

SUMMARY
Degradation of Gram-positive bacterial cell wall peptidoglycan in macrophage and dendritic cell phagosomes leads to activation of the NLRP3 inflammasome, a cytosolic complex that regulates processing and secretion of interleukin (IL)-1β and IL-18. While many inflammatory responses to peptidoglycan are mediated by detection of its muramyl dipeptide component in the cytosol by NOD2, we report here that NLRP3 inflammasome activation is caused by release of N-acetylglucosamine that is detected in the cytosol by the glycolytic enzyme hexokinase. Inhibition of hexokinase by N-acetylglucosamine causes its dissociation from mitochondria outer membranes, and we found that this is sufficient to activate the NLRP3 inflammasome. In addition, we observed that glycolytic inhibitors and metabolic conditions affecting hexokinase function and localization induce inflammasome activation. While previous studies have demonstrated that signaling by pattern recognition receptors can regulate metabolic processes, this study shows that a metabolic enzyme can act as a pattern recognition receptor.
Graphical abstract
INTRODUCTION
Macrophages and dendritic cells play essential roles in initiating inflammation by releasing cytokines and chemokines in response to pathogen-associated molecular patterns (PAMPs) detected by innate immune receptors. Surface receptors, such as surface Toll-like receptors (TLRs) and C-type lectin receptors, detect extracellular PAMPs (Kumar et al., 2011). In addition, microbes internalized by phagocytes are enzymatically degraded, releasing small molecules that are screened for potential danger by a panel of intracellular innate immune receptors, such as intracellular TLRs and Nod-like receptors. We and others have found that degradation of Staphylococcus aureus in phagosomes is a key factor in determining the types and amounts of inflammatory cytokines produced following phagocytosis (Ip et al., 2010; Wolf et al., 2011; Müller et al., 2015). In particular, we noted that production of interleukin (IL)-1β and IL-18 required the degradation of S. aureus cell wall peptidoglycan (PGN) and that this response is suppressed when the organism modifies its PGN to become resistant to degradation (Shimada et al., 2010).
IL-1β and IL-18 play essential roles in controlling bacterial infections, in part, by recruiting neutrophils to sites of infection and polarizing T cell responses. Unlike many other cytokines, IL-1β and IL-18 are transcribed as pro-cytokines in the cytosol. Signaling to multiprotein complexes known as inflammasomes activates caspase-1 to process and secrete the cytokines (Lamkanfi and Dixit, 2014; Martinon et al., 2004). While there are several varieties of inflammasomes, the one responsible for responding to PGN is defined by the presence of NOD-like receptor family, pyrin domain-containing 3 (NLRP3). The mechanism by which NLRP3 is activated by PGN is not known. It is generally thought that all of the immunomodulatory activity of the S. aureus PGN comes from the degradative release of muramyl dipeptide (MDP), which is detected by cytosolic NOD2 receptor. However, we observed that NLRP3 inflammasome activation in response to S. aureus PGN was not affected by the loss of NOD2 (Shimada et al., 2010). Thus, the fragment of PGN that must be generated through degradation to activate the inflammasome and how it is sensed have not been established.
Diverse particulate stimuli that activate the NLRP3 inflammasome have been identified, including crystals such as silica, alum, asbestos, uric acid, and cholesterol (Dostert et al., 2008; Duewell et al., 2010; Hornung et al., 2008; Martinon et al., 2006). Like PGN particles, phagocytosis of these crystals is a necessary step in the process leading to inflammasome activation. For crystalline particles, which are non-degradable and non-microbial, it has been suggested that disruption of the phagosomal compartment leads to NLRP3 inflammasome activation (Hornung et al., 2008). However, we have previously observed that phagosomes containing PGN remain intact, and this, together with the observation that lysosomal degradation is necessary, suggests the existence of an alternative mechanism for specifically sensing PGN degradation products.
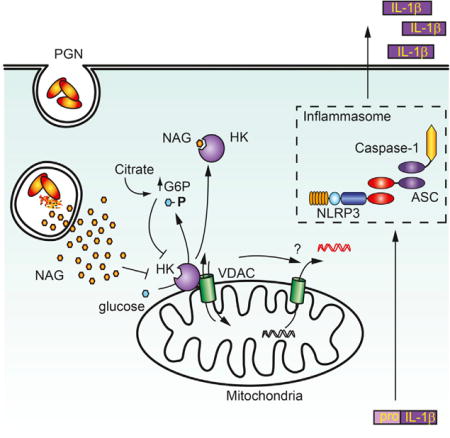
In this study, we have identified N-acetylglucosamine (NAG), a sugar subunit of the backbone of PGN, as an activator of the NLRP3 inflammasome. Anthrax bacteria specifically de-acetylate NAG in PGN, and we show that this PGN becomes a poor activator of IL-1β secretion in vitro and in vivo. Mechanistically, we observed that purified NAG and NAG released upon degradation of PGN in phagosomes are detected via inhibition of the glycolytic enzyme hexokinase, resulting in its dissociation from the mitochondrial outer membrane. Using a peptide that competes with hexokinase for binding to mitochondria and induces its dissociation from the outer membrane, we observed that hexokinase dissociation alone is sufficient to induce NLRP3 inflammasome activation. These conclusions are further supported by the observation that specific metabolic perturbations that affect hexokinase function also induce inflammasome activation. Together, the data suggest a model in which hexokinase effectively acts as a pattern recognition receptor, alerting the cell to degradation of bacterial PGN in phagosomes and activating an inflammatory response via disruption of the glycolytic pathway and mitochondrial function.
RESULTS
PGN-Induced NLRP3 Inflammasome Activation Is Independent of Potassium Efflux and Pyroptosis
We and others have shown that phagocytosis of Gram-positive PGN by bone marrow-derived macrophages (BMDMs) stimulates secretion of IL-1β via the NLRP3 inflammasome (Figures 1A and 1B) (Martinon et al., 2004; Shimada et al., 2010), a process that, as we have previously shown, requires degradation (Shimada et al., 2010). The diminished IL-1β production by Nlrp3−/−BMDMs in response to PGN are not a consequence of differential phagocytosis or lysosomal enzyme activity, which are equivalent in wild-type and Nlrp3−/−BMDMs (Figures S1A–S1C). In the process of evaluating inflammasome activation in response to PGN, we observed some behaviors inconsistent with current models of mechanisms of activation. First, it has been suggested that efflux of cytosolic potassium is essential for NLRP3 inflammasome activation (Muñoz-Planillo et al., 2013). While we confirmed that IL-1β secretion triggered by ATP or nigericin in lipopolysaccharide (LPS)-primed macrophages is strongly inhibited by extracellular potassium, we found that PGN-induced IL-1β secretion is not affected by extracellular potassium (Figure 1C). We also observed no effect of extracellular potassium on IL-1β secretion in response to whole ΔoatA S. aureus (Figure 1D), a strain that makes a PGN that is highly sensitive to phagosomal degradation and that we have previously shown to be a strong activator of the NLRP3 inflammasome (Shimada et al., 2010). Second, unlike many inflammasome activators, PGN-induced caspase-1 activation does not result in pyroptosis, as measured by the release of lactate dehydrogenase (LDH) (Figure 1E), annexin V staining (Figure S1D), or propidium iodide uptake (Figure S1E). We only observe background levels of cell death over 3 days in macrophages stimulated with PGN (Figure 1E). Compared to classic NLRP3 activators like ATP and nigericin, PGN-induced inflammasome activation occurs over a longer time period; for example, in Figure 1A, PGN induces much less IL-1β over 6 hr than ATP triggers in 2 hr. Given these unique features of PGN-induced activation, we set out to determine how PGN is sensed by macrophages.
Figure 1. PGN Activates the NLRP3 Inflammasome Independent of Potassium Efflux and Cell Death.
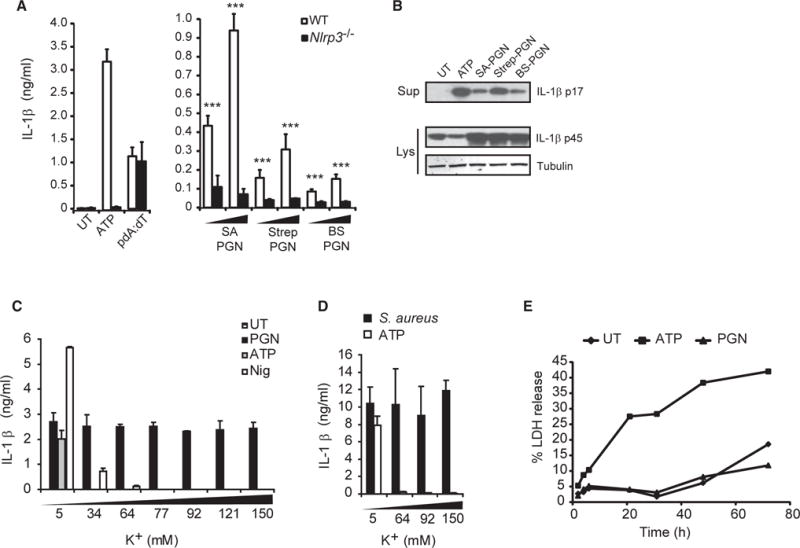
(A) LPS-primed BMDMs from wild-type and Nlrp3−/−mice were untreated (UT) or stimulated with 5 mM ATP for 2 hr or pdA:dT or PGN from ΔoatA S. aureus (SA), Streptomyces (Strep), or B. subtilis (BS) at 20–40 μg/ml, and IL-1β was assayed in the supernatant after 6 hr.
(B) Immunoblot of mature IL-1β in supernatants (Sup) or pro-IL-1β in cell lysates (Lys) of wild-type macrophages stimulated as in (A). (C and D) LPS-primed BMDMs were stimulated in the presence of increasing concentrations of extracellular KCl with (C) 20 μg/ml PGN 6 hr, 5 mM ATP 2 hr, 10 μg/ml nigericin (Nig) 2 hr, or (D), S. aureus (ΔoatA) 6 hr.
(E) LPS-primed BMDMs were stimulated with PGN (20 μg/ml) or ATP (5 mM), and release of lactate dehydrogenase (LDH) into supernatants was measured at indicated times and shown as percentage of maximum at each time point.
Error bars indicate SD. ***p < 0.001.
See also Figure S1.
NAG Is the Minimal Inflammasome-Activating Component of PGN
PGN, which makes up as much as 80% of the dry weight of typical Gram-positive bacteria, is a polysaccharide of repeating units of N-acetylmuramic acid (NAM; MurNAc) and NAG (GlcNAc) cross-linked by short amino acid side chains (Figure 2A).
Figure 2. NAG Is the NLRP3 Inflammasome-Activating Component of PGN.
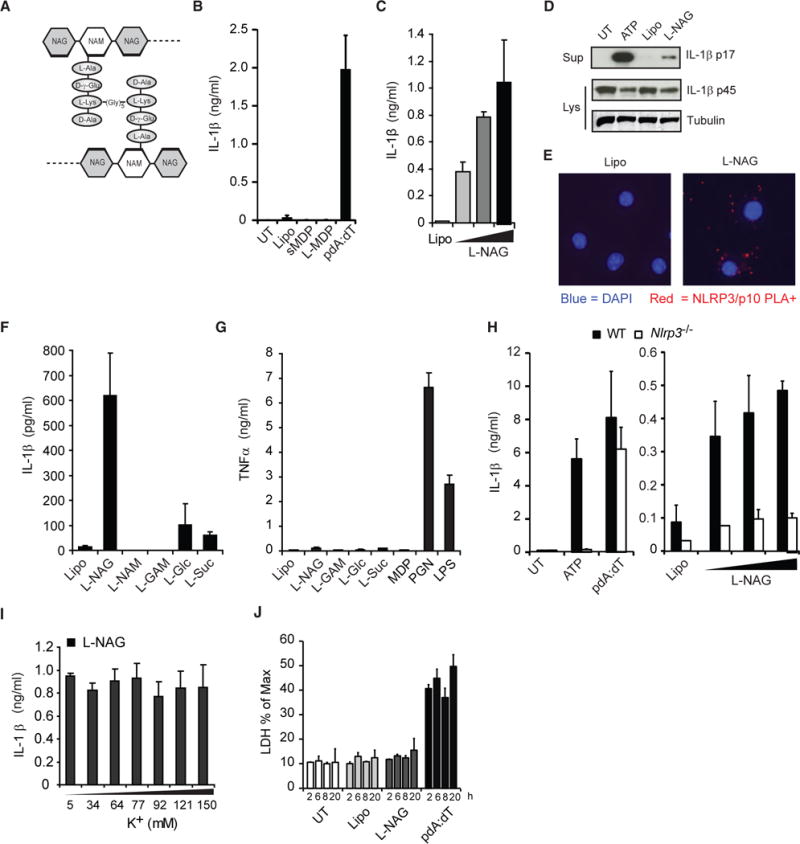
(A) Schematic diagram of PGN structure.
(B) LPS-primed BMDMs were stimulated with lipofectamine-complexed MDP (L-MDP), soluble MDP (sMDP, 10 μg/ml), pdA:dT, or lipofectamine alone (Lipo) for 6 hr. UT, untreated.
(C) Cells were stimulated for 6 hr with lipofectamine complexes containing increasing amounts of NAG.
(D) IL-1β processing was assessed by immunoblot as in (C). Sup, supernatant; Lys, lysate.
(E) Proximity ligation assay (for association of NLRP3 with caspase-1) of LPS-primed BMDMs treated for 3 hr with lipofectamine alone (Lipo) or complexed with NAG (L-NAG) (experiment was performed 2×).
(F) Cells were stimulated for 6 hr with different sugars complexed with lipofectamine (NAM, N-acetylmuramic acid; GAM, glucosamine; Gluc, glucose; and Suc, Sucrose).
(G) Unprimed BMDMs were treated with lipofectamine-complexed sugars as in (F), and TNF-α was measured in the supernatant after 6 hr.
(H) LPS-primed BMDMs from wild-type (WT) and Nlrp3−/−mice were stimulated as described above.
(I) LPS-primed BMDMs were stimulated with lipofectamine-complexed NAG in the presence of increasing concentrations of extracellular KCl for 6 hr.
(J) LPS-primed BMDMs, stimulated as described above, were assessed for LDH release at indicated times.
Error bars indicate SD. ***p < 0.001, Student’s t test.
See also Figure S2.
MDP, the NOD2-activating fragment of PGN, has been suggested to stimulate IL-1β release under certain conditions (Faustin et al., 2007; Ferwerda et al., 2008; Hsu et al., 2008; Marina-García et al., 2008; Martinon et al., 2004; Pan et al., 2007). However, when we treated macrophages with soluble MDP or lipofectamine-complexed MDP (to deliver it to the cytosol), they did not mimic the response to PGN that we saw (Figure 2B). Furthermore, as noted earlier, we have previously shown that PGN-induced IL-1β secretion is not blocked in macrophages lacking NOD2 (Shimada et al., 2010). Together, the data suggest that some other lysosomal degradation product of PGN must be detected.
Therefore, we examined the inflammasome-activating capacity of other potential PGN degradation products. We observed that the NAG sugar subunit from the backbone of PGN becomes a potent activator of IL-1β processing and secretion in LPS-primed macrophages when it is complexed with lipofectamine to deliver it to the cytosol (Figure 2C). We found that delivery of NAG to the cytosol is important to its function, since soluble NAG only induced IL-1β when added to culture medium at high concentrations (Figure S2A). The IL-1β detected in response to lipofectamine-complexed NAG was confirmed to be cleaved IL-1β p17 by immunoblot (Figure 2D), and inflammasome assembly was detected by NLRP3 and caspase-1 p10 proximity ligation (Figure 2E). In contrast, when we exposed LPS-primed cells to lipofectamine-complexed NAM (the other sugar subunit of the PGN backbone) or other sugars like glucosamine (GAM), glucose, or sucrose, they triggered little or no IL-1β secretion (Figure 2F). Lipofectamine-complexed NAG alone was unable to induce tumor necrosis factor α (TNF-α) in un-primed cells (Figure 2G), indicating that NAG does not reproduce the priming activity of PGN. In addition to mouse macrophages, PGN and NAG activate IL-1β secretion in mouse dendritic cells, as well as human macrophages and dendritic cells primed with LPS (Figures S2B–S2D). Like PGN, NAG inflammasome activation is dependent on the NLRP3 inflammasome (Figure 2H), independent of potassium efflux (Figure 2I), and does not induce pyroptosis (Figure 2J).
Acetylation of PGN Is Necessary for Its Inflammasome-Activating Potential
We reasoned that, if NAG is the critical component of PGN involved in NLRP3 inflammasome activation, PGN without NAG should not stimulate the inflammasome. PGN produced by Bacillus anthracis (anthrax) is unusual in that as much as 88% of its NAG is de-acetylated to GAM due to the expression of PGN NAG deacetylase activity (Zipperle et al., 1984). Thus, anthrax PGN contains little NAG, although it can be chemically re-acetylated in vitro (Zipperle et al., 1984). Native NAG-deficient anthrax PGN induces little or no inflammasome activation, while re-acetylated anthrax PGN strongly induces activation from LPS-primed (Figure 3A) or PAM3CSK4-primed (Figure S3A) macrophages. Native anthrax PGN is internalized by macrophages slightly less efficiently than the re-acetylated PGN in vitro (Figures 3B and 3C), although this difference cannot account for the profound lack of IL-1β secretion. The re-acetylated anthrax PGN-induced IL-1β was completely NLRP3 dependent (Figure 3D). The level of IL-1β produced by the re-acetylated PGN-stimulated macrophages is sufficient to induce potent inflammatory immune responses in vivo, since we observed that neutrophil recruitment to the peritoneum of mice injected with these macrophages was largely, although not entirely, NLRP3 dependent (Figure S3B).
Figure 3. Acetylation of NAG Is Necessary for Inflammasome Activation by PGN.
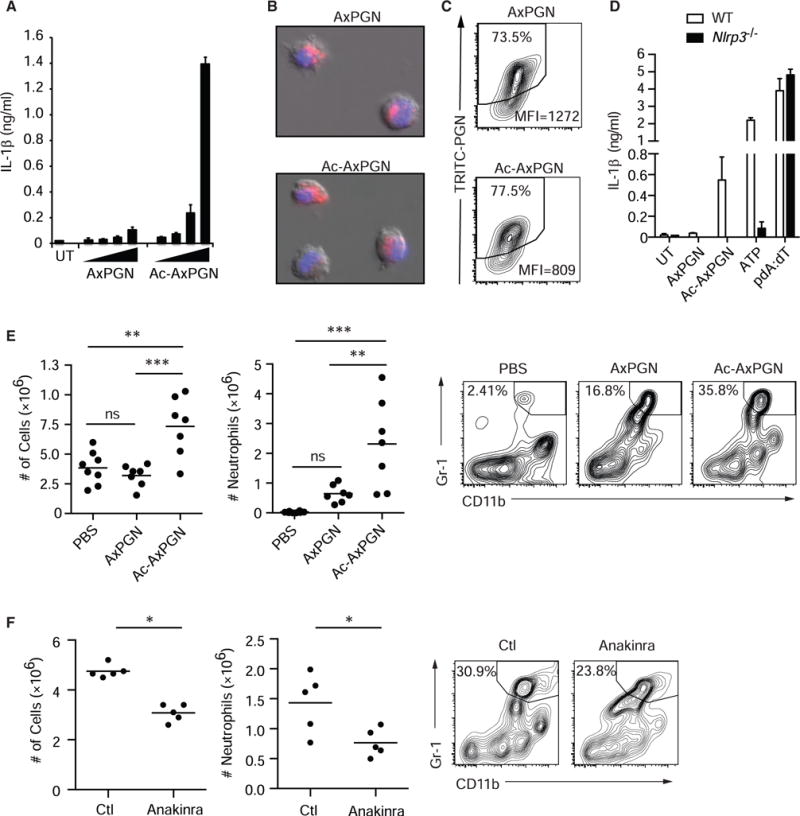
(A) LPS-primed BMDMs were treated with the increasing doses of native (AxPGN) or re-acetylated (Ac-AxPGN) anthrax PGN (20–160 μg/ml) for 6 hr, and IL-1β in the supernatant was measured by ELISA. UT, untreated.
(B) TRITC-labeled native or re-acetylated anthrax PGN (40 μg/ml) was incubated with LPS-primed BMDMs for 1 hr, and internalization was confirmed by fluorescence microscopy.
(C) TRITC-labeled native or re-acetylated anthrax PGN internalization by BMDMs was measured by flow cytometry after 6 hr.
(D) LPS-primed BMDMs from wild-type (WT) and Nlrp3−/−mice were stimulated with 80 μg/ml AxPGN for 6 hr, 80 μg/ml Ac-AxPGN for 6 hr, 5 mM ATP for 2 hr, or pdA:dT for 6 hr, and IL-1β was measured by ELISA.
(E) Mice were injected i.p. with PBS (n = 8) or 10 μg of AxPGN (n = 7) or Ac-AxPGN (n = 7). After 4 hr, cells in the peritoneal lavage were harvested and analyzed by flow cytometry. Total cells (left panel) and neutrophils (middle and right panels) were increased in response to re-acetylation of AxPGN. ns, not significant.
(F) Mice were injected i.p. with 10 μg Ac-AxPGN without (Ctl) (n = 5) or with 25 mg/kg anakinra (n = 5) and assayed as in (E) (experiment was performed 1×).
Error bars indicate SD. **p % 0.01; ***p < 0.001, one-way ANOVA and Newman-Keuls multiple comparison test (E) and unpaired Student’s t test (F).
See also Figure S3.
Re-acetylated anthrax PGN was significantly more inflammatory upon direct intraperitoneal injection into mice than native anthrax PGN, as measured by neutrophil infiltration (Figure 3E). PGN induces inflammation via a wide variety of mediators (e.g., cytokines, chemokines, and complement). We observed that the IL-1β receptor antagonist anakinra partially inhibited re-acetylated anthrax PGN-induced neutrophil infiltration (Figure 3F), indicating that IL-1β production plays an important role in the overall response.
PGN and NAG Inhibit Hexokinase and Induce Its Dissociation from Mitochondria
In order to determine how NAG activates the NLRP3 inflammasome, we began by investigating how PGN and NAG relate to mechanisms previously implicated in NLRP3-induced IL-1β secretion. As noted earlier, they do not require potassium efflux or induce pyroptosis. However, PGN (Figure 4A) and NAG (Figure S4A) both trigger the appearance of mtDNA in the cytosol. DNA release from the mitochondria has been associated with NLRP3 activation in response to other stimuli (Shimada et al., 2012; Zhong et al., 2016; Zhou et al., 2011), and while its exact relationship to PGN-induced NLRP3 inflammasome regulation is not clear, the observations prompted us to investigate further how PGN-derived NAG could affect mitochondria.
Figure 4. Hexokinase Is the Receptor that Detects NAG for Inflammasome Activation.
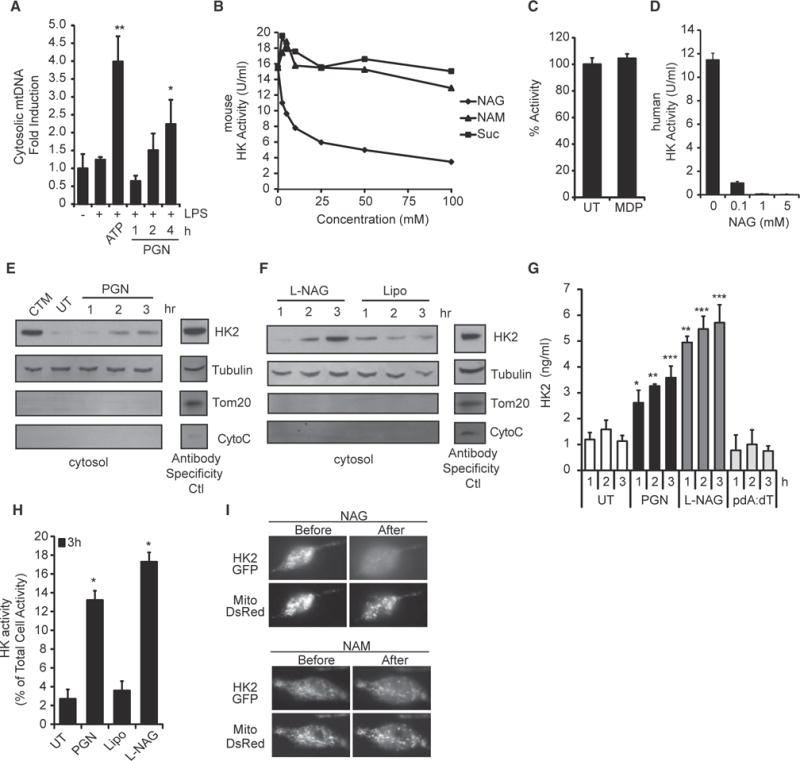
(A) LPS-primed BMDMs were stimulated with ATP (5 mM) for 2 hr or PGN (40 μg/ml) as indicated, and the presence of mtDNA in the cytosolic fraction was measured by RT-PCR.
(B and C) Hexokinase (HK) activity in purified mouse macrophage mitochondria was assessed in the presence of increasing concentrations of (B) NAG, NAM, sucrose, or (C) MDP (25 mM). UT, untreated.
(D) NAG inhibits the activity of purified human hexokinase.
(E and F) Hexokinase in the cytosol fraction of LPS-primed BMDMs stimulated for the indicated times with PGN (40 μg/ml, from S. aureus) or lipofectamine-complexed NAG was detected by immunoblot. Clotrimazole (CTM) treatment was used as a positive control for hexokinase release from mitochondria, and mitochondrial markers Tom20 and cytochrome c (CytoC) were included to control for mitochondrial integrity. Control lysates were included (Antibody Specificity Ctl) to confirm antibody staining for each marker (non-continuous lane from the same gel). Lipo, lipofectamine.
(G) LPS-primed BMDMs were stimulated as indicated PGN (40 μg/ml), and hexokinase 2 in the cytosol was determined by ELISA.
(H) LPS-primed BMDMs were stimulated as indicated, and cytosolic hexokinase enzyme activity was measured.
(I) LPS-primed BMDMs expressing hexokinase 2 fused to GFP (HK2-GFP) and DsRed targeted to mitochondria (Mito-DsRed) were microinjected with NAG or NAM as indicated. Association of hexokinase with mitochondria was visualized before and 1 min after injection (n = 10 cells assessed for each sugar).
Error bars indicate SD. *p % 0.05; **p % 0.01; ***p % 0.001, Student’s t test.
See also Figure S4.
Early biochemical studies attempting to identify the cytosolic enzyme responsible for phosphorylation of glucose, the first step in glycolysis, noted that NAG could competitively inhibit the process (Spiro, 1958). The enzyme inhibited by NAG was later identified as hexokinase (Wilson et al., 2011). Using purified mouse macrophage mitochondria and recombinant human hexokinase, we confirmed that NAG is a dose-dependent inhibitor of hexokinase enzymatic activity (Figures 4B and 4D), while other breakdown products of PGN, including NAM and MDP, do not inhibit hexokinase (Figures 4B and 4C).
As a competitive inhibitor, NAG, by definition, competes with glucose for binding to the active site of the enzyme. Binding of glucose to hexokinase has been directly characterized by solving the crystal structure of hexokinase bound to glucose, and NAG binding to the active site has been modeled (Aleshin et al., 1998a, 1998b; Madej et al., 2014) (MMDB: 43169). Consistent with these structural studies and with previous enzymatic studies and our current data (Figure 4B), we have further observed NAG directly binding to hexokinase by protein thermal shift assay (Figure S4B). Even though NAG binds to hexokinase, the enzyme cannot phosphorylate NAG (Figure S4C), which is consistent with its role as a competitive inhibitor. Acetylation of NAG is important for its role as a hexokinase inhibitor, because de-acetylated NAG, i.e., GAM, is phosphorylated by hexokinase (Figure S4C). Because GAM is phosphorylated by hexokinase, though less efficiently, it is not perceived as an inhibitor, suggesting why GAM does not activate the NLRP3 inflammasome (Figure 2F), and anthrax PGN, which is naturally de-acetylated, is less inflammatory than re-acetylated anthrax PGN (Figure 3). To further examine the role of acetylation, we solubilized native and re-acetylated anthrax PGN, as well as S. aureus PGN, with macrophage lysosomal extracts and observed that degradation products derived from PGNs rich in NAG, re-acetylated anthrax and S. aureus PGN, partially inhibited hexokinase activity (Figure S4D), while native anthrax PGN with low NAG content did not.
NAG inhibition of hexokinase was of particular interest, because the enzyme (hexokinase I and/or II, depending on the cell type) associates with the mitochondrial outer membrane through an interaction with the voltage-dependent anion channel (VDAC). This interaction with the VDAC is involved in regulation of glycolysis, mitochondrial stability, ROS production, and permeability transition pore formation (Pastorino and Hoek, 2008). Previous investigators had noted that the knockdown of the VDAC somehow blocks NLRP3 inflammasome activation, but how this related to microbial sensing or whether this has any relationship to cellular metabolism has not been clear (Zhou et al., 2011). Previous studies on cellular metabolism and regulation of apoptosis have observed that the association of hexokinase with the VDAC is closely regulated by signaling and feedback inhibition (Pastorino and Hoek, 2008). We hypothesized that the release of NAG after phagocytosis and degradation of PGN could affect mitochondria by influencing hexokinase association with mitochondrial VDAC.
To test this hypothesis, we measured the ability of PGN and NAG to induce release of hexokinase from mitochondria. We observed that macrophages stimulated with PGN or NAG showed elevated cytosolic levels of hexokinase by immunoblot (Figures 4E and 4F). The increased hexokinase in the cytosol is not a result of generalized loss of mitochondrial integrity; we did not detect increased cytosolic levels of other mitochondria proteins, including Tom20 or cytochrome c (Figures 4E and 4F), and mitochondrial membrane potential was not affected (Figures S4E and S4F). Thus, while acute activation of the NLRP3 inflammasome by some stimuli such as ATP may cause severe cell damage and mitochondrial disruption, PGN and NAG appear to induce a physiologically tolerable level of hexokinase release that does not involve degradation of mitochondria or collapse of total cellular mitochondrial function.
To more quantitatively measure hexokinase dissociation from mitochondria, we measured hexokinase release into the cytosol by ELISA (Figure 4G) and enzyme activity (Figure 4H) and observed increases in response to PGN and NAG. We observed hexokinase dissociation from mitochondria after phagocytosis of a variety of Gram-positive bacteria (Figure S4G). Overall expression of hexokinase was not affected (Figure S4H). Hexokinase release in response to PGN is upstream of NLRP3, since we observed normal hexokinase release into the cytosol in NLRP3-deficient macrophages (Figure S4I). To more directly evaluate the effect of NAG on hexokinase, we microinjected sugars directly into the cytosol of primary BMDMs expressing GFP-tagged hexokinase, which localizes to mitochondria (Figure 4I). While injection of NAM had no effect on hexokinase localization, injection of NAG induced dissociation of hexokinase from mitochondria within minutes, confirming that free NAG in the cytosol is sufficient to trigger hexokinase release. Mitochondria remain intact, as observed by the expression of DsRed tagged with a mitochondrial localization sequence.
Hexokinase Dissociation from Mitochondria Is Sufficient to Trigger NLRP3 Inflammasome Activation and IL-1β Production
We predicted that, if hexokinase inhibition and release from the mitochondria constitute an initiating step in NLRP3 inflammasome activation by PGN, then forcing hexokinase to dissociate from the VDAC would be sufficient to trigger IL-1β secretion. Previous metabolic studies have characterized the binding site between hexokinase II and the VDAC and have shown that a peptide derived from hexokinase II (HKVBD) can be used to block binding and cause dissociation of the enzyme from the VDAC (Chiara et al., 2008; Majewski et al., 2004; Pastorino et al., 2002). When we treated LPS-primed macrophages with a cell-permeable version of this peptide, hexokinase rapidly dissociated from mitochondria, as measured by immunoblot of the cytosol fraction (Figure 5A). We also directly observed HKVBD-peptide-induced dissociation of GFP-tagged hexokinase 2 from macrophage mitochondria by microscopy within minutes of exposure to the peptide (Figure 5B). When we treated LPS-primed BMDMs with the hexokinase dissociation peptide, we observed dose-dependent release of mature IL-1β by ELISA (Figure 5C; Figure S5A), as well as IL-18 (Figure 5D). IL-1β processing and secretion in response to the HKVBD peptide were faster than PGN (Figures S5B and S5C) and NLRP3 dependent (Figure 5E). Inflammasome activation was confirmed by observation of cleaved IL-1β p17 and caspase-1 p10 in the supernatant of treated BMDMs (Figure 5F). Proximity ligation assay revealed direct association of NLRP3 and caspase-1, evidence of initial inflammasome assembly, within minutes of exposure to the peptide (Figure 5G). To determine whether hexokinase dissociation from the VDAC on mitochondrial membranes is sufficient to activate inflammatory responses in vivo, we injected mice intraperitoneally with cell-permeable control or HKVBD peptides. The HKVBD peptide was sufficient to induce inflammation and recruitment of inflammatory cells (Figures 5H and S5D), and this response was reduced in mice deficient in caspase-1 and -11 (Figure 5I).
Figure 5. Hexokinase Dissociation from Mitochondria Is Sufficient to Activate the NLRP3 Inflammasome.
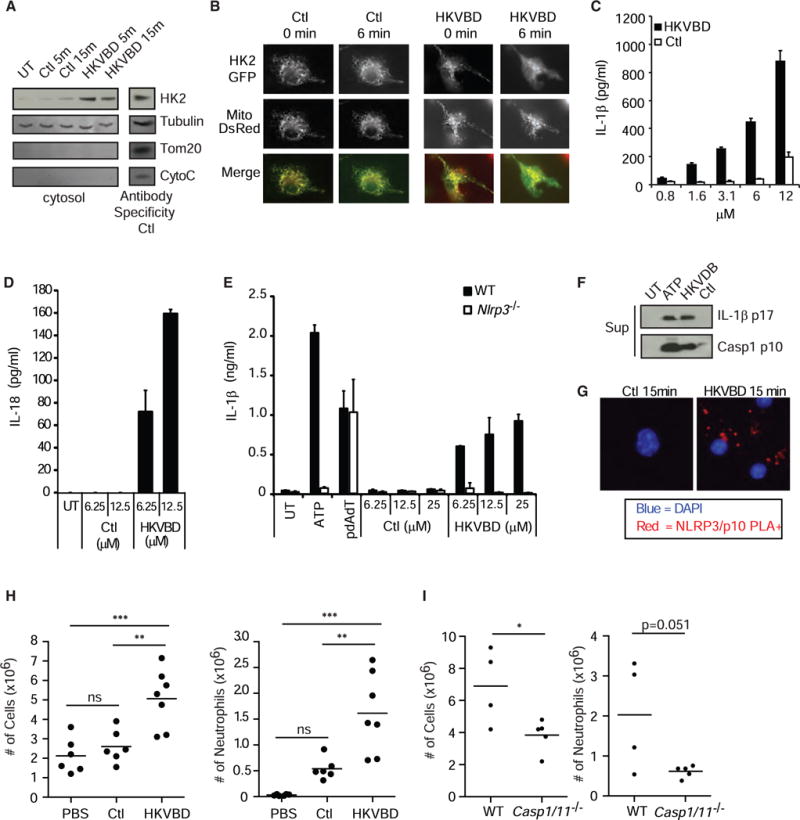
(A) LPS-primed BMDMs were treated with cell-permeable hexokinase dissociation peptide (HKVBD) or scrambled control peptides (Ctl) fused to TAT peptide (20 μM) for the indicated times, and the amount of hexokinase in the cytosolic fraction was determined by immunoblot. Control lysate was included on each gel (Antibody Specificy Ctl) to confirm antibody staining for each marker (non-continuous lane from the same gel). UT, untreated.
(B) LPS-primed BMDMs expressing HK2-GFP and mitochondria-localized DsRed (Mito-DsRed) were imaged following treatment with HKVBD and Ctl peptides fused to TAT (20 μM) to assess hexokinase redistribution.
(C and D) LPS-primed BMDMs were treated with HKVBD or control peptides fused to cell-permeable antennapedia peptide; IL-1β (C) and IL-18 (D) were measured by ELISA after 2 hr.
(E) LPS-primed BMDMs from wild-type and Nlrp3−/−mice were stimulated with 5 mM ATP, pdA:dT, HKVBD or control peptides fused to antennapedia peptide, and IL-1β was measured in the supernatant after 2 hr.
(F and G) LPS-primed BMDMs were treated with HKVBD or control peptides fused to TAT peptide; (F) cleaved IL-1β and caspase-1 were detected by immunoblot at 2 hr, and (G) inflammasome assembly was observed by NLRP3 and caspase-1 p10 proximity ligation (PLA,red). Nucleiwere stained withDAPI(blue). Sup, supernatant. (H and I) Indicated mice were injected i.p. with 500 μl of PBS (n = 6), 240 μM HKVBD (n = 7), or control peptide fused to TAT peptide (n = 6); peritoneal cavities were lavaged after 4 hr; total cells were counted; and neutrophil content was determined by flow cytometry (both experiments were each done 1×). ns, not significant. Error bars indicate SD. *p % 0.05; **p % 0.01; ***p % 0.001, Student’s t test (C–E and H) and one-way ANOVA and Newman-Keuls multiple comparison test (G).
See also Figure S5.
While NAG inhibits hexokinase activity and, therefore, triggers hexokinase dissociation from mitochondria, HKVBD peptide induces hexokinase dissociation but does not inhibit hexokinase activity (Figure S5E). Therefore, we conclude that hexokinase dissociation, rather than inhibition, is the important upstream step in inflammasome activation. HKVBD peptide can be toxic to the cells during extended exposure, but during the short exposure in which IL-1β is induced, we observed only a small amount of increased cell death (Figure S5F). Consistent with PGN and NAG, we observed an increase in cytosolic mtDNA when we stimulated cells with HKVBD peptide (Figure S5G). Since HKVBD-peptide-induced hexokinase release is sufficient to activate the NLRP3 inflammasome, and since PGN degradation leads to hexokinase release, we conclude that this mechanism is sufficient to explain how PGN activates the NLRP3 inflammasome.
Metabolic Conditions that Result in Hexokinase Inhibition Lead to Inflammasome Activation
Glycolysis is regulated, in part, by feedback inhibition of hexokinase by its enzymatic product glucose-6-phosophate (G6P). High levels of G6P trigger the release of hexokinase from mitochondria and, thus, reduce the rate of further G6P production (Gerber et al., 1974; Pastorino et al., 2002). Therefore, we predicted that excess G6P would activate the NLRP3 inflammasome. Indeed, when we treated primed BMDMs and bone marrow-derived dendritic cells (BMDCs) (data not shown) with lipofectamine-complexed G6P, we observed IL-1β secretion by ELISA (Figure 6A) and production of cleaved IL-1β p17 and caspase-1 p10 in the supernatant (Figure 6B). This activation was NLRP3 dependent (Figure S6A). Consistent with its role as a hexokinase inhibitor, we observed increased hexokinase in the cytosol following G6P treatment (Figure 6C). 2-deoxyglucose (2-DG) is a glycolytic inhibitor that is commonly used in studies of cell metabolism. It competes with glucose in the glycolytic pathway. When we treated primed BMDMs with 2-DG, we observed dose-dependent induction of IL-1β by ELISA (Figure 6D), as well as cleaved IL-1β p17 and caspase-1 p10 in the supernatant (Figure 6E), as has recently been reported by others (Nomura et al., 2015). However, 2-DG does not function as an inhibitor of hexokinase like NAG or G6P. Instead, 2-DG is metabolized by hexokinase to 2-deoxyglucose-6-phosphate (2-DG6P) (Figure S4C), which cannot be utilized by downstream glycolytic enzymes (Wick et al., 1957). The result is a buildup of 2-DG6P that inhibits hexokinase like G6P but is sensitive to the presence of glucose, hexokinase’s preferred substrate. Thus, while 2-DG can trigger inflammasome activation in primed macrophages in media containing glucose, it is more effective in the absence of glucose (Figure 6F) and is completely dependent on NLRP3 (Figure S6B). Consistent with our previous observations, 2-DG treatment leads to an increase in hexokinase in the cytosol of BMDMs (Figure 6G). Lastly, we treated primed cells with citrate, a natural intermediate in the tricarboxylic acid (TCA) pathway that inhibits phosphofructokinase when it accumulates. Buildup of citrate thus backs up the glycolytic pathway and naturally elevates cytosolic G6P levels (Berg et al., 2002). As expected, treating primed cells with citrate triggered NLRP3-dependent IL-1β production and caspase-1 cleavage (Figures 6H and S6C). While these metabolic stresses can cause cell death under certain conditions, we observed IL-1β production under conditions that do not cause substantial cell death (Figure S6D). These data suggest an intriguing relationship between cellular metabolism and inflammatory signaling.
Figure 6. Metabolic Perturbations Affecting Hexokinase Activate the NLRP3 Inflammasome.
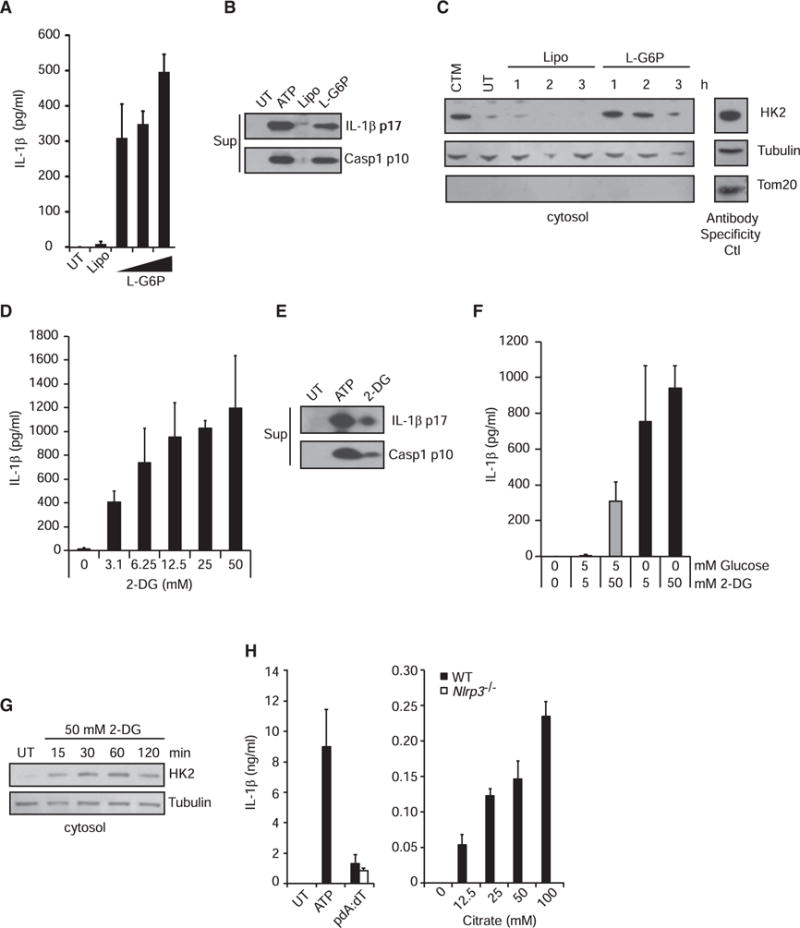
(A and B) LPS-primed BMDMs were treated with increasing concentrations of lipofectamine-complexed glucose-6-phosphate (G6P) for 6 hr. IL-1β was measured in the supernatant by ELISA (A), and cleaved IL-1β and caspase-1 were detected by immunoblot (B). UT, untreated; Lipo, lipofectamine.
(C) Hexokinase was detected in the cytosolic fraction following treatment with lipofectamine-complexed G6P for the indicated times. Control lysate was included on each gel (Antibody Specificity Ctl) to confirm antibody staining for each marker (non-continuous lane from the same gel). CTM, clotrimazole.
(D and E) LPS-primed BMDMs were treated with increasing concentrations of 2-deoxyglucose (2-DG) for 6 hr. IL-1β was measured in the supernatant (Sup) by ELISA (D), and cleaved IL-1β and caspase-1 were detected by immunoblot (E).
(F) LPS-primed BMDMs were treated with 2-DG in the presence or absence of glucose for 6 hr, and IL-1β was measured in the supernatant by ELISA.
(G) Hexokinase was measured in the cytosolic fraction following treatment with 2-DG in the absence of glucose.
(H) LPS-primed wild-type or Nlrp3−/−BMDMs were treated with increasing concentrations of sodium citrate for 6 hr, and IL-1β was measured in the supernatant by ELISA.
Error bars indicate SD. *p % 0.05; **p % 0.01; ***p % 0.001, Student’s t test.
See also Figure S6.
DISCUSSION
While the original evolutionary role of phagocytosis was to eat and degrade other microbes to obtain nutrients, this study suggests that mammalian phagocytes have adapted the cell’s metabolic machinery for utilizing these nutrients to detect the presence of microbial-derived sugars and metabolic perturbation as danger signals. In this study, we have shown that the NAG subunit of the sugar backbone of bacterial PGN induces inflammasome activation by inhibiting hexokinase, the first step in glycolysis. The inhibition of hexokinase results in hexokinase dissociation from the mitochondria, which, as we have observed, is sufficient to initiate an NLRP3 inflammasome-activating cascade in the cell. This model is supported by the observation that several metabolic perturbations that inhibit hexokinase function, such as treatment with glucose-6-phosphate, 2-deoxyglucose, or citrate, all lead to inflammasome activation. How exactly hexokinase release from the mitochondrial outer membrane promotes NLRP3 inflammasome activation remains to be understood.
Mitochondrial dynamics have been broadly implicated in regulation of the NLRP3 inflammasome, including studies demonstrating a role for mitochondrial movement along microtubules (Misawa et al., 2013), regulation of mitochondrial fission and growth (Park et al., 2015), mitophagy (Zhong et al., 2016), and release of mtDNA into the cytosol (Shimada et al., 2012; Zhong et al., 2016) in modulating inflammasome activation. NAG inhibition of hexokinase was particularly interesting, because hexokinases I and II, the primary isoforms that regulate glycolysis, are known to associate with the VDAC in the mitochondrial outer membrane (John et al., 2011; Pastorino and Hoek, 2008; Pastorino et al., 2002; Rasola et al., 2010). The VDAC is known to regulate mitochondrial ROS (reactive oxygen species) production (da-Silva et al., 2004), is a suggested component of the mitochondrial permeability transition pore that can release large molecules (including mtDNA) into the cytosol (Rasola et al., 2010; Tomasello et al., 2009), and is localized to regions enriched for cardiolipin (Sun et al., 2012) an NLRP3 activator. The interaction of hexokinase with the VDAC on the outer membrane of mitochondria provides hexokinase with preferential access to newly produced ATP transported from the matrix by the VDAC (Pastorino and Hoek, 2008). Hexokinase inhibition and dissociation from the mitochondria constitute an essential step in regulation of the rate of glycolysis (da-Silva et al., 2004; Pastorino and Hoek, 2008). Excess glucose-6-phosphate generated by hexokinase leads to feedback inhibition of hexokinase and its dissociation from mitochondria, slowing glycolysis. In addition, the interaction of hexokinase with the VDAC protects cells from mitochondrial ROS production (da-Silva et al., 2004) and suppresses pro-apoptotic interactions between the VDAC and Bcl-family members (Bax, Bid, etc.), which promotes sustained opening of the mitochondrial permeability transition pore (Chiara et al., 2008; Majewski et al., 2004; Pastorino and Hoek, 2008; Pastorino et al., 2002; Rasola et al., 2010). Each of these processes have been previously implicated in NLRP3 inflammasome regulation, but how they relate to microbial sensing has not been understood (Shimada et al., 2012; Zhou et al., 2011).
At first thought, NAG would seem to be a poor candidate to be a PAMP detected by the innate immune system, since it is not unique to bacteria. However, free NAG is not generally found in the cytosol of mammalian cells and is primarily generated only in small amounts following degradation of glycosylated proteins. In biosynthetic pathways, uridine diphosphate (UDP)-NAG is synthesized directly from glycolytic intermediates and utilized in glycosylation processes without existing as free NAG. In contrast, during degradation of particulate PGN in phagosomes, unusually large amounts of NAG can be expected to become available. The presence of a transporter that moves NAG from lysosomes into the cytosol has been biochemically documented (Jonas and Jobe, 1990; Jonas et al., 1989), although the molecular identity of the transporter is not yet known. We interpret the data to suggest that macrophages have adapted to use their ancient and essential metabolic glycolysis pathway to directly sense unusually high levels of bacteria-derived NAG, generated only upon phagocytosis of bacteria. Interestingly, unlike many strong NLRP3 inflammasome activators, PGN does not stimulate pyroptosis. It is possible that this resistance to cell damage is a result of the lower level of prolonged inflammasome activation stimulated by PGN, as compared to strong acute NLRP3 activators such as ATP. This is consistent with the idea that, from the perspective of mounting an effective host defense, macrophage cell death would seem to be an inappropriate response to detection of PGN.
Though many studies over the years have suggested that innate immune signaling has important effects on metabolism, these studies have not implicated metabolic enzymes themselves as sensors of non-self (Haneklaus and O’Neill, 2015; Wen et al., 2012). Our findings suggest a novel area for crosstalk between metabolism and innate immune signaling. The observation that modulation of a metabolic process can directly induce inflammation may have profound implications for diseases as wide ranging as diabetes, obesity, atherosclerosis, or inflammatory bowel disease, which have been linked to inflammasome activation (Wen et al., 2012).
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 mice were purchased from Jackson Laboratory. Nlrp3−/−mice (Mariathasan et al., 2006) were obtained from Dr. K. Fitzgerald (University of Massachusetts), and Casp1−/−mice also deficient in caspase-11 (Kuida et al., 1995) were obtained from R.A. Flavell (Yale University). Mice were housed in specific pathogen-free conditions in the Cedars-Sinai animal facility, and all animal experiments were conducted according to Cedars-Sinai Medical Center Institutional Animal Care and Use Committee guidelines.
Cell Preparation and Stimulation/Lipofectamine Complexing
BMDMs and dendritic cells were grown as described previously (Wolf et al., 2011), using 10% L-cell conditioned media or 50 ng/ml recombinant human macrophage colony-stimulating factor (M-CSF) or murine granulocyte-macrophage (GM)-CSF, respectively. Cells were plated at 1 × 105 in a 96-well plate and primed with LPS (50–100 ng/ml) for 3–4 hr, followed by treatment with PGN (20–40 μg/ml) for 6 hr or ATP (5 mM) or nigericin (10 μM) for 2 hr. Lipofectamine-complexed stimuli were prepared by mixing pdA:dT (1 μg/ml) or sugars (1 M) prepared in Opti-MEM with 2–4 μl lipofectamine (Invitrogen) per 100 μl for 30 min at room temperature. Cells were stimulated with 10 μl of the mix per well. All sugar solutions were adjusted to a pH ~7.4 prior to mixing with lipofectamine. Infection of cells with ΔoatA S. aureus and other bacteria was done as described previously (Shimada et al., 2010; Wolf et al., 2011). Cell supernatants were analyzed by ELISA for IL-1β and TNFα (BioLegend) and IL-18 (MBL International).
Anthrax PGN
PGN from Bacillus anthracis Sterne stain was purified at the University of Oklahoma Health Sciences Center, as previously described (Iyer and Coggeshall, 2011; Langer et al., 2008). Anthrax PGN was re-acetylated according to previously published methods (Vollmer and Tomasz, 2000). AxPGN and Ac-AxPGN were labeled with TRITC (Biotium) or Alexa Fluor 647 in PBS for 1 hr at 37°C and washed to remove unreacted fluorophore. Labeled PGN was added to BMDMs and spun down briefly, and cells were allowed to internalize PGN for 1 hr and washed. Cells were either imaged or treated with PBS + proteinase K (1 U/ml) to remove unbound particles and analyzed by flow cytometry to determine the degree of phagocytosis.
Intraperitoneal Injections of PGN and HKVBD
C57BL/6 mice (n = 7 per group) were injected intraperitoneally (i.p.) with 500 μl sterile PBS alone or containing 10 μg/ml AxPGN or Ac-AxPGN or 240 μM TAT-fused HKVBD or scramble peptide. For adoptive transfer experiments, wild-type and Nlrp3−/−BMDMs were primed with 100 ng/ml PAM3CSK4 for 4 hr and then given 80 μg/ml Ac-AxPGN for 1 hr. Cells were washed 3× with PBS and counted, and 1 × 105 cells in 500 μl sterile PBS were injected i.p. Mice were rested for 4 hr before being euthanized, and the peritoneal cavity was lavaged 2× with 5 ml of cold sterile PBS + 2 mM EDTA. Cells were counted; stained to assess cell types using antibodies against CD11b, CD11c, Gr-1, CD3, and CD19 (BioLegend); and analyzed by flow cytometry using FlowJo software.
Cell Fractionation and Hexokinase Detection
Cells were grown in six-well plates and stimulated as described earlier. Mitochondria and cytosol were separated using protocols previously described (Pastorino et al., 2002), with some modification. Cells were lifted in 600 μl of cell disruption buffer (20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 250 mM sucrose, Roche cOmplete Protease Inhibitor) and incubated on ice for 5 min. The cells were disrupted by passing 30 times through a 22-gauge needle. In some cases, cytosol was isolated by adding 50 μg/ml digitonin to the disruption buffer and incubating with rocking for 10 min. Lysates were then centrifuged at 10,000 × g for 10 min, and the supernatant was designated cytosol. For hexokinase assays, the pellet containing mitochondria was washed and resuspended in mitochondria suspension media (20 mM HEPES, 1.5 mM MgCl2, 250 mM sucrose). Cytosolic hexokinase was measured by either immunoblot or mouse hexokinase 2 ELISA (Novateinbio) on equivalent amounts of protein lysate.
Proximity Ligation Assay
BMDMs were plated on glass coverslips and primed with LPS for 4 hr. Cells were stimulated for designated times with the indicated stimuli, fixed with 4% paraformaldehyde, permeablized using 0.1% Triton X-100, and stained with anti-NLRP3 (Cryo-2) (AdipoGen) and anti-caspase-1 p10 (Santa Cruz Biotechnology). The Duolink In Situ PLA Kit was used according to the manufacturer’s instructions (Olink Biosciences).
Statistical Analysis
All experiments were conducted with triplicate measurements a minimum of three times unless otherwise stated in the figure legends. Experiments were analyzed using GraphPad Prism software or Microsoft Excel. The Grubbs test was used to identify and exclude a single outlier (Figure 3E).
Supplementary Material
Highlights.
Peptidoglycan-derived N-acetylglucosamine activates the NLRP3 inflammasome
N-acetylglucosamine in the cytosol is detected by hexokinase
Hexokinase release from mitochondrial outer membranes triggers NLRP3 activation
Metabolic conditions affecting hexokinase activity trigger inflammasome formation
Acknowledgments
This study was funded by grants from the NIH (GM085796 to D.M.U., T32AI089553 to A.J.W., AI067995 to M.A., and AI062629 to K.M.C.). Further support came from the Janis and William Wetsman Family Chair in Inflammatory Bowel Disease at Cedars-Sinai Medical Center (D.M.U.). Thanks to the laboratory of Dr. Robin Shaw for use of their FemtoJet microinjection system.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.05.076.
An audio PaperClip is available at http://dx.doi.org/10.1016/j.cell.2016.05.076#mmc3.
AUTHOR CONTRIBUTIONS
Studies were designed by A.J.W. and D.M.U. and performed by A.J.W., C.N.R., C.B., M.L.W., and K.S. Microinjection experiments were performed by A.J.W. and W.L. Anthrax PGN was prepared by K.M.C. and N.I.P. Further advice and conceptual development were provided H.C.C. and M.A. The manuscript was prepared by A.J.W. and D.M.U.
References
- Aleshin AE, Zeng C, Bartunik HD, Fromm HJ, Honzatko RB. Regulation of hexokinase I: crystal structure of recombinant human brain hexokinase complexed with glucose and phosphate. J Mol Biol. 1998a;282:345–357. doi: 10.1006/jmbi.1998.2017. [DOI] [PubMed] [Google Scholar]
- Aleshin AE, Zeng C, Bourenkov GP, Bartunik HD, Fromm HJ, Honzatko RB. The mechanism of regulation of hexokinase: new insights from the crystal structure of recombinant human brain hexokinase complexed with glucose and glucose-6-phosphate. Structure. 1998b;6:39–50. doi: 10.1016/s0969-2126(98)00006-9. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L, editors. Biochemistry. Fifth. New York: W. H. Freeman; 2002. The glycolytic pathway is tightly controlled. Section 16.2. http://www.ncbi.nlm.nih.gov/books/NBK21154/ [Google Scholar]
- Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da-Silva WS, Gómez-Puyou A, de Gómez-Puyou MT, Moreno-Sanchez R, De Felice FG, de Meis L, Oliveira MF, Galina A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem. 2004;279:39846–39855. doi: 10.1074/jbc.M403835200. [DOI] [PubMed] [Google Scholar]
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Ferwerda G, Kramer M, de Jong D, Piccini A, Joosten LA, Devesaginer I, Girardin SE, Adema GJ, van der Meer JW, Kullberg BJ, et al. Engagement of NOD2 has a dual effect on proIL-1beta mRNA transcription and secretion of bioactive IL-1beta. Eur J Immunol. 2008;38:184–191. doi: 10.1002/eji.200737103. [DOI] [PubMed] [Google Scholar]
- Gerber G, Preissler H, Heinrich R, Rapoport SM. Hexokinase of human erythrocytes. Purification, kinetic model and its application to the conditions in the cell. Eur J Biochem. 1974;45:39–52. doi: 10.1111/j.1432-1033.1974.tb03527.x. [DOI] [PubMed] [Google Scholar]
- Haneklaus M, O’Neill LA. NLRP3 at the interface of metabolism and inflammation. Immunol Rev. 2015;265:53–62. doi: 10.1111/imr.12285. [DOI] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, Eckmann L, Powell JJ, Nizet V, Dixit VM, Karin M. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci USA. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip WK, Sokolovska A, Charriere GM, Boyer L, Dejardin S, Cappillino MP, Yantosca LM, Takahashi K, Moore KJ, Lacy-Hulbert A, Stuart LM. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J Immunol. 2010;184:7071–7081. doi: 10.4049/jimmunol.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer JK, Coggeshall KM. Cutting edge: primary innate immune cells respond efficiently to polymeric peptidoglycan, but not to peptidoglycan monomers. J Immunol. 2011;186:3841–3845. doi: 10.4049/jimmunol.1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE. 2011;6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas AJ, Jobe H. N-acetyl-D-glucosamine countertransport in lysosomal membrane vesicles. Biochem J. 1990;268:41–45. doi: 10.1042/bj2680041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas AJ, Speller RJ, Conrad PB, Dubinsky WP. Transport of N-acetyl-D-glucosamine and N-acetyl-D-galactosamine by rat liver lysosomes. J Biol Chem. 1989;264:4953–4956. [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Langer M, Malykhin A, Maeda K, Chakrabarty K, Williamson KS, Feasley CL, West CM, Metcalf JP, Coggeshall KM. Bacillus anthracis peptidoglycan stimulates an inflammatory response in monocytes through the p38 mitogen-activated protein kinase pathway. PLoS ONE. 2008;3:e3706. doi: 10.1371/journal.pone.0003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej T, Lanczycki CJ, Zhang D, Thiessen PA, Geer RC, Marchler-Bauer A, Bryant SH. MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014;42:D297–D303. doi: 10.1093/nar/gkt1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Marina-García N, Franchi L, Kim YG, Miller D, McDonald C, Boons GJ, Núñez G. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J Immunol. 2008;180:4050–4057. doi: 10.4049/jimmunol.180.6.4050. [DOI] [PubMed] [Google Scholar]
- Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- Müller S, Wolf AJ, Iliev ID, Berg BL, Underhill DM, Liu GY. Poorly cross-linked peptidoglycan in MRSA due to mecA induction activates the inflammasome and exacerbates immunopathology. Cell Host Microbe. 2015;18:604–612. doi: 10.1016/j.chom.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflam-masome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura J, So A, Tamura M, Busso N. Intracellular ATP decrease mediates NLRP3 inflammasome activation upon nigericin and crystal stimulation. J Immunol. 2015;195:5718–5724. doi: 10.4049/jimmunol.1402512. [DOI] [PubMed] [Google Scholar]
- Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, Kobayashi KS, Bertin J, Grant EP, Coyle AJ, et al. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82:177–183. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- Park S, Won JH, Hwang I, Hong S, Lee HK, Yu JW. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci Rep. 2015;5:15489. doi: 10.1038/srep15489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr. 2008;40:171–182. doi: 10.1007/s10863-008-9148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- Rasola A, Sciacovelli M, Pantic B, Bernardi P. Signal transduction to the permeability transition pore. FEBS Lett. 2010;584:1989–1996. doi: 10.1016/j.febslet.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Götz F, et al. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe. 2010;7:38–49. doi: 10.1016/j.chom.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro RG. The effect of N-acetylglucosamine and glucosamine on carbohydrate metabolism in rat liver slices. J Biol Chem. 1958;233:546–550. [PubMed] [Google Scholar]
- Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem. 2012;287:40652–40660. doi: 10.1074/jbc.M112.419721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, Thoraval D, Crouzet M, Ichas F, De Pinto V, De Giorgi F. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 2009;19:1363–1376. doi: 10.1038/cr.2009.98. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Tomasz A. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J Biol Chem. 2000;275:20496–20501. doi: 10.1074/jbc.M910189199. [DOI] [PubMed] [Google Scholar]
- Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases–did Warburg miss inflammation? Nat Immunol. 2012;13:352–357. doi: 10.1038/ni.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick AN, Drury DR, Nakada HI, Wolfe JB. Localization of the primary metabolic block produced by 2-deoxyglucose. J Biol Chem. 1957;224:963–969. [PubMed] [Google Scholar]
- Wilson MH, Edsell ME, Davagnanam I, Hirani SP, Martin DS, Levett DZ, Thornton JS, Golay X, Strycharczuk L, Newman SP, et al. Cerebral artery dilatation maintains cerebral oxygenation at extreme altitude and in acute hypoxia–an ultrasound and MRI study. J Cereb Blood Flow Metab. 2011;31:2019–2029. doi: 10.1038/jcbfm.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Arruda A, Reyes CN, Kaplan AT, Shimada T, Shimada K, Ar-diti M, Liu G, Underhill DM. Phagosomal degradation increases TLR access to bacterial ligands and enhances macrophage sensitivity to bacteria. J Immunol. 2011;187:6002–6010. doi: 10.4049/jimmunol.1100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, et al. NF-kB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- Zipperle GF, Jr, Ezzell JW, Jr, Doyle RJ. Glucosamine substitution and muramidase susceptibility in Bacillus anthracis. Can J Microbiol. 1984;30:553–559. doi: 10.1139/m84-083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.