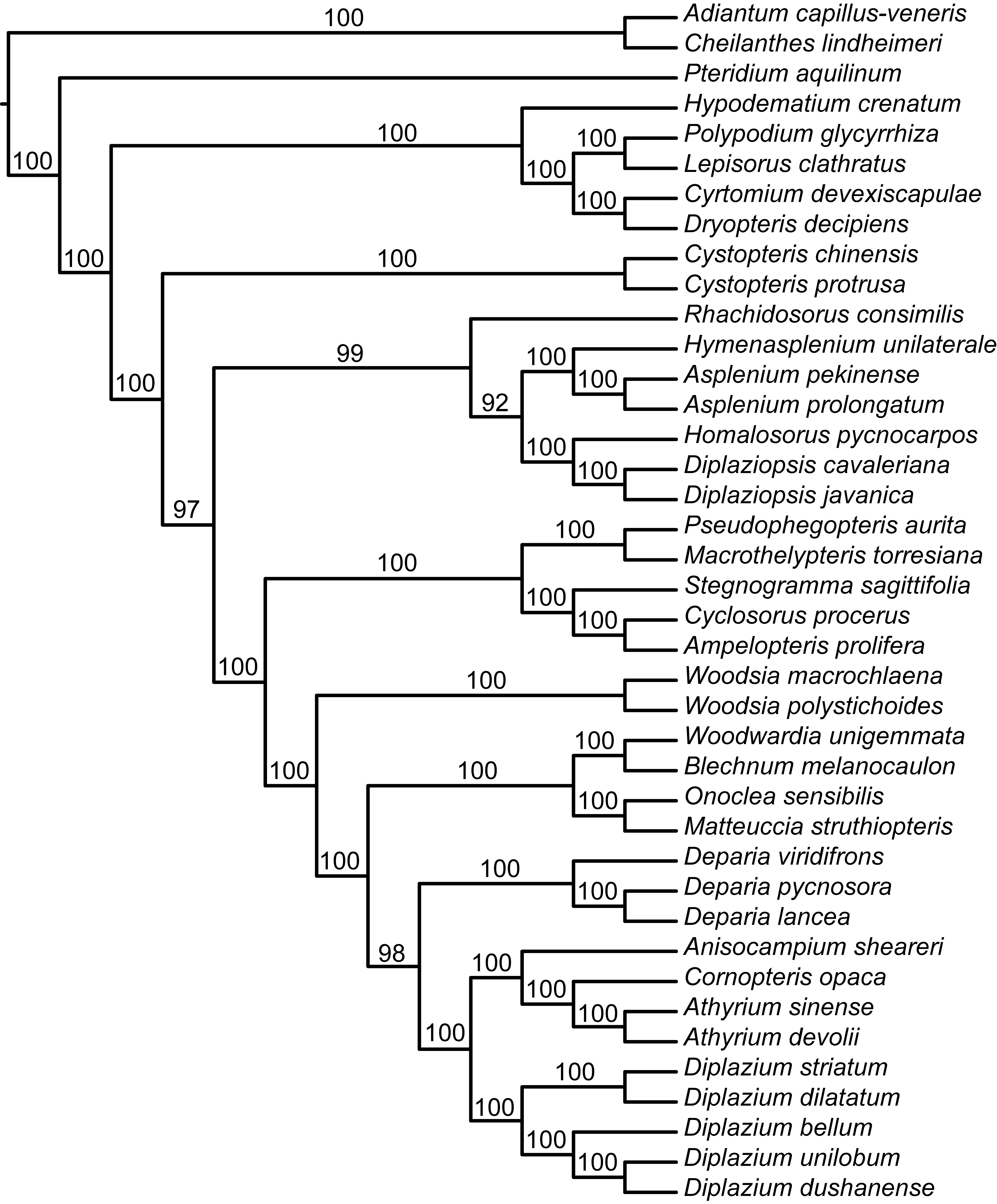
The results from different data matrices, partitioning strategies, and analytical methods were largely congruent (supplementary fig. S2, Supplementary Material online). The ML analyses of the 88- and 83-gene matrices partitioned by gene had the lowest AICc scores, respectively (table 2). Among ML trees based on different data sets (the complete plastome, 88-gene, 83-gene, codon positions 1 + 2, codon position 3, and 14-gene with Pi > 0.1), we found that patristic distances were highly correlated (coefficient = 1.0). ML trees were highly consistent with results from BI analyses. The ML and BI analyses resolved a strongly supported phylogenetic framework with nearly all deep nodes supported by 100% in BS values (BSs) and 1.0 in PP values (PPs) (fig. 2a). Notably, all analyses showed Rhachidosoraceae as sister to the clade including Diplaziopsidaceae and Aspleniaceae with strong support of 100% or 98% BSs and 1.0 PPs, and the sister relationship of Diplaziopsidaceae to Aspleniaceae also received support with BSs ≥ 69% and PPs = 1.0. Furthermore, the monophyly of Athyriaceae was strongly supported in all analyses with BSs of 99–100% and PPs of 1.0.
Table 2.
Comparison of GTR + I + G Partition Models from Maximum Likelihood Analysis of the Data Set
| GTR Model Partition | No. Partitions | −logeL* | No. Free Parameters | AICc |
|---|---|---|---|---|
| 88 genes | ||||
| Unpartitioned | 1 | 770062.3769 | 86 | 1540296.92 |
| PartitionFinder | 13 | 765521.0473 | 194 | 1531430.92 |
| Gene partitioned | 88 | 764509.8944 | 869 | 1530774.41 |
| 83 genes | ||||
| Unpartitioned | 1 | 597995.6031 | 86 | 1196163.42 |
| PartitionFinder | 11 | 595226.2139 | 176 | 1190805.32 |
| Gene partitioned | 83 | 594484.0772 | 824 | 1190635.76 |
Note.—−logeL, the likelihood; AICc, corrected Akaike Information Criterion; AICc = 2(−logeL) + 2K(n/(n − K − 1)); “n,” number of sites in the alignment; “K,” number of free model parameters.
Fig. 2.—

The ML phylogram of 40 species based on 83-gene matrix and codon partitioned strategy and Bayesian divergence time estimation based on 83-gene matrix with unpartitioned strategy. (a) Maximum likelihood bootstrap values (BSs) are 100% and Bayesian posterior probabilities (PPs) are 1.0, unless otherwise indicated. Numbers above the branches indicate BSs and PPs based on whole plastome matrix, and BSs (unpartitioned, ParititionFinder, gene partitioned) and PPs based on 88-gene matrix, while numbers below the branches as BSs (unpartitioned, codon partitioned, ParititionFinder, gene partitioned) and PPs based on 83-gene matrix; (b) chronogram with secondary calibration nodes indicated by numbers; (c) bar chart indicating stem clade ages and HPD intervals of each family of eupolypods II. Blue bars indicate 95% highest posterior density (HPD) intervals of the age estimates; grey bars indicate the time-scale of eupolypod II radiation.
Our Bayesian divergence time showed that most families within eupolypods II evolved within the period ranging from 65 to 95 Ma (fig. 2b and c). Bayesian analysis under the RLC model in Beast, revealed a mean value of 33 substitution rate shifts (fig. 3 andsupplementary fig. S3, Supplementary Material online). Our test showed that there was similar heterogeneity within all markers, and we detected no outliers (supplementary fig. S4, Supplementary Material online). Rates accelerated in Thelypteridaceae, Woodsiaceae, Onocleaceae and Blechnaceae, and decelerated in Cystopteridaceae. Aspleniaceae and Athyriaceae experienced both accelerations and decelerations. Comparison of runs with priors only versus with sequence data indicated that the 95% HPD of 30–36 rate changes was significantly different from the 95% HPD of 0–5 for prior only (supplementary fig. S3, Supplementary Material online).
Fig. 3.—

Relative plastid substitution rates among clades/branches of the eupolypod II ferns, based on the “83-gene matrix,” resulting from the random local clock analyses in BEAST v1.8.1. Numbers above the branches indicate relative, median rates with no measured units, scaled by dividing all rates by that of the crown node of eupolypods II. Colors of branches represents to relative rate. Light red indicates a relative rate of 0, whereas a trend of blue color marks a relative rate approaching 3.0.
The sliding window analysis revealed that genes located in SC region are more variable than in the IR regions (fig. 4). However, it was interesting that variability of genes without introns was generally higher than genes with intron regions. For eupolypod II ferns, ndhF, rpoC2 and ycf1 have Pi > 0.15, among which the two genes with heterogeneous base compositional frequencies, ndhF and ycf1, had the highest and second highest Pi, respectively. Other genes having Pi > 0.1 were ccsA, cemA, chlN, clpP, matK, ndhA, ndhD, petB, rpl2, rpl16, and rps16, and these were mainly located in SC regions. Only one gene with Pi > 0.1, ycf2, was located in the IR region. The most commonly used plastid gene markers, such as atpA, atpB, rbcL, had very low Pi value (< 0.1).
Fig. 4.—

Comparison of variability in nonoverlapping 1,000-bp sliding windows for eupolypod II ferns. Genes with interests are indicated, that is, genes with Pi > 0.1 are marked in black and genes with Pi < 0.1 are marked in red.
Discussion
Deep Phylogenetic Relationships within Eupolypod II Ferns
The plastome phylogenies presented here represent the most robust phylogenetic framework to-date of eupolypods II. Nearly all deep nodes have strong support (BSs = 100% and PPs = 1.0; fig. 2a) even under different partitioning strategies and phylogenetic methods. The current results help to verify the phylogenetic position and delimitation of Cystopteridaceae, Diplaziopsidaceae, Aspleniaceae, Thelypteridaceae, Woodsiaceae, Blechnaceae and Onocleaceae proposed in previous studies (Schuettpelz and Pryer 2007; Kuo et al. 2011; Rothfels et al. 2012a,b). Our results unambiguously show the position of Rhachidosoraceae and Athyriaceae, which have been extremely difficult to place within eupolypods II (Wei et al. 2010; Kuo et al. 2011; Rothfels et al. 2012b). Rothfels et al. (2012b) report a similar topology to ours based on five chloroplast markers and a different sampling strategy. However, the position of Rhachidosorus and the circumscription of Athyriaceae (BS = 63% for Rhachidosorus, BS = 75% for athyrioids) are not well supported, which left the deep relationships of eupolypod II unresolved. They hypothesized that weakly or unresolved relationships were a result of rapid radiation in the eupolypods, and noted that their phylogenetic results bore a classic footprint of rapid radiation: short branches at deep nodes compared with long ones within crown clades. Our results support the hypothesis of Rothfels et al. (2012b) of a deep, rapid radiation in eupolypods II. We found short branches at deep nodes, and observed that the evolution of most families occurred during a 30-Myr period between 65 and 95 Ma (fig. 2). Nevertheless, we also found that our robust plastome provided a strong phylogenetic signal even for short branches. In particular, the plastomes provide nearly 9 times the PICs of the 25 nuclear genes used in smaller sample size of nuclear genes used by in Rothfels et al. (2012b) (26,000 PICs vs. less than 3,000 PICs). We have achieved improved phylogenetic resolution, particularly for Rhachidosoraceae as sister to the clade containing Diplaziopsidaceae and Aspleniaceae (BSs > 98% and PPs = 1.0) and found strong support for the monophyly of Athyriaceae (BSs > 99% and PPs = 1.0).
More recently, Rothfels et al. (2015) reported low support for most deep nodes of eupolypods II based on a topology reconstructed from 25 low-copy nuclear genes. The topology from Rothfels et al. (2015) differs from our topologies by resolving Thelypteridaceae as sister to Aspleniaceae with 46% BS support and rendering Athyriaceae paraphyletic by including Blechnaceae and Onocleaceae with BS as 80%, which was one of the highest supports among nodes in the nuclear phylogeny. One possible explanation for the incongruence between reconstructed phylogenies in that study and ours may be incomplete lineage sorting among nuclear genes, given the fact that nuclear genes may experience 4 times slower coalescence than plastid genes (Moore 1995; Rothfels et al. 2015). However, other biological processes, such as hybridization, introgression and polyploidization, could also contribute to phylogenetic discordance, because reticulate evolution has been frequently observed in several families of leptosporangiate ferns (Wagner 1954; Werth et al. 1985; Kato and Kramer 1990; Schneider et al. 2004b; and references therein). Thus, the incongruence between topologies in the Rothfels et al. (2015) study and ours merits further investigation to elucidate the evolutionary processes within eupolypods II, even though the incongruence is soft (i.e., not well supported in Rothfels et al. 2015).
Although morphological stasis is well-known within eupolypod II ferns (Rothfels et al. 2012a,b; Sundue and Rothfels 2014), a few obvious morphological synapomorphies of Rhachidosoraceae and Aspleniaceae can be recognized. For example, both families possess single-sided “asplenioid” sori and clathrate scales, while all the species of Athyriaceae share J-shaped “athyrioid” or back-to-back “diplazioid” sori and non-clathrate scales (Sundue and Rothfels 2014). Previous taxonomic treatments based on morphology regard Rhachidosorus as members of athyrioid ferns, but the characters supporting this relationship, such as leaf shape, rachis-costa architecture, and bisection of stipes (two hippocampus-shaped vascular bundles), may be homoplastic (Ching 1964; Kato 1977; Chu and He 1999).
Substitution Rate Heterogeneity within Eupolypod II Ferns
Our Bayesian analysis based on RLC model revealed about 33 rate shifts among the eupolypod II clades (fig. 3 andsupplementary fig. S3, Supplementary Material online), and all genes showed similar numbers of rate shifts indicating that substitution rate heterogeneity is likely a result of variation in the whole plastome rather than certain genes (supplementary fig. S4, Supplementary Material online). It is notable that only the Rhachidosoraceae-Aspleniaceae and Athyriaceae clades have experienced significant relative rate accelerations and decelerations (fig. 3). Coincidentally, the systematic problems of these two lineages are the most difficult to resolve as indicated in previous studies (Schuettpelz and Pryer 2007; Wei et al. 2010; Kuo et al. 2011; Li et al. 2011; Rothfels et al. 2012b). Given that no significant unequal base frequencies are found in most chloroplast genes within these two lineages (except ndhF and ycf1, results not shown), we propose that the unusual rate heterogeneity probably contributed to their weak resolutions and poor supports at deep nodes (Rosenberg and Kumar 2003; Zhong et al. 2011). Our study successfully resolves the deep phylogenetic relationships involving these problematic lineages, and also demonstrates the power of character-rich data set from plastid genome to overcome the systematic problems caused by rapid radiation tangling together with substitution rate heterogeneity.
Chloroplast Gene Variability and Phylogenetically Informative Markers
Our results demonstrate that the gene number, gene order, and GC content of the plastid genome are largely consistent among eupolypods II and other polypod ferns (table 1) (Wolf et al. 2003, 2011; Der 2010; Lu et al. 2015). The genes do not exhibit rearrangements or segmental inversions that have been found in other ferns and lycophytes, i.e., Huperiza–Isoëtes–Selaginella clade and tree ferns (Gao et al. 2009; Karol et al. 2010; Zhong et al. 2014). However, the chloroplast genes do show high levels of base variability among different lineages (fig. 4). For example, genes such as clpP, matK, ndhA, ndhF, rpoC2, rps16 and ycf1 from the SC region, have strikingly higher PICs than the genes from the IR regions. One probable reason for this phenomenon is that genes that are translocated to the IR region undergo a reduction in substitution rate (Li et al. 2016). Moreover, we find that some frequently used plastid markers, e.g., atpA, atpB, rbcL, and rps4, are among the least informative genes (fig. 4). Therefore, it is not surprising that previous phylogenetic studies based on these markers produced low resolution, especially for deep relationships among families with fast rates and short internodes (Schuettpelz and Pryer 2007; Wei et al. 2010, 2013; Kuo et al. 2011; Li et al. 2011; Rothfels et al. 2012b). By contrast, our sliding window analysis find 14 genes in the SC region with the highest Pi (> 0.1) values (fig. 4), and the phylogenetic reconstruction based on these genes recovered a strongly supported topology with high patristic distance correlations to the whole plastome, 88- and 83-gene trees (coefficient = 1.0 in Mesquite) (supplementary figs. S2 and S5, Supplementary Material online). We suggest that genes with high Pi are suitable markers to elucidate the relationships for fern lineages (e.g., at familial or generic levels in our study) which have experienced rapid radiation, as long as care is taken to check for and accommodate base compositional heterogeneity.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This study was supported by grants from the National Science Foundation of China Grants (Nos. 31600175 and 31470317), and the External Cooperation Program of Bureau of International Co-operation Chinese Academy of Sciences (No. GJHZ201321), and the Shanghai Municipal Administration of Forestation and City Appearances Grant (No. G152420). The authors are grateful to Dr Harald Schneider, Dr Josmaily Lóriga, Dr Rui Zhang and Mr Yu-Dong Wu for providing materials. We thank Dr Craig F. Barrett for help and suggestions on beast analyses, and Dr Ronald L. Viane for helpful suggestions to improve the language of the original manuscript. The authors also thank the three anonymous referees and GBE editors for their critical and suggestive comments that greatly improved our manuscript.
Literature Cited
- Akaike H. 1974. A new look at the statistical model identification. IEEE Trans Autom Control 19:716–723. [Google Scholar]
- Barrett CF, et al. 2016. Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytol. 209:855–870. [DOI] [PubMed] [Google Scholar]
- Burnhan KP, Anderson DR.. 2002. Model selection and multimodel inference: a practical information-theoretic approach. 2nd ed New York: Springer. [Google Scholar]
- Ching R-C. 1964. On some confused genera of the family Athyriaceae. Acta Phytotax Sin. 9:41–84. [Google Scholar]
- Chu W-M, He Z-R.. 1999. Monomelangium Hayata, Allantodia R. Br., Callipteris Bory, Diplazium Sw In: Chu W-M, editor. Flora Reipublicae Popularis Sinicae, vol. 3(2). Beijing: Science Press; p. 346–349, 365–499. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2012. JModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der JP. 2010. Genome perspectives on evolution in bracken fern. PhD thesis, Utah: Utah State University.
- Doyle JJ, Doyle JL.. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15. [Google Scholar]
- Drummond AJ, Suchard MA.. 2010. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 8:114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A.. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MAR, Suchard MA.. 2008. Bayesian analysis of elapsed times in continuous-time Markov chains. Can J Stat. 36:355–368. [Google Scholar]
- Gao L, Yi X, Yang Y-X, Su Y-J, Wang T.. 2009. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol Biol. 9:130.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98. [Google Scholar]
- He L-J, Zhang X-C.. 2012. Exploring generic delimitations within the fern family Thelypteridaceae. Mol Phylogenet Evol. 65:757–764. [DOI] [PubMed] [Google Scholar]
- Ho SYW, Jermiin LS.. 2004. Tracing the decay of the historical signal in biological sequence data. Syst Biol. 53:623–637. [DOI] [PubMed] [Google Scholar]
- Holttum RE. 1982. Thelypteridaceae In: van Steenis CGGJ, Holttum RE, editors. Flora Malesiana. Series II. Pteridophyta. Vol. 1, part 5. The Hague: Martinus Nijhoff: p. 1959–1982. [Google Scholar]
- Hurvich CM, Tsai C-L.. 1993. A corrected Akaike information criterion for vector autoregressive model selection. J Time Ser Anal. 14:271–279. [Google Scholar]
- Jian S, et al. 2008. Resolving an ancient, rapid radiation in Saxifragales. Syst Biol. 57:38–57. [DOI] [PubMed] [Google Scholar]
- Karol KG, et al. 2010. Complete plastome sequences of Equisetum arvense and Isoëtes flaccida: implications for phylogeny and plastid genome evolution of early land plant lineages. BMC Evol Biol. 10:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M. 1977. Classification of Athyrium and allied genera of Japan. Bot Mag. 90:23–40. [Google Scholar]
- Kato M, Kramer KU.. 1990. Subfamily Athyrioideae In: Kramer KU, Green PS, editors. The families and genera of vascular plants, Vol. I–Pteridophytes and gymnosperms. Berlin: Springer-Verlag; p. 130–144. [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L-Y, Li F-W, Chiou W-L, Wang C-N.. 2011. First insights into fern matK phylogeny. Mol Phylogenet Evol. 59:556–566. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SY, Guindon S.. 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29:1695–1701. [DOI] [PubMed] [Google Scholar]
- Li C-X, Lu S-G, Sun X-Y, Yang Q.. 2011. Phylogenetic positions of the enigmatic asiatic fern genera Diplaziopsis and Rhachidosorus from analyses of four plastid genes. Am Fern J. 101:142–155. [Google Scholar]
- Li F-W, Kuo L-Y, Pryer KM, Rothfels CJ.. 2016. Genes translocated into the plastid inverted repeat show decelerated substitution rates and elevated GC content. Genome Biol Evol. 8:2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J.. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. [DOI] [PubMed] [Google Scholar]
- Lu J-M, Zhang N, Du X-Y, Wen J, Li D-Z.. 2015. Chloroplast phylogenomics resolves key relationships in ferns. J Syst Evol. 53:448–457. [Google Scholar]
- Maddison WP, Maddison DR.. 2015. Mesquite: a modular system for evolutionary analysis. Version 3.10. http://mesquiteproject.org.
- Miller MA, Pfeiffer W, Schwartz T.. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE). LA: New Orleans.
- Moore WS. 1995. Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution 49:718–726. [DOI] [PubMed] [Google Scholar]
- Mynssen CM, Vasco A, Moran RC, Sylvestre LS, Rouhan G.. 2016. Desmophlebiaceae and Desmophlebium: a new family and genus of eupolypod II ferns. Taxon 65:19–34. [Google Scholar]
- PPG I. 2016. A community-derived classification for extant lycophytes and ferns. J Syst Evol. 54:563–603. [Google Scholar]
- Rambaut A. 2009. FigTree v.1.4. http://tree.bio.ed.ac.uk.
- Rambaut A, Drummond AJ.. 2009. Tracer v.1.6. http://beast.bio.ed.ac.uk.
- Ronquist F, et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg MS, Kumar S.. 2003. Heterogeneity of nucleotide frequencies among evolutionary lineages and phylogenetic inference. Mol Biol Evol. 20:610–621. [DOI] [PubMed] [Google Scholar]
- Rothfels CJ, et al. 2012a. A revised family-level classification for eupolypod II ferns (Polypodiidae: Polypodiales). Taxon 61:515–533. [Google Scholar]
- Rothfels CJ, et al. 2012b. Overcoming deep roots, fast rates, and short internodes to resolve the ancient rapid radiation of eupolypod II ferns. Syst Biol. 61:490–509. [DOI] [PubMed] [Google Scholar]
- Rothfels CJ, et al. 2015. The evolutionary history of ferns inferred from 25 low-copy nuclear genes. Am J Bot. 102:1–19. [DOI] [PubMed] [Google Scholar]
- Sano R, Takamiya M, Ito M, Kurita S, Hasebe M.. 2000. Phylogeny of the lady fern group, tribe Physematieae (Dryopteridaceae), based on chloroplast rbcL gene sequences. Mol Phylogenet Evol. 15:403–413. [DOI] [PubMed] [Google Scholar]
- Schneider H, et al. 2004a. Ferns diversified in the shadow of angiosperms. Nature 428:553–557. [DOI] [PubMed] [Google Scholar]
- Schneider H, et al. 2004b. Chloroplast phylogeny of asplenioid ferns based on rbcL and trnL-F spacer sequences (Polypodiidae, Aspleniaceae) and its implication for the biogeography of these ferns. Syst Bot. 29:260–274. [Google Scholar]
- Schuettpelz E, Pryer KM.. 2007. Fern phylogeny inferred from 400 leptosporangiate species and three plastid genes. Taxon 56:1037–1050. [Google Scholar]
- Schuettpelz E, Pryer KM.. 2009. Evidence for a Cenozoic radiation of ferns in an angiosperm-dominated canopy. Proc Natl Acad Sci U S A. 106:11200–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y-Z, Wei R, Zhang X-C, Xiang Q-P.. 2015. Molecular phylogeny of the cliff ferns (Woodsiaceae: Polypodiales) with a proposed infrageneric classification. PLoS One 10:e0136318.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC. 2013. The interaction between base compositional heterogeneity and among-site rate variation in models of molecular evolution. ISRN Evol Biol. 2013 e391561. [Google Scholar]
- Smith AR. 1990. Thelypteridaceae In: Kramer KU, Green PS, editors. The families and genera of vascular plants. Berlin: Springer-Verlag. [Google Scholar]
- Smith AR, et al. 2006. A classification for extant ferns. Taxon 55:705–731. [Google Scholar]
- Soubrier J, et al. 2012. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol Biol Evol. 29:3345–3358. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Sundue MA, Rothfels CJ.. 2014. Stasis and convergence characterize morphological evolution in eupolypod II ferns. Ann Bot. 113:35–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. 2002. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4.0b10. Sunderland (MA: ): Sinauer Associates. [Google Scholar]
- Talavera G, Castresana J.. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56:564–577. [DOI] [PubMed] [Google Scholar]
- Tryon RM, Tryon AF.. 1982. Ferns and allied plants with special reference to tropical America. New York: Springer. [Google Scholar]
- Wagner WH. 1954. Reticulate evolution in the Appalachian aspleniums. Evolution 8:103–118. [Google Scholar]
- Wang M-L, Chen Z-D, Zhang X-C, Lu S-G, Zhao G-F.. 2003. Phylogeny of the Athyriaceae: evidence from chloroplast trnL-F region sequences. Acta Phytotax Sin. 41:416–426. [Google Scholar]
- Wei R, Schneider H, Zhang X-C.. 2013. Toward a new circumscription of the twinsorus-fern genus Diplazium (Athyriaceae): a molecular phylogeny with morphological implications and infrageneric taxonomy. Taxon 62:441–457. [Google Scholar]
- Wei R, Zhang X-C, Qi X-P.. 2010. Phylogeny of Diplaziopsis and Homalosorus based on two chloroplast DNA sequences: rbcL and rps4+rps4-trnS IGS. Acta Bot Yunnan. S17:46–54. [Google Scholar]
- Werth CR, Guttman SI, Eshbaugh WH.. 1985. Recurring origins of allopolyploid species in Asplenium. Science 228:731–733. [DOI] [PubMed] [Google Scholar]
- Wolf PG, Rowe CA, Sinclair RB, Hasebe M.. 2003. Complete nucleotide sequence of the chloroplast genome from a leptosporangiate fern, Adiantum capillus-veneris L. DNA Res. 10:59–65. [DOI] [PubMed] [Google Scholar]
- Wolf PG, et al. 2011. The evolution of chloroplast genes and genomes in ferns. Plant Mol Biol. 76:251–261. [DOI] [PubMed] [Google Scholar]
- Wu Z-Q, Ge S.. 2012. The phylogeny of the BEP clade in grasses revisited: evidence from the whole-genome sequences of chloroplasts. Mol Phylogenet Evol. 62:573–578. [DOI] [PubMed] [Google Scholar]
- Wyman SK, Jansen RK, Boore JL.. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255. [DOI] [PubMed] [Google Scholar]
- Xi Z-X, et al. 2012. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc Natl Acad Sci U S A. 109:17519–17524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, et al. 2014. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat Commun. 5:4956.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E.. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wen J, Zimmer EA.. 2016. Another look at the phylogenetic position of the grape order Vitales: chloroplast phylogenomics with an expanded sampling of key lineages. Mol Phylogenet Evol. 101:216–223. [DOI] [PubMed] [Google Scholar]
- Zhang N, Zeng L, Shan H, Ma H.. 2012. Highly conserved low-copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. New Phytol. 195:923–937. [DOI] [PubMed] [Google Scholar]
- Zhong B-J, et al. 2011. Systematic error in seed plant phylogenomics. Genome Biol Evol. 3:1340–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B-J, Fong R, Collins LJ, McLenachan PA, Penny D.. 2014. Two new fern chloroplasts and decelerated evolution linked to the long generation time in tree ferns. Genome Biol Evol. 6:1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.