

Abstract
One hallmark of neuroinflammation is the activation of microglia, which triggers the production and release of reactive oxygen species (ROS), nitrate, nitrite, and cytokines. N-acetyl cysteine (NAC) is a free radical scavenger that is involved in the intracellular and extracellular detoxification of ROS in the brain. However, the clinical application of NAC is limited by its low bioavailability and short half-life. Herein, NAC was conjugated to a polymer through disulfide bond to form a NAC-prodrug nanoparticle (NAC-NP). Dynamic light scattering found that the NAC-NP has a size around 50 nm. In vitro study revealed that the release of NAC from NAC-NP is responsive to its environmental redox potential. In order to mimic neuroinflammation in vitro, microglial cells were stimulated by LPS (Lipopolysaccharide) and the effect of NAC-NP on activated microglia was investigated. The study found that the morphology as well as the expression of microgliosis marker lba-1 of the cells treated with NAC-NPs and LPS were close to those of control cells, indicating that NAC-NPs can inhibit the activation of microglia stimulated by LPS. Compared with free NAC, the production of ROS, NO3-, NO2-, tumor necrosis factor-α (TNF-α), and interleukin (IL)-1β from the LPS-stimulated microglia was considerably decreased when the cells were pretreated with NAC-NP. Furthermore, LPS-induced microglial phagocytocis of neurons was inhibited in the presence on NAC-NPs. These results indicated that NAC-NPs are more effective than free NAC in reversing the effect of LPS on microglia and subsequently protecting neurons.
Keywords: redox potential sensitive, N-acetyl cysteine, prodrug, anti-oxidant, anti-inflammatory, nanoparticle
Graphical Abstract
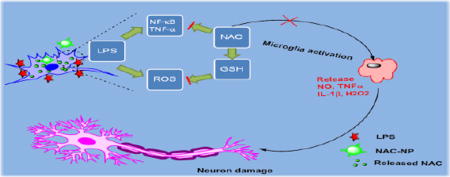
1. Introduction
Microglia are macrophage-like cells that reside in the brain and serve as the first line of active immune defense in the central nervous system (CNS). Microglia scavenge the CNS for invading bacteria, plaques, and damaged neurons. It typically exists in a ramified form, a resting state, while transforming into activated form when it is exposed to inflammatory stimuli.1,2 The level of microglia transformation depends on the intensity and the type of the stimuli, which is accompanied not only with changes in morphology but also with differentiated expressions of several proteins.2 Some of the typical morphological changes and protein expressions (e.g. lba-1, MHC II, CD11b, and CD68) of the activated microglia have been used as markers of microglia activation. Signaling and gene expression changes also result in the release of pro- or anti-inflammatory factors, reactive oxygen species (ROS)3 and nitric oxide (NO).4 The activation of microglia in response to infectious agents, amyloid endogenous proteins, and neuronal death cause the release of cytotoxic factors that lead to neuroinflammation.5 The release of cytotoxic factors such as TNF-α, NO, IL-1β, and ROS have been implicated in the onset of neurodegenerative diseases.6
N-acetyl L-cysteine (NAC) is an anti-oxidant and anti-inflammatory drug that has been explored in the treatment of many neurological diseases.7 NAC is a scavenger of free radicals as well as a precursor of the antioxidant, glutathione, that is implicated not only in the intracellular but also in the extracellular detoxification of ROS in the brain.8 There are many ongoing and completed clinical trials for the use of NAC in neurological disorders mainly focused on Alzheimer9 and Parkinson disease.7 It was revealed that the GSH level was elevated in the human brain in response to intravenous administration of NAC.10 Due to its hydrophobicity and free sulfhydryl group, which can be easily oxidized, the bioavailability and stability of NAC is limited. The oral bioavailability of NAC varies between 6 −10%.11 When it is administered intravenously, the formation of the disulfide bond between NAC and human serum albumin also limits its bioavailability.12 A pharmacokinetic study showed that in order to reach a concentration of 10 mM, the loading dose of NAC must be 5010 mg/kg. Such a high dose is not conducive to intravenous administration.13 Therefore, there is an urgent need for improving the bioavailability of NAC.
Various approaches have been explored to meet that need. Research showed that the esterification of its carboxyl group to produce N-acetylcysteine ethyl ester (NACET) increases its lipophilicity. However, studies found that only low concentrations can be achieved in plasma when it is administered orally.14 Later, another derivative, N-acetylcysteine amide (NACA), was synthesized to improve its lipophilicity, membrane permeability, and antioxidant properties. These properties were improved after the amidation of NAC,15 while achieving a high NAC plasma concentration remains a challenge. Conjugation of NAC to nanoparticles can increase the bioavailability and stability of NAC by increasing the intracellular level of NAC and by decreasing their interaction with serum proteins. A more recent study showed that dendrimer-NAC conjugates were able to inhibit the nitrite production on LPS activated microglia16 and had much higher anti-oxidant and anti-inflammatory effect when compared to free NAC in vitro.17
Glutathione (GSH) concentration is much higher in the cytoplasm (1–10 mM) than that in the plasma (2 µM). The use of disulfide bond for GSH mediated intracellular release has already been studied18 and proved to be very effective in reducing off-targeting effect.19 In our previous studies, we have reported the fabrication of nanoparticles using Poly[(2-(pyridin-2-yldisulfanyl)-co-[poly(ethylene glycol)]] (PDA-PEG), which is sensitive to both high redox potential and acidic pH.20 Due to the aboudance of pyridine-2-thiol groups in the PDA-PEG polymer and its amphiphilic property, we postulate that PDA-PEG could be a better carrier for NAC than the existing dendrimer-NAC system. For the present study, we developed a drug delivery system based on a NAC conjugated PDA-PEG polymer through disulfide bonds. Since disulfide bonds can only be cleaved by elevated GSH level, which ensure the stability of NAC nanoparticles in the plasma while promptly releasing NAC intracellularly.
2. Materials and methods
2.1. Materials
N-acetyl cysteine (NAC), Lipopolysaccharides from Escherichia coli (LPS), 2-mercaptoethenol, DL-dithiothreitol (DTT), tris(2-carboxyethyl)phosphine (TCEP), 2, 2-Azobisisobutyronitrile (AIBN), Poly(ethylene glycol)methacrylate (Mn = 360 Da) and Thiazolyl Blue Tetrazolium Bromide (MTT) were purchased from Sigma–Aldrich. Aldrithiol-2 and Silica gel (Spherical, 100 µm) were purchased from Tokyo Chemical Industry Co. Interleukin-1 beta ELISA kit, Iba-1 antibody and Alexa Fluor 568 to Rabbit IgG secondary Antibody were purchased from abcam, Nitrate/nitrite colorimetric assay kit was purchased from Cayman chemical and TNF alpha ELISA kit was purchased from eBioscience. Cell tracker dyes were purchased from Thermo Fisher Scientific. Penicillin (10,000 U/mL), streptomycin (10,000 µg/mL), 0.25% trypsin-EDTA, Dulbecco's Modified Eagle Medium (with L-glutamine), DMEM/F12 (1:1) (with L-glutamine), MEM-Non–essential amino acids 100X, sodium pyruvate 100 mM and fetal bovine serum (FBS) were obtained from Gibco.
2.2. NAC-PDA-PEG polymer synthesis
PDA-PEG polymer was synthesized by free radical polymerization as described previously.20 For NAC-polymer preparation, PDA-PEG polymer (20 mg) was dissolved in 1 ml DMSO and 678 µg in 67.8 µl of NAC were added. The reaction was stirred overnight at room temperature. Then the mixture was dialyzed against DMSO using dialysis tube MWCO 1 kDa. The concentration of NAC in the polymer after dialysis was quantified using HPLC. HPLC was carried out with Waters model 2695 attached to a Waters 2996 photodiode array detector and column (C18, dimensions of 25 cm × 4.6 mm) using acetonitrile-water (both 0.14% TFA by weight) as the mobile phase. Gradient method used for analysis was (100:0) Water: Acetonitrile to (60:40) Water-Acetonitrile in 25 min followed by returning to initial conditions in 5 min. The flow rate was 1 mL/min. The eluted samples were detected at 210 nm.
2.3. Nanoparticle fabrication
Nanoparticles were prepared by crosslinking reaction of NAC-polymer via disulfide bonds cleavage followed by aerial oxidation. TCEP was used in the fabrication of NPs to generate free thiol groups in the polymer. An amount of TCEP equal to 10% of total pyridine thione was added and then the mixture was dropped into ddH2O under stirring to form crosslinking through disulfide bonds by aerial oxidation. Briefly, 298 µg TCEP in 20 µl DMSO were added in 0.5 ml of 20 mg/ml polymer solution in DMSO and vortexed vigorously. Then this mixture was immediately added in 5 ml ddH2O under stirring conditions for 4 hours at room temperature. The final solution was loaded into a dialysis bag (MWCO: 1000 Da) and dialyzed against PBS 7.4 for 24 hours. The control NPs were fabricated following the same protocol except replacing NAC-conjugated polymer with PDA-PEG.
2.4. NAC conjugation and release
The yield of NAC conjugation to the polymer was estimated after cleavage of NAC in the presence of 200 mM DTT. The release of conjugated NAC was studied in PBS 10 mM pH 7.4 in the presence of 0.02 mM, 2 mM and 10 mM DTT for 4 hours, 24 hours and 48 hours. NAC conjugation to PDA-PEG polymer and NAC release in the presence of DTT were determined using HPLC as described above.
2.5. Stability of NAC-NP in serum containing medium
To investigate the stability of NAC-NP in serum, 1.5 mg/ml free NAC and NAC-NP were incubated in presence of 80% FCS for 24 hours under shaking at 37 °C. After that, acetonitrile: serum solution at a ratio 3:1 (v/v) was used for protein precipitation. The collected supernatant was collected, precessed with DTT to liberate NAC from NAC-NP, and analyzed using the NAC HPLC method described above.
2.6. Antioxidant effect of NAC-NP evaluated by DPPH scavenging activity
DPPH radical scavenging activities of NAC, NAC-NP or control NP were evaluated as described elsewhere with slight modifications.21 Three-milliliter of 2,2-diphenyl-1-picrylhydrazyl (DPPH) solution (0.1 mM) in methanol was added to 1 ml of various concentrations of NAC, NAC-NP, or control NP. The mixture was vortexed and allowed to stand in dark at room temperature for 1 hour and the absorbance was measured at 517 nm. The DPPH scavenging activity was calculated by the following equation:
Where, Asample is the absorbance of each sample in presence of DPPH, Ablank is the absorbance in the absence of DPPH reagent and AControl is the absorbance of the reaction mixture in the absence of any sample.
2.7. Cell culture
Immortalized mouse microglial cells BV-2 cells were provided by Dr. Grace Y. Sun (University of Missouri) (passage 10). DMEM cell media supplemented with 10% FBS and 1% penicillin-streptomycin was used to culture BV-2 cells. SH-SY5Y cells were obtained from ATCC (passage 23) and cultured in DMEM/F12 (1:1) media supplemented with 10% FBS and 1% penicillin-streptomycin, 1× Non Essential amino acids and 1mM Sodium Pyruvate. In most studies that LPS was used for activation of microglia cells, serum free media was adopted either because the presence of serum could affect the inflammation markers22 or the differentiation of microglia.23 Therefore, serum free medium was used in the experiments carried out below.
2.8. Cellular uptake of NAC-NPs
To monitor NAC-NPs entering BV-2 cells, fluorescence dye Cy3 was conjugated to the PDA-PEG polymer through a two-step modification following our reported method.24 Briefly, PDA-PEG was reacted with cysteamine first through thiol-disulfide exchange reaction and then coupled with Cy3-NHS to yield Cy3-PDA-PEG polymer. After that, Cy3 labeled NAC-NPs and PDA-PEG NPs were fabricated as described above. BV-2 cells were seeded in 35 mm glass bottom Petri dishes at the density of 105 cells/ml and incubated for 24 hours. The medium was removed and 100 µL of fresh serum free medium containing either NAC-NPs or PDA-PEG NPs at the Cy3 concentration of 2 nM were added to the cells for 3 hours. The medium was changed and cells were washed with cold PBS for 3 times. Cells were stained with 1 µg/ml Hoechst and then imaged with a Carl Zeiss LSM 700 confocal microscope.
2.9. The protective effect of NAC-NPs
BV-2 cells were seeded in 24-well plates at the density of 105 cells /ml and incubated for 24 hours. The medium was removed and 100 µL of fresh serum free medium was added. LPS (100 ng/mL) and 0.25 and 2.5mM of either free NAC or NAC-NPs were added to the cells for 3 hours. The medium was changed and 100 µL of fresh, serum free medium containing 100 ng/mL of LPS was added and incubated for an additional 72 hours. The morphology of the cells after treatment was observed under an optical microscope. Untreated BV-2 cells exhibit the typical ramified morphology of resting microglia but activated microglia have a greatly enlarged cell body. For cell viability assay, 100 µl of MTT (0.5 mg/ml) were added for 4 hours. The formazan crystals in each well were dissolved in dimethyl sulfoxide (DMSO), and the absorbance was measured at 590 nm using a microplate reader.
2.10. Immunocytochemistry
After treatment of BV-2 cells with 100 ng/ml LPS and 0.25 or 2.5 mM of either free NAC or NAC-NPs for 3 hours the medium was removed and replaced with 100 µL of fresh serum free medium, containing 100 ng/mL of LPS. The cells were incubated for another 24 hours. After treatment microglia cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% triton for 5 minutes. For blocking, 3% normal donkey serum with 1% bovine serum albumin in PBS was used for 30 minutes. Then the cells were incubated with lba-1 antibody diluted 1:250 in 1% BSA in PBS for 2 hours at room temperature. After 2 washes with PBS and another step of blocking with 3% BSA in PBS for 30 minutes, the cells were incubated with a secondary Alexa Fluor 568 conjugated anti-rabbit IgG for 1 hour at room temperature. Then the nucleus was stained with 1 µg/ml Hoechst for 5 min. The staining of Iba-1 was visualized by fluorescence microscopy (Carl Zeiss LSM 700 confocal microscope).
2.11. NO production
BV-2 cells were seeded in 96 well plates at the density of 30,000 cells/well and incubated for 24 hours. The medium was removed and 100 µL of fresh serum free medium was added. LPS (100 ng/mL) and 0.25 and 2.5mM of free NAC or NAC-NPs were added to the cells for 3 hours. The medium was changed and 100 µL of fresh, serum free medium containing 100 ng/mL of LPS was added. The cells were incubated for 72 hours, and the culture medium was removed for analysis. The formation of NO was measured in the culture media. The nitrite concentration was determined by using the Griess reagent system (Cayman) that uses sulfanilamide (Reagent 1) and N-(1-Naphthyl)-ethylene diamine (Reagent 2). The supernatants (100 µl) from BV-2 cells exposed to different treatments were incubated with 50 µl of Griess reagent 1 and 50 µl of Griess reagent 2 for 10 min at room temperature. The absorbance of the resulting solution at 540 nm was measured, and the nitrite concentration was determined by comparing it with a standard curve generated with nitrite standards of the Nitrite kit. The total Nitrite and Nitrate concentration was estimated using the same kit but in this case the nitrate reductase enzyme and cofactors were added to each well for 1 hour prior to the addition of Griess reagents.
2.12. ROS production
ROS formation was estimated for cells receiving the same treatment as described above using Carboxy-H2DCFDA as an indicator. This compound is non-fluorescent and accumulates within cells upon deacetylation. After that, dichlorodihydrofluorescein reacts with ROS to form fluorescent dichlorofluorescein (DCF). BV-2 cells exposed to different treatments were washed with PBS and then loaded with carboxy-H2DCFDA at a final concentration of 5 µM in a culture medium supplemented with 10% serum for 30 min. Cells treated with H2O2 at 20 and 50 mM were used as positive controls. Cellular fluorescence was monitored using a microplate reader and visualized by confocal microscopy at the excitation of 480 nm and emission of 530 nm.
2.13. Superoxide detection
Cells receiving the same treatment as described above were also tested for superoxide (O2-) production selectively using hydroethidine. Hydroethidine can be oxidized by superoxide but not by hydroxyl radical, singlet O2, H2O2, or nitrogen radicals. Hydroethidine exhibits blue-fluorescence in the cytosol until oxidized. Upon oxidized, it intercalates within the cell's DNA, staining the nucleus a bright red-fluorescence. LPS (100 ng/mL) and 0.25 and 2.5 mM of free NAC or NAC-NPs were added to the cells for 3 hours. The medium was replaced with a fresh, serum free medium containing 100 ng/mL of LPS and the cells were incubated for 24 hours. Then the Cells were treated with 20 µM hydroethidine for 30 minutes and fixed with 4% paraformaldehyde for 10 min. The formation of ethidium bromide by superoxide was visualized using confocal microscopy.
2.14. TNF-α release
TNFα secretion was measured using a mouse TNF alpha ELISA kit (eBioscience) according to the manufacturer's instructions. Capture antibody was added in each well of a 96-well ELISA plate and incubated overnight at 4 °C. The plate was washed with buffer and then incubated with the supernatant (100 µl) from BV-2 cells exposed to different treatments for 2 hours at room temperature followed by 5 washes. Biotin-conjugated antibody was applied to each well and incubated for 30 min at room temperature followed by 5 washes. Diluted avidin-HRP was added, and the plate was incubated at room temperature for 30 min. After washing the plate, substrate solution was added to each well and the color was developed in the dark for 5 minutes at room temperature. The stop solution was added to each well, and the plate was read at 450 nm using a microplate reader. The concentration of TNFα was calculated using a mouse TNF alpha standard.
2.15. IL-1β release
IL-1β secretion was measured using IL-1β mouse ELISA kit (abcam) according to the manufacturer's instructions. In brief, the plate was incubated with the supernatant (100 µl) from BV-2 cells exposed to different treatments for 2.5 hrs at room temperature followed by 4 washes. Biotinylated IL-1β detection antibody was added to each well and incubated for 1 hour at room temperature with gentle shaking followed by 4 washes. Diluted HRP-streptavidin was added, and the plate was incubated at room temperature for 45 min. After washing the plate, TMB substrate reagent was added to each well and the color was developed in the dark for 30 minutes at room temperature. The stop solution was added to each well, and the absorbance of each well was immediately read at 450 nm using a microplate reader. The concentration of IL-1β was calculated using mouse IL-1β standard.
2.16. Co-culture of BV2 with neurons
Human neuroblastoma cell line SH-SY5Y (ATCC CRL-2266) was cultured in DMEM/F12 (1:1) (with L-glutamine), supplemented with 0.1mM MEM-Non–essential amino acids, 1mM sodium pyruvate and 10% FBS and differentiated with gradual serum starvation. After that, BV2 cells were seeded into the wells where SH-SY5Y cells were grown. Both cell lines were stained using cell tracker dyes according to manufacturer’s protocol. LPS (100 ng/mL) and 0.25 and 2.5 mM of free NAC or NAC-NPs were added to the co-culture for 3 hours. The medium was changed and 100 µL of fresh, serum free medium containing 100 ng/mL of LPS was added. The cells were incubated for 72 hours before imaged by Carl Zeiss LSM 700 confocal microscope. The percentage of SH-SY5Y cells engulfed by BV2 cells was calculated by the following equation: Engulfed cell % = the number of SH-SY5Y cells overlapped with BV2 cells/total number of SH-SY5Y cells in each images. For each treatment, at least three images were analyzed.
2.17. Statistical Analysis
The results were reported as the mean ± standard deviation of three different studies. Student's two-tailed t-test assuming equal variance was used to determine statistical significance (p < 0.05) of the experimental data.
3. Results and Discussion
3.1. Preparation and characterization of NAC conjugated nanoparticles (NAC-NP)
The PDA-PEG polymer (Mw: 41.8 kDa, PDI 1.21) was prepared following our reported method.20 NAC was conjugated to the polymer through thiol-disulfide exchange reaction as shown in Figure 1A. The formed disulfide bond was utilized to ensure the stability of the prodrug while intracellularly releasing NAC after it enters cells. The replacement of pyridine-2-thiol of the polymer by NAC was first estimated by measuring the absorbance of cleaved pyridine-2-thiol at 375 nm. The conjugation efficiency of NAC onto the polymer was estimated using HPLC and found to be 95% (Figure S1). The conjugation of NAC to the polymer was also confirmed by 1H NMR as evidenced by the emerging peak at 4.81 ppm and the weaken of peaks in the range from 7 to 8.5 ppm (Figure S2).
Figure 1.

Preparation and characterization of NAC-NPs. Scheme for the synthesis of NAC-PDA-PEG polymer (A). TEM image (B) and size distribution of NAC-NP (C). The release kinetics of NAC from nanoparticle in presence of 0.02 mM, 2 mM and 10 mM DTT (D). The scale bar is 100 nm in (B). Data expressed as mean ± SD.
Dynamic light scattering (DLS) was used to investigate the physicochemical characteristics of the nanoparticles. The size of the NAC-NP was ~50 nm with a polydispersity index (PI) lower than 0.4, indicating a relatively narrow size distribution (Figure 1C). The nanoparticles were also visualized by TEM, which further confirmed the small size, narrow size distribution, and spherical shape of the nanoparticles (Figure 1B).
3.2. Release kinetics of NAC-NPs
The release of NAC from NAC-NPs was evaluated in releasing media supplemented with 0.02, 2, and 10 mM of DTT to mimic different physiological environments, plasma and cytosol. Figure 1D showed that NAC-NPs at 10 mM DTT concentration were able to release more than 20% of NAC within 4 hours and almost 80% of conjugated NAC within 24 hours. To the contrary, NAC-NPs released less than 10% within 24 hours at 0.02 mM DTT condition, the indicating that NAC-NPs are stable during blood circulation while promptly releasing NAC intracellularly.
3.3. Stability of NAC-NP in serum containing medium
To investigate the stability of free NAC and NAC-NP in serum, HPLC method was employed. After 24 hours of incubation, the quickly diminished NAC peak (only 18.3% retained) in Figure 2B suggests that free NAC is not stable during circulation in the blood stream. In contrast, for NAC-NP, more than 76.4% of NAC was retained after a same period of incubation (Figure 2D), which proved that NAC-NPs are much more stable than free NAC in a serum containing environment.
Figure 2.

HPLC spectra of free NAC and NAC-NP before and after incubating with serum containing medium. The red dashed circles indicate the location of NAC peak.
3.4. NAC-NP antioxidant activity
In order to evaluate the in-vitro free radical scavenging activity of NAC, NAC-NPs and control-NPs, a modified DPPH assay was used.21 A dose-dependent scavenging activity was observed for free NAC, NAC-NPs, and control NPs in Figure 3, while the scavenging activity of free NAC was higher than that of NAC-NP when the reaction mixture was measured after 1 h. After 24 hours of incubation, the activity difference between free NAC and NAC-NPs was significanty decreased, indicating that the reaction is time dependent. Since the released NAC from NAC-NP is negligible in non-reducing environment within 24 hours, we postulate this time dependent antioxidant activity was caused by the the steric hindrance effect in the nanoparticle, due to which the diffusion of the reactant was the rate limiting step for the reaction. In addition, Figure 3 also revealed that the antioxidant effect of conjugated NAC was not affected a lot by its conjugation to the polymer through the disulfide bond and the release of NAC was not nessasary for its functionality as a radical scavenger. It is worth noting that control NPs also have certain level of antioxidant activity.
Figure 3.

The DPPH scavenging activity of NAC, NAC-NP, and control NP. The measurements were carried out 1 hour (A) and 24 hours (B) after the co-incubation.
3.5. Cellular uptake of NAC-NPs
To observe the cellular uptake of NAC-NPs, nanoparticles were labeled with Cy3 fluorescence dye following our reported method.24 The red fluorescence signals in Figure 4 proved that both PDA-PEG NPs and NAC-NPs can effectively enter BV-2 cells. There was no difference between NAC-NPs and PDA-PEG NPs in entering BV-2 cells (p=0.12, not statistically significant), suggesting that the substitution of pyridine-2-thiol with NAC did not significantly slow down the cellular uptake process.
Figure 4.

Confocal images of BV-2 cells after co-incubated with NAC-NPs. Cells were treated with control NPs and NAC-NPs at the Cy3 concentration of 2 nM for 3 hours and then imaged with a Carl Zeiss LSM 700 confocal microscope.
3.6. The protective effect of NAC-NPs for LPS stimulated microglia
To study the protective effect of NAC-NPs on LPS stimulated microglia, the morphological changes and viability of BV-2 cells after receiving LPS and NAC treatment was investigated. Although microglial cells in vitro usually do not have the ramified structure, which is typical in the CNS, the morphological changes can still be observed after treatment with LPS.25
As shown in Figure 5A, non-treated BV-2 cells were mostly round with bright refringency, which is the typical ramified morphology of resting microglia. Whereas, LPS treatment activates cells, with short thick processes and with an enlarged cell body which are characteristics of activated microglia. However, pretreatment with NAC-NP not only reversed the effect of LPS on the morphology of microglial cells, but also increased significantly the survival rate of the cells in presence of LPS (Figure 5B). The viability of BV-2 cells treated with LPS only was less than 30%, but for the cells pretreated with 0.25 mM and 2.5 mM of NAC-NP the viability was significantly increased to 57% and 69%, respectively. Pretreatment with free NAC increased the cell viability to 53%, which only happened at the high dose of NAC.
Figure 5.

Protective effect of free NAC and NAC-NPs on LPS-activated microglia. LPS-induced morphological changes (A) and cell viability in BV-2 cells (B), as well as the expression of microgliosis marker lba-1 (C), and the quantitative Iba-1 (Cy3) intensity as compared with its corresponding nuclear signal (D). BV-2 cells were co-treated with LPS (100 ng/ml) and NAC for 3 hours and followed by additional 72 hours of LPS treatment. Data expressed as mean ± SD (* P < 0.01).
3.7. Activation of microglia
Iba-1 protein is upregulated upon the activation of microglia due to inflammation, which is frequently used to discriminate between surveilling and activated microglia. After the stimulation of LPS, strong red fluorescence signals appeared in the cells (Figure 5C), suggesting the activation of microglia. As expected, both free NAC and NAC-NPs decreased the intensity of Iba1 marker. Whereas, for cells treated with NAC-NPs, the intensities of lba-1 marker are close to those of non-activated cells, indicating the strong suppressive effect for microglia activation.
3.8. Antioxidant effect of NAC-NPs
The antioxidant effect of NAC-NPs was first studied by estimating the release of Nitrite (NO) and the production of ROS in microglial cells that have been activated by LPS. BV-2 cells were pretreated with either free NAC or NAC-NPs for 3 hours in the presence of 100 ng/ml LPS and then for further cultured for 72 hours in the presence of LPS only.
LPS induced the production of nitrite by BV-2 microglial cells and the release of NO to the culture media reached at a concentration of 15 µM after 72 hours (Figure 6A). Pre-treatment of cells with 0.25 mM or 2 mM of free NAC significantly reduced nitrite production almost by 22 and 50 % of that of LPS, respectively; while pre-treatment of cells with 0.25 mM and 2 mM NAC-NP decreased the release of nitrite by 59 and 77% of that of LPS, respectively. For cells pretreated with control-NP (without NAC) and LPS, NO release was decreased in a dose-dependent way. This indicates that the polymer itself may have an anti-inflammatory activity. Although the exact mechanism of the anti-inflammatory activity of pyridinethione remains unclear, the potential anti-inflammatory activity of pyridine derivatives has already been reported.26 Krejčová et. al studied the anti-inflammatory and neurological activities of pyridine thione and four related sulfur-containing pyridine N-oxides, which are prominent constituents of plant Allium stipitatum, and found that 2-[(Methylthio)methyldithio]pyridine N-oxide had very high anti-inflammatory effects. Measurements of nitrite and nitrate levels (Figure 6A and B) showed that the ratio of nitrite to nitrate was not significantly affected by treatment with NAC-NPs (Figure 6C), ruling out that the reduction of nitrite release in NAC-NPs treated cells was not due to increased conversion to nitrate.
Figure 6.

Effect of NAC and NAC-NPs on the release of NO by LPS-activated microglia. Release of nitrite (A), nitrate plus nitrite (B), and nitrate/nitrite ratio (C). BV-2 cells were co-treated with LPS (100 ng/ml) and/or 0.25 or 2 mM free NAC or NAC-NPs for 3 hours first followed by additional 72 hours of LPS treatment. The concentrations of the control-NP is equivalent to NAC-NPs providing 0.25 or 2 mM NAC. Nitrite and nitrate production was estimated in culture media using Griess reagent. Data expressed as mean ± SD (* P < 0.01).
3.9. The effect of NAC-NPs on ROS production
ROS production and release also plays an important role in neuroinflammation, oxidative stress, and the pathogenesis of neurodegenerative diseases. We examined the effect of NAC-NPs on the production of ROS induced by LPS in immortalized microglial cells. Microglia cells treated as described above were tested for the production of ROS using two different methods. DCFH-DA reagent was used for detection of total ROS including superoxide radical (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (.OH), while dihydroethidium was used as a superoxide indicator. Superoxide is the most well-known of ROS as it is mainly produced during the natural pathway of oxidative phosphorylation.27
As others reported, LPS induced the production of ROS in BV-2 cells (Figure 7A). The addition of NAC could decrease intracellular ROS level induced by LPS. To our surprise, NAC-NPs also exhibited much stronger ROS attenuating effect than free NAC, which could be due to the better cell membrane penetration ability of the NAC-NP. NAC-NP achieved 100% inhibition for LPS induced ROS at the concentration of 0.25 mM, suggesting NAC-NP has stronger potency than a reported NAC-dendrimer conjugate.17 The quenched green fluoresence in NAC-NP treated cells also proved its high efficacy (Figure 7B).
Figure 7.

Effect of NAC and NAC-NPs on the production of ROS by LPS-activated microglia. ROS generation was measured by DCFH-DA fluorescence intensity (A) and visualized by confocal microscopy (B). The nuclei were stained with DAPI (blue) and the ROS was detected by DCFH-DA (green). The production of superoxide was detected by hydroethidine and visualized by confocal microscopy as indicated by the converting blue fluorescent signal to red (C). BV-2 cells were pre-treated with 2 mM free NAC or NAC-NPs in the presence of 100 ng/ml LPS for 3 hours followed by additional 24 hours of LPS (100 ng/ml) treatment. Data expressed as mean ± SD (* P < 0.01).
LPS can trigger the production of superoxide inside microglia cells. Hydroethidine is a superoxide indicator, which remains in the cytosol and exhibits blue fluorescence. However, once it is oxidized, it enters nucleus and intercalates with DNA, staining the nucleus with a bright red fluorescent signal. Cells treated as describe above were incubated with hydroethidine to study their superoxide production. As shown in Figure 7C, cells treated with LPS exhibited a very bright red signal in their nuclei and faint blue signal in their cytoplasma, indicating the production of superoxide anions. The decreased red nuclear signal in LPS-activated cells pretreated with free NAC and NAC-NPs proved that both treatments can abolish the intracellular superoxide level.
3.10. The anti-inflammatory effect of NAC-NPs
LPS not only triggers the production and release of NO and ROS for microglia, it also results in the secretion of cytokines such as TNFα and IL-1β. Therefore, the levels of these pro-inflammatory indicators were measured to investigate the anti-inflammatory effect of free NAC and NAC-NPs.
As shown in Figure 8A, LPS induced the release of TNF-α by BV-2 cells and reached at a concentration of 1.2 ng/ml after 72 hours of exposure. Pre-treatment of cells with 2.5 mM of free NAC reduced TNF-α release to 50 % of that of LPS treatment. Interestingly, pre-treatment of cells with 0.25 mM NAC-NPs decreased the release of NAC to 10% of that of LPS treatment.
Figure 8.

Effect of NAC and NAC-NPs on cytokines release by LPS-activated microglia. TNF-α (A) and IL-1β (B) release from BV-2 cells that were pre-treated with 0.25 mM and 2 mM free NAC or NAC-NP for 3 hours in the presence of 100 ng/ml LPS followed by additional 72 hours of LPS (100 ng/ml) treatment. Data expressed as mean ± SD (* P < 0.01).
Figure 8B showed that LPS-stimulated microglia released IL-1β reached at a concentration of 750 pg/ml after 72 hours of exposure. The IL-1β release was decreased to 477 pg/ml for cells pretreated with 2.5 mM of free NAC. As expected, pre-treatment of cells with 0.25 mM NAC-NPs decreased the IL-1β release to 268 pg/ml, suggesting that NAC-NP has a much stronger anti-inflammatory effect than free NAC.
3.11. The neuroprotective effect of NAC-NPs
We have studied the effects of free NAC and NAC-NP on microglia upon LPS stimulation by quantifying the production and release of ROS and NO and release of cytokines TNF-α and IL-1β, and revealed that NAC-NPs has anti-oxidant and anti-inflammatory effects. In addition, NAC-NPs showed a better protective effect for microglia upon the co-culture with LPS as evidenced by higher microglia viability. Recent studies indicate that activated microglia “eat” viable brain neurons referred as ‘phagoptosis’.28,29 To study the neuroprotective effect of NAC-NPs we used a co-culture system treated with LPS as a tool for neuroinflammation.30
Activation of BV-2 cells with LPS increases phagocytosis of neurons.2, 29, 31 Furthermore, research found that the suppression of microglia is neuroprotective.32 In order to investigate the neuroprotective effect of NAC-NPs, we used a co-culture of BV2 cells with a neuroblastoma cell line, SHSY5Y. It was found that LPS induced the total phagocytosis of SHSY5Y cells by BV2 cells (Figure 9), which is evidenced by nearly 100% overlap of red and green fluorescence signals. When the cells were co-treated with free NAC and LPS, there were less red signals overlapped with the green ones, indicating that fewer SHSY5Y cells were engulfed by BV-2 cells. To our surprise, only a few all red signals were separated from green ones in the cells treated with both NAC-NPs and LPS, suggesting that fewer phagocytosis of SHSY5Y cells in this cell co-culture system. Furthermore, the spread structure of the NAC-NPs treated SHSY5Y cells indicates the healthiness of the cells. The activated phagocytic microglia is the maximum immune responsive form. After engulfing a certain amount of material, the phagocytic microglia becomes unable to phagocytose any further materials. Consequently, the final form of microglia after phagocytosis is known as a granular corpuscle due to its “grainy” appearance.33 Based on the evidences collected above, we can conclude that NAC-NPs inhibit the activation of microglia due to their anti-oxidant and anti-inflammatory effects, and subsequently protect the viability of neurons and microglia upon the challenge of LPS.
Figure 9.

The neuroprotective effect of NAC-NPs under a co-culture model. Confocal images of BV-2 cells (green) with SHSY5Y cells (red) receiving different treatments (A) and the percentage of engulfed SHSY5Y cells under co-culture (B). LPS-induced microglia phagocytosis of SHSY5Y cells visualized by confocal microscopy after 24 hours of co-culture of BV-2 cells with SHSY5Y cells. BV-2 cells were treated with either free NAC or NAC-NPs at the NAC equivalent concentration of 2.5 mM in the presence of LPS 100 ng/ml for 24 hours. Arrows indicate the overlapping of red and green signals. Data expressed as mean ± SD (* P < 0.05).
4. Discussion
NAC is an promising drug for the treatment of many neurological diseases owing to its excellent anti-oxidant and anti-inflammatory effects. However, due to its hydrophobicity and free sulfhydryl group, NAC shows a low bioavailability when administrated orally.11 Since NAC can react with serum albumin through the formation of disulfide bonds, the administration of NAC by an alternative intravenous route requires extreme high loading dose to achieve a therapeutical concentration (10 mM), which makes the application of NAC for the treatment of neurological diseases impractical.12 The aim of this research is to develop a NAC delivery system for enhanced anti-oxidant and anti-inflamatory efficacy by limiting the formation of the disulfide bond between NAC and human serum albumin through an intracellularly releasable NAC-NP. Although the intracellular release of NAC requires the consumption of GSH and yields oxidized GSH, GSSG, the total GSH (GSSG/GSH) amount is unchanged during the releasing process. Since NAC is a precurcor for the synthesis of GSH, the intracellular delivery of NAC will result in the increase of total GSH (GSSG/GSH). In addition, Figure 2 revealed that, to certain extent, the anti-oxidant effect NAC-NP is not dependent on the consumption of GSH to release free NAC. Furthermore, the PDA in the control NP also displays anti-oxidant effect. Altogether, the enhanced anti-oxidant effect of NAC-NP is the combination effects of the increased total GSH (GSSG/GSH) level and the anti-oxidant effect of PDA-PEG.
Because of the above mentioned benefits of the PDA-PEG, NAC-NP exhibits a much stronger inhibitory effect on the activation of microglia upon the LPS stimulation and subsequently protects the neuron cells. Since chronic neural inflammation is the main cause of Alzheimer’s disease, while microglia activation is crucial for the disease development, a system could effectively inhibit the activation of microglia and protect the neuron from apoptosis will shed light on the control and cure of Alzheimer’s disease. Although NAC-NP exhibits excellent anti-oxidant and anti-inflammatory effects for microglia, as well as protective effect for neurons in vitro, how to direct the NAC-NP across the blood brain barrier remains a challenge. One potential solution for that is to incorporate brain targeting ligands, such as TAT peptide and transferrin,34–35 to the NAC-NP system, which is an ongoing research for the application of NAC-NP in our group.
5. Conclusions
In summary, we developed a redox potential sensitive N-acetyl cysteine-prodrug nanoparticles for releasing NAC intracellularly. The antioxidant and anti-inflammatory effects of these nanoparticles were tested in vitro using LPS-activated microglia cells. Our studies revealed that NAC-NPs can more effectively decrease the LPS-induced ROS and NO release from microglia than free NAC. Furthermore, NAC-NPs treatment significantly attenuated the release of pro-inflammatory cytokines, TNF-α and IL-1β, in LPS-stimulated microglia. The pretreatment of NAC-NPs reversed the neuronal phagocytosis and death mediated by LPS-activated microglia in the co-culture of microglia and neurons. Due to the strong anti-oxidant and anti-inflammatory effects as well as neuroprotective properties, NAC-NPs could be a very promising tool in fighting against central nervous system diseases, such as Alzheimer’s disease.
Acknowledgments
The authors want to thank the ASPIRE award from the Office of the Vice President for Research of The University of South Carolina and National Institutes of Health (1R01AG054839-01A1, 1R15CA188847-01A1, and5P20GM109091-02) for financial support of the research.
Footnotes
ASSOCIATED CONTENT
Supporting Information. The HPLC and 1H NMR spectra of the NAC-PDA-PEG polymer. This material is available free of charge via the Internet at http://pubs.acs.org.”
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interests.
References
- 1.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19(8):312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 2.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol. Rev. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 3.Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987;223(2):284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- 4.Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur. J. Neurosci. 2001;13(3):529–538. doi: 10.1046/j.1460-9568.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- 5.Ransohoff RM. Microgliosis: the questions shape the answers. Nat. Neurosci. 2007;10(12):1507–1509. doi: 10.1038/nn1207-1507. [DOI] [PubMed] [Google Scholar]
- 6.Block ML, Hong J. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog. Neurobiol. 2005;76(2):77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Shahripour RB, Harrigan MR, Alexandrov AV. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;4(2):108–122. doi: 10.1002/brb3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dringen R. Hirrlinger, Glutathione pathways in the brain. J. Biol. Chem. 2003;384(4):505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 9.Remington R, Chan A, Paskavitz J, Shea TB. Efficacy of a vitamin/nutriceutical formulation for moderate-stage to later-stage Alzheimer's disease: a placebo-controlled pilot study. Am. J. Alzheimers Dis. Other. Demen. 2009;24(1):27–33. doi: 10.1177/1533317508325094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmay MJ, Terpstra M, Coles LD, Mishra U, Ahlskog M, Öz G, Cloyd JC, Tuite P. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. J. Clin. Neuropharmacol. 2013;36(4):103–6. doi: 10.1097/WNF.0b013e31829ae713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borgström L, Kågedal B, Paulsen O. Pharmacokinetics of N-acetylcysteine in man. Eur. J. Clin. 1986;31(2):217–22. doi: 10.1007/BF00606662. [DOI] [PubMed] [Google Scholar]
- 12.Harada D, Anraku M, Fukuda H, Naito S, Harada K, Suenaga A, Otagiri M. Kinetic studies of covalent binding between N-acetyl-L-cysteine and human serum albumin through a mixed-disulfide using an N-methylpyridinium polymer-based column. Drug Metab. Pharmacokinet. 2004;19(4):297–302. doi: 10.2133/dmpk.19.297. [DOI] [PubMed] [Google Scholar]
- 13.Hong SY, Gil HW, Yang JO, Lee EY, Kim HK, Kim SH, Chung YH, Lee EM, Hwang SK. Effect of high-dose intravenous N-acetylcysteine on the concentration of plasma sulfur-containing amino acids. Korean J. Intern. Med. 2005;20(3):217–223. doi: 10.3904/kjim.2005.20.3.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giustarini D, Milzani A, Dalle-Donne I, Tsikas D, Rossi R. N-Acetylcysteine ethyl ester (NACET): a novel lipophilic cell-permeable cysteine derivative with an unusual pharmacokinetic feature and remarkable antioxidant potential. Biochem. Pharmacol. 2012;84(11):1522–33. doi: 10.1016/j.bcp.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Grinberg L, Fibach E, Amer J, Atlas D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med. 2005;38(1):136–45. doi: 10.1016/j.freeradbiomed.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 16.Navath RS, Kurtoglu YE, Wang B, Kannan S, Romero R, Kannan RM. Dendrimer-drug conjugates for tailored intracellular drug release based on glutathione levels. Bioconjug Chem. 2008;19(12):2446–2455. doi: 10.1021/bc800342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Navath RS, Romero R, Kannan S, Kannan R. Anti-inflammatory and anti-oxidant activity of anionic dendrimer-N-acetyl cysteine conjugates in activated microglial cells. Int. J. Pharm. 2009;377(1–2):159–168. doi: 10.1016/j.ijpharm.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giri S, Trewyn BG, Stellmaker MP, Lin VS. Stimuli-responsive controlled-release delivery system based on mesoporous silica nanorods capped with magnetic nanoparticles. Angew. Chem. Int. Ed. Engl. 2005;44(32):5038–5044. doi: 10.1002/anie.200501819. [DOI] [PubMed] [Google Scholar]
- 19.Wang YC, Wang F, Sun TM, Wang J. Redox-responsive nanoparticles from the single disulfide bond-bridged block copolymer as drug carriers for overcoming multidrug resistance in cancer cells. Bioconj. Chem. 2011;22(10):1939–1945. doi: 10.1021/bc200139n. [DOI] [PubMed] [Google Scholar]
- 20.Bahadur KCR, Xu P. Multicompartment intracellular self-expanding nanogel for targeted delivery of drug cocktail. Adv. Mater. 2012;24(48):6479–6483. doi: 10.1002/adma.201202687. [DOI] [PubMed] [Google Scholar]
- 21.Ates B, Abraham L, Ercal N. Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC) Free Radic Res. 2008;42(4):372–7. doi: 10.1080/10715760801998638. [DOI] [PubMed] [Google Scholar]
- 22.Mochida S, Matsura T, Yamashita A, Horie S, Ohata S, Kusumoto C, Nishida T, Minami Y, Inagaki Y, Ishibe Y, Nakada J, Ohta Y, Yamada K. Geranylgeranylacetone ameliorates inflammatory response to lipopolysaccharide (LPS) in murine macrophages: Inhibition of LPS binding to the cell surface. J. Clin. Biochem. Nutr. 2007;41:115–123. doi: 10.3164/jcbn.2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harry GJ, Kraft AD. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology. 2012;33(2):191–20. doi: 10.1016/j.neuro.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He H, Altomare D, Ozer U, Xu H, Creek K, Chen H, Xu P. Cancer cell-selective killing polymer/copper combination. Biomater. Sci. 2016;4(1):115–20. doi: 10.1039/c5bm00325c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.abd-el-Basset E, Fedoroff S. Effect of bacterial wall lipopolysaccharide (LPS) on morphology, motility, and cytoskeletal organization of microglia in cultures. J. Neurosci. Res. 1995;41(2):222–37. doi: 10.1002/jnr.490410210. [DOI] [PubMed] [Google Scholar]
- 26.Krejčová P, Kučerová P, Stafford GI, Jäger AK, Kubec R. Antiinflammatory and neurological activity of pyrithione and related sulfur-containing pyridine N-oxides from Persian shallot (Allium stipitatum) J. Ethnopharmacol. 2014;154(1):176–82. doi: 10.1016/j.jep.2014.03.066. [DOI] [PubMed] [Google Scholar]
- 27.Kerksick C, Willoughb D. The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2005;2:38–44. doi: 10.1186/1550-2783-2-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown GC, Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol. Neurobiol. 2010;41(2–3):242–247. doi: 10.1007/s12035-010-8105-9. [DOI] [PubMed] [Google Scholar]
- 29.Neher JJ, Neniskyte U, Brown GC. Primary phagocytosis of neurons by inflamed microglia: potential roles in neurodegeneration. Front Pharmacol. 2012;3:27. doi: 10.3389/fphar.2012.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gresa-Arribas N, Viéitez C, Dentesano G, Serratosa J, Saura J, Solà C. Modelling neuroinflammation in vitro: a tool to test the potential neuroprotective effect of anti-inflammatory agents. PLoS One. 2012;7:e45227. doi: 10.1371/journal.pone.0045227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: 'phagoptosis'. Trends Biochem. Sci. 2012;37(8):325–32. doi: 10.1016/j.tibs.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Peng B, Xiao J, Wang K, So KF, Tipoe GL, Lin B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J. Neurosci. 2014;34(24):8139–50. doi: 10.1523/JNEUROSCI.5200-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell GV. The compound granular corpuscle or gitter cell: a review, together with notes on the origin of this phagocyte. Tex. Rep. Biol. Med. 1962;20:338–51. [PubMed] [Google Scholar]
- 34.Morshed RA, Muroski ME, Dai Q, Wegscheid ML, Auffinger B, Yu D, Han Y, Zhang L, Wu M, Cheng Y, Lesniak MS. Cell-penetrating peptide-modified gold nanoparticles for the delivery of doxorubicin to brain metastatic breast cancer. Mol. Pharm. 2016;13(6):1843–54. doi: 10.1021/acs.molpharmaceut.6b00004. [DOI] [PubMed] [Google Scholar]
- 35.Zheng C, Ma C, Bai E, Yang K, Xu R. Transferrin and cell-penetrating peptide dual-functioned liposome for targeted drug delivery to glioma. Int. J. Clin. Exp. Med. 2015;8(2):1658–68. [PMC free article] [PubMed] [Google Scholar]