

Clostridium difficile
is the leading cause of hospital-acquired diarrhea and pseudomembranous colitis worldwide. The organism produces two homologous toxins, TcdA and TcdB, which enter and disrupt host cell function by glucosylating and thereby inactivating key signalling molecules within the host. As a toxin-mediated disease, there has been a significant interest in identifying small molecule inhibitors of the toxins’ glucosyltransferase activities. This study was initiated as part of an effort to identify the mode of inhibition for a small molecule inhibitor of glucosyltransferase activity called apigenin. In the course of trying to get co-crystals with this inhibitor, we determined five different structures of the TcdA and TcdB glucosyltransferase domains and made use of a non-hydrolyzable UDP-glucose substrate. While we were able to visualize apigenin bound in one of our structures, the site was a crystal packing interface and not likely to explain the mode of inhibition. Nevertheless, the structure allowed us to capture an apo-state (one without the sugar nucleotide substrate) of the TcdB glycosyltransferase domain that had not been previously observed. Comparison of this structure with structures obtained in the presence of a non-hydrolyzable UDP-glucose analogue have allowed us to document multiple conformations of a C-terminal loop important for catalysis. We present our analysis of these five new structures with the hope that it will advance inhibitor design efforts for this important class of biological toxins.
Introduction
Clostridium difficile is a spore-forming anaerobe that produces two large, homologous toxins. The toxins, TcdA and TcdB, are the primary virulence factors for C. difficile infection (CDI) and are part of the large clostridial toxin (LCT) family. Members of the LCT family share sequence homology, domain organization, and common origins (Hofmann et al., 1995). In addition to TcdA and TcdB, the LCTs include virulence factors produced by the pathogens C. sordellii (TcsL and TcsH), C. novyi (Tcnα), and C. perfringens (TpeL) (Ziegler et al., 2008). Each LCT contains four domains: a glycosyltransferase domain (GTD), autoprotease domain (APD), delivery domain, and combined repetitive oligopeptides (CROPs) domain. The toxins bind and enter host cells through receptor-mediated endocytosis. During endosomal acidification, a conformational change in the delivery domain facilitates translocation of the GTD and APD across the endosomal membrane into the cell cytosol (Barth et al., 2001). The APD is activated by inositol hexakisphosphate (IP6) and cleaves the GTD at its C-terminus, thus releasing the GTD into the cell (Reineke et al., 2007). The LCT-GTDs catalyze the transfer of a sugar from uridine diphosphate (UDP) to a regulatory domain of host cell GTPases (Just et al., 1995). The TcdA and TcdB GTDs target Rho family GTPases—including RhoA, Rac1, and Cdc42 (Busch et al., 1998). These GTPases are essential regulators of focal adhesions, actin organization, cell morphology, and migration. Glycosylation by TcdA and TcdB GTDs leads to loss of focal adhesions, F-actin depolymerization, and apoptotic cell death (Jank and Aktories, 2008; Nagahama et al., 2012; Ziegler et al., 2008).
Along with the primary sequence homology of the holotoxins, the GTD structures of TcdA, TcdB, TcsL, and Tcnα reveal that the LCT-GTDs also share structural homology (D’Orzo et al., 2012; Pruitt et al., 2012; Reinert et al., 2005; Ziegler et al., 2008). The GTDs can be organized into four domains, which includes a membrane localization domain (MLD) (Figure 1, yellow), the glycosyltransferase-A fold (Figure 1, blue), a globular subdomain (Figure 1, orange), and two helical clusters (Figure 1, green). An important element within LCT-GTDs is a conserved tryptophan, which resides on a flexible loop at the GTD C-terminus, with proximity to UDP-glucose (Figure 1, inset, magenta). Mutation of this tryptophan affects catalysis, but not UDP-glucose binding. Specifically, in TcdB-GTD W520A, the kcat of glucosyltransfer is reduced over 800-fold compared to wildtype, while the UDP-glucose Km varies only slightly (Jank et al., 2007). The first structures of TcdB-GTD were obtained by including UDP-glucose and cofactor Mn2+ in the crystallization conditions (Reinert et al., 2005). The electron density maps revealed TcdB bound to UDP and glucose, indicating that the substrate had been hydrolyzed. In contrast, no hydrolysis was seen in structures emerging from co-crystallization of the TcdA GTD with UDP-glucose and Mn2+ (Pruitt et al., 2012). These observations are consistent with kinetic data indicating that in the absence of target GTPases, TcdB-GTD will hydrolyze UDP-glucose with a five-fold higher Vmax compared to TcdA-GTD (Chaves-Olarte et al., 1997; Ciesla and Bobak, 1998). To date, only two GTDs from the LCT family have crystal structures under apo conditions: TcdA-GTD and Tcna-GTD (Pruitt et al., 2012; Ziegler et al., 2008).
Figure 1.
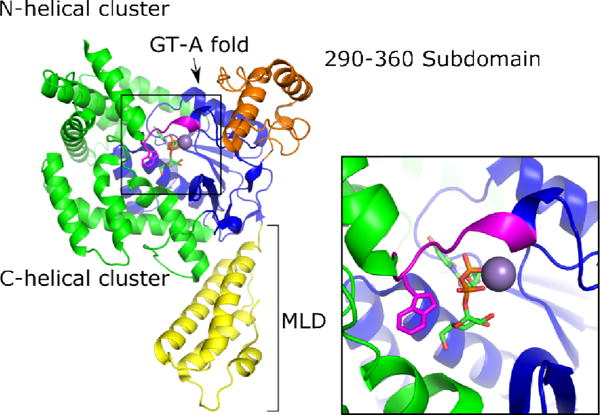
Cartoon representation of TcdA-GTD bound to UDP-glucose and Mn2+ with the membrane localization domain (MLD) in yellow, the 290–360 domain in orange, the glycosyltransferase (GT) type A fold in blue, the N and C-terminal helical clusters in green, and the conserved tryptophan loop in magenta.
To better understand the structural changes associated with substrate binding, we set out to investigate, 1) an intact substrate bound to TcdB-GTD, 2) an apo form of TcdB-GTD, and 3) the effects and/or binding sites of the small molecule inhibitor apigenin. In this study, we present crystal structures of TcdA and TcdB-GTDs in complex with UDP-2-deoxy-2-fluoroglucose (U2F), a non-hydrolyzable UDP-glucose analogue. Additionally, we show that apigenin can bridge two TcdB-GTD chains, giving rise to a new crystal form and space group, one that allows visualization of TcdB-GTD in an apo-like form. Together, these new structures provide insight into the range of flexibility associated with the catalytic tryptophan loop when moving from apo to UDP-glucose bound conditions.
Materials and methods
Synthesis of UDP-2-deoxy-2-fluoroglucose
U2F was synthesized through Vanderbilt’s Small Molecule Synthesis Core according to published methods (Gibson et al., 2004). Sample purity was determined by liquid chromatography mass spectrometry, and the final structure was validated by 1H/13C nuclear magnetic resonance. Samples of dry U2F were resuspended in 20 mM HEPES pH 8, 50 mM NaCl and stored at −20 °C.
Purification and Crystallization of TcdA-GTD
TcdA-GTD was purified according to published methods using the Bacillus megaterium expression system transformed with pBL161(Pruitt et al., 2012). Pure TcdA-GTD aliquots in 20 mM HEPES pH 8, 150 mM NaCl were flash-frozen with liquid N2 and stored at −80 °C. Prior to crystallization, aliquots were thawed rapidly, subjected to size-exclusion chromatography (SEC) in the same buffer (20 mM HEPES pH 8, 150 mM NaCl), and concentrated using Amicon Ultra centrifugal filters with a 50 kDa cutoff. For co-crystallization with UDP-glucose or U2F, a final concentration of 1 mM of the UDP-conjugate was used. All protein solutions also included 2–10 mM MnCl2. All TcdA GTD co-crystals were obtained by hanging drop diffusion with a 1:1 ratio of 5–12 mg/mL protein solution to mother liquor comprised of 0.1 M HEPES pH 7–8.5, 0.2 M L-proline, and PEG 3350 10–25%. DMSO stocks of 0.1 M apigenin were diluted into concentrated protein solutions to obtain final concentrations of 0.5–10 mM. We observed significant compound precipitation at these concentrations; however, the total protein concentration was not affected. Large, hexagonal rods grew within 24–48 h at 21 °C. Crystals were transferred into mother liquor containing 15–20% glycerol, including 0.5–10 mM apigenin (where indicated) mounted on cryo-loops and flash-frozen in liquid N2.
Purification and Crystallization of TcdB-GTD
TcdB-GTD was purified similar to previous methods (Pruitt et al., 2012). The sequence corresponding to TcdB-GTD1–543 was copied from the parent plasmid pBL149 and cloned into pET28a+ using restriction sites BamHI 5′ and XhoI 3′ to yield pBL720. Transformed E. Coli (BL21-Star) glycerol stocks were grown in LB-Miller broth at 37 °C. At OD600 = 0.6, protein expression was induced with 1 mM IPTG, and the temperature was reduced to 18 °C. After 16–18 h, cultures were pelleted and frozen. After thawing, cell pellets were lysed using an Emulsiflex C3 at 18,000 psi and centrifuged at 16,000 x g to remove membranes and insoluble components. Lysis buffer was composed of 50 mM HEPES pH 8, 500 mM NaCl, and 2 mM imidazole. Approximately 10 μg of DNAse-I (Sigma-Aldrich), 1 mM PMSF, 1 mM Leupeptin, and 1 mM Pepstatin were added to lysis buffer. The lysate was purified by successive immobilized metal affinity, anion exchange, and SEC. Aliquots of concentrated protein were frozen in liquid N2 and stored at −80 °C.
For crystallization, aliquots were thawed, purified using SEC (20 mM HEPES pH 8, 150 mM NaCl), and re-concentrated. Apigenin was dissolved in 100% DMSO at a stock concentration of 0.1 M. Aliquots were stored at −20 °C until used. The DMSO apigenin solution was added to concentrated protein to obtain final concentrations of 0.5–10 mM. We observed significant compound precipitation at these concentrations; however, the total protein concentration was not affected. TcdB-GTD crystals with apigenin were obtained by hanging drop diffusion with a 1:1 ratio of 8–15 mg/mL protein solution (20 mM HEPES pH 8, 150 mM NaCl, 1–10 MnCl2) to mother liquor comprised of 0.1 M HEPES pH 7.5–8.5, 0.2 M MgAcetate, and PEG 3350 5–25%. Rhombohedral crystals grew within 24–48 h only in the presence of apigenin. Crystals were transferred into mother liquor containing 10–20% glycerol, 0.5–10 mM apigenin (where indicated) mounted on cryo-loops and flash-frozen in liquid N2. TcdB-GTD + U2F co-crystals were obtained by hanging-drop diffusion using conditions previously reported with a mother liquor of 0.1 M MES pH 6–6.5, 0.2 (NH4)2SO4, and PEG 8K 16–34% (Reinert et al., 2005). This was mixed 1:1 with TcdB-GTD at 10-15 mg/mL in 20 mM HEPES pH 8, 150 mM NaCl, 1 mM U2F, and 1–10 mM MnCl2. Short bars and biaxial clover crystals formed within 72 h at 21 °C. Crystals were transferred into mother liquor containing 15–20% glycerol, including 0.5–10 mM apigenin (where indicated) mounted on cryo-loops and flash-frozen in liquid N2.
Data collection and refinement
Data were collected from single crystals on LS-CAT beamlines 24 ID-D, F, and G at the Advanced Photon Source (Argonne National Laboratory, IL) at 100 K. Data were indexed and scaled using HKL2000 or the program suite xia2 (Kabsch, 2010; Otwinowski and Minor, 1997). TcdA-GTD data collected from crystals without UDP substrates were phased using PHENIX-Phaser-MR with coordinates from Protein Data Bank (PDB) 3SS1, while substrate-bound crystals were phased with PDB 3SRZ. TcdB-GTD data were phased using PHENIX-Phaser-MR with coordinates from PDB 2BVL (Adams et al., 2010; Pruitt et al., 2012; Reinert et al., 2005). Models were iteratively built with Coot and refined via PHENIX with 4–5 TLS groups per chain (26, 27). Final coordinates for TcdA-GTD co-crystallized with U2F and apigenin (5UQK) and U2F alone (5UQL) were deposited into the PDB. In TcdB-GTD datasets indexed as C2221 two chains exhibiting non-crystallographic symmetry (NCS) were identified in the asymmetric unit (ASU). Alternatively, crystals grown under previously reported conditions were indexed as P41212and contained a single chain in the ASU. NCS torsion-angle restraints were used in refining the C2221 structure. RMSD values were calculated and figures were generated using PyMol (Schrödinger, 2015). Final coordinates for TcdB-GTD co-crystallized with U2F (5UQN), U2F and apigenin (5UQM), and apigenin alone (5UQL) were deposited into the PDB.
Results
TcdA-GTD and TcdB-GTD in complex with U2F
UDP-2-deoxy-2-fluoroglucose (U2F) and related UDP-fluoro-sugars have been used to crystallize a number of diverse glycosyltransferases (Gibson et al., 2004; Hiromoto et al., 2013; Persson et al., 2001; Truman et al., 2009). The U2F as a substrate renders hydrolysis or transfer by glycosyltransferases unfavorable. The GTDs of TcdA and TcdB were crystallized in the presence of U2F, and structures were determined at 2.0 and 2.06 Å, respectively (Table 1). Published structures of the GTDs bound to UDP-glucose were used as models for molecular replacement (3SRZ for TcdA and 2BVL for TcdB), and the refined U2F structures aligned to their starting models with Cα RMSD values of 0.25 Å2 (TcdA) and 0.44 Å2 (TcdB).
Table 1.
X-ray statistics for TcdA and TcdB-GTD structures. Values in parenthesis refer to the highest resolution shell.
| Protein complex | TcdB-GTD U2F | TcdA-GTD U2F | TcdB-GTD U2F+ apigenin | TcdA-GTD U2F + apigenin | TcdB-GTD apigenin |
|---|---|---|---|---|---|
| 5UQN | 5UQL | 5UQM | 5UQK | 5UQT | |
| Data processing | xia2 | HKL2000 | xia2 | HKL2000 | xia2 |
| Space group | P41212 | P65 | P41212 | P65 | C2221 |
| Cell dimensions | |||||
| a, b, c (Å) | 62.3, 62.3, 327.6 | 142.1, 142.1, 63 | 61.9, 61.9, 326.0 | 142.8, 142.8, 66 | 82, 154.8, 271.3 |
| Resolution (Å) | 54.19–2.06 | 46.5–1.97 | 42.28–2.03 | 50–1.85 | 41–2.75 |
| I/σI | 23.31 (3.75) | 21.35 (1.55) | 23.89 (2.91) | 28.4 (1.32) | 21.33 (1.4) |
| Completeness (%) | 97.8 (93.3) | 100 (99) | 99.3 (96.2) | 100 (98) | 99.8 (99.9) |
| Redundancy | 11.3 (10.3) | 8.2 (7) | 11.3 (9.3) | 7.4 (6.8) | 6.6 (6.8) |
| Wavelength (Å) | 0.97856 | 0.97872 | 0.97856 | 0.97856 | 0.97856 |
| Unique observations | 41228 | 51462 | 42362 | 65449 | 45388 |
| Refinement | |||||
| Resolution (Å) | 54.19–2.06 | 46.5–1.97 | 42.2–2.03 | 31.4–1.85 | 44.4–2.75 |
| No. of reflections | 41070 | 51428 | 42208 | 65417 | 45276 |
| Rwork/Rfree | 0.236/0.203 | 0.204/0.184 | 0.228/0.197 | 0.207/0.182 | 0.236/0.196 |
| No. of atoms | 4789 | 5002 | 4768 | 5058 | 8842 |
| Avg. protein B-factor (Å2) | 43.54 | 39.63 | 42.69 | 52.58 | 102.66 |
| RMSD values | |||||
| Bond lengths (Å) | 0.008 | 0.004 | 0.014 | 0.021 | 0.007 |
| Bond angles (degrees) | 1.019 | 0.665 | 1.264 | 1.598 | 0.906 |
| Ramachandran plot (%) | |||||
| Most favored | 97.4 | 98.52 | 97.59 | 97.01 | 96.83 |
| Allowed | 2.41 | 1.48 | 2.41 | 2.99 | 3.08 |
| Disallowed | 0.19 | 0 | 0 | 0 | 0.09 |
The electron density clearly shows the presence of U2F within the GTD active sites of TcdA (Figure 2A) and TcdB (Figure 2B). The TcdB-GTD + U2F structure gave us an opportunity to visualize any changes from TcdB-GTD bound to hydrolyzed UDP and glucose. The primary changes between an intact substrate are found in the position of glucose when still bound to UDP or after hydrolysis. We found that while the glucose ring tilts slightly, the residues which interact with glucose do not significantly change upon substrate hydrolysis (Figure 3). Overall, active site residues of both TcdA and TcdB-GTD bound to U2F closely match the UDP-glucose structure of TcdA and the UDP + glucose structure of TcdB. Comparing TcdA-GTD+U2F to the structure co-crystallized with UDP-glucose, we observe nearly identical positions of residues in the active site.
Figure 2.
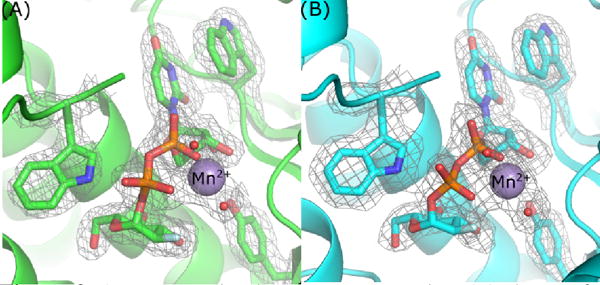
2mFo-DFc density map contoured at 1.0 sigma of select aromatic groups and U2F in the active site of TcdA-GTD (A) and TcdB-GTD (B).
Figure 3.
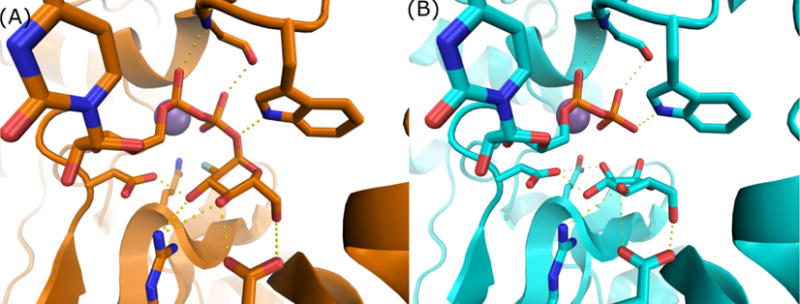
Comparisons between U2F (A) and UDP + glucose (B) co-crystal structures of TcdB-GTD demonstrate highly similar active site conformation and interactions.
The UDP moiety is held in place by a combination of hydrogen bonds, aromatic interactions, and a Mn2+ cofactor (Shown for TcdB in Figure 4, but also true in TcdA). Several lines of evidence support the assignment of this cofactor as a Mn2+. First, all five crystal forms were grown in the presence of Mn2+. While two of these crystal forms also had Mg2+ in the crystallization buffer, the peak height for the cofactor omit map was at least twice as intense as that of a carbonyl oxygen, suggesting Mn2+ rather than Mg2+. In addition, the bond lengths for the Mn–O bond in the highest resolution structure (5UQL) were 2.21 Å for the Mn-Glu and 2.10 Å for the Mn-Asp interactions, consistent with high resolution Mn–O bonds in the protein data bank. Finally, the B-factors for the Mn2+ atom was in line with the overall B-factor for its structure. A conserved tryptophan (TcdA W101, TcdB W102) anchors the uracil ring with aromatic π-stacking (Figure 4A,4B). In the context of TcsL, the analogous TcsL-GTD W102A mutant has a 2000-fold higher Km and a 16-fold lower kcat compared to wild-type TcsL-GTD (Busch et al., 2000). The W102Y TcsL-GTD mutant was slightly more active than the W102A mutant with only a 21-fold increase in Km and four-fold reduction in kcat (Busch et al., 2000). This suggests that the aromatic nature of the tryptophan is significant for interaction with the uracil ring. The fact that W102A TcdB-GTD also has significantly impaired glycosyltransferase and hydrolase activities suggests that this role will be common across the LCT family of GTDs (Busch et al., 2000; Spyres et al., 2003).
Figure 4.
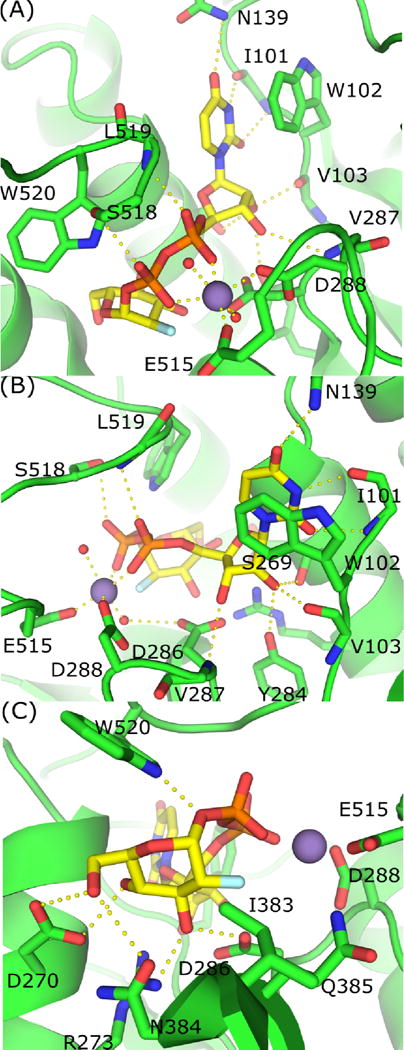
Coordination of UDP-2-deoxy-2-fluoroglugose by TcdB-GTD. (A) π-stacking and hydrogen bonding between UDP, Mn2+ and TcdB-GTD. (B) Alternate view of interactions between active site residues and UDP and Mn2+. (C) Interactions between active site residues and 2-deoxy-2-fluoroglucose.
The U2F of each structure interacts with other conserved elements within the GTD active sites. The uracil C2 and C4 carbonyls form hydrogen bonds with the I101 backbone amine and N139 amide nitrogens, respectively (shown for TcdB, Figure 4A, 4B). The backbone carbonyl of I101 forms a third hydrogen bond with uracil N3, and the V287 backbone amine forms a hydrogen bond with the ribose 3′-hydroxyl. The ribose 2′-hydroxyl interacts via hydrogen bonds to the V103 backbone carbonyl, Y284 sidechain hydroxyl group, and S269 sidechain hydroxyl group (Figure 4B). In TcdB-GTD, it was demonstrated that Y284A constructs have over 1000-fold lower glucosyltransferase activity as compared to wild-type. This interaction is also observed in crystal structures of TcdA-GTD, which suggests Y283 plays a similarly important role by coordinating with the ribose 2′-hydroxyl in TcdA (Jank et al., 2007; Pruitt et al., 2012).
A key element in glycosyltransferases is the conserved DxD motif (D286 and D288 in TcdB), where two aspartates bind Mn2+, which in turn supports the interactions with the nucleotide diphosphates (Figure 4B). Mutation of both DxD aspartates to asparagines in TcdA-GTD and TcsL-GTD reduces glycosyltransfer by three orders of magnitude, demonstrating the essential role of this network of interactions (Busch et al., 1998; Teichert et al., 2006). The octahedral Mn2+ coordination system is completed by the diphosphate backbone of the substrate and two additional water molecules (Figure 4A, 4B). Typically, retaining glycosyltransferases have aspartates from the DxD motif directly interact with the metal cofactor (Breton et al., 2006). However, we note that within the LCT family, the Mn2+ is directly coordinated by D288 and E515 while D286 coordinates Mn2+ indirectly through a water molecule. This LCT architecture is similar to the galactosyltransferase LgtC from Neisseria meningitidis (Persson et al., 2001).
Apigenin and the crystallization of TcdB-GTD in an apo-like conformation
Tam et al. recently reported that select flavonoids such as phloretin and apigenin inhibit cell rounding caused by TcdA and TcdB (Tam et al., 2015). In vitro tests with purified TcdA and TcdB-GTD indicated that these flavonoids were significantly more potent in inhibiting glucosyltransferase compared to hydrolytic activities. The inhibition was independent of UDP-glucose concentration, suggesting that phloretin and apigenin function as noncompetitive inhibitors of the GTDs. Apigenin is approximately five-fold more effective at inhibiting the glucosyltransferase activity and has comparable solubility with phloretin. To better understand the effects and mechanism of action for these inhibitors, we sought to identify the apigenin binding site(s) in TcdA and TcdB-GTD crystal structures. We performed a series of screens and crystal soaking experiments with apigenin. These included multi-factorial broad screens as well as the replication of previously reported crystallization conditions. Since there was no information as to which conformation apigenin would bind, we set up apo conditions along with trials that contained U2F. This allowed sampling of conditions with TcdB-GTD bound to a substrate, without the complication of hydrolysis.
We obtained structures of TcdA and TcdB-GTD in complex with U2F grown in the presence of apigenin (Table 1). For TcdB-GTD, no differences were observed between the structure containing U2F and the crystal grown in the presence of U2F and apigenin. We observed high occupancy of U2F within the active site. This is consistent with apigenin inhibiting glucosyltransferase activity without competing for UDP-glucose binding. However, there was no unaccounted density which would represent one or more copies of apigenin within the structure.
Despite success in obtaining a high resolution dataset (1.85 Å), we again did not observe apigenin in the TcdA-GTD + U2F + apigenin structure. However, we observed an apo conformation of the W519 loop, despite the presence of U2F in the active site (Figure 5, orange). This everted apo conformation was also observed in the recent TcdA1–1832 crystal structure (Chumbler et al., 2016). We considered that apigenin may interact with W519 in TcdA-GTD in a manner that stabilized this apo conformation. We analyzed Polder maps (a difference map omitting bulk solvent) to scan small areas (up to five contiguous residues) with moderate to high solvent exposure, including the W519 loop. While we did not locate apigenin, the Polder maps indicated positive density similar to an intermediate position of W519 (Figure 5, cyan) from TcdA-GTD bound to UDP (D’Orzo et al., 2012).
Figure 5.
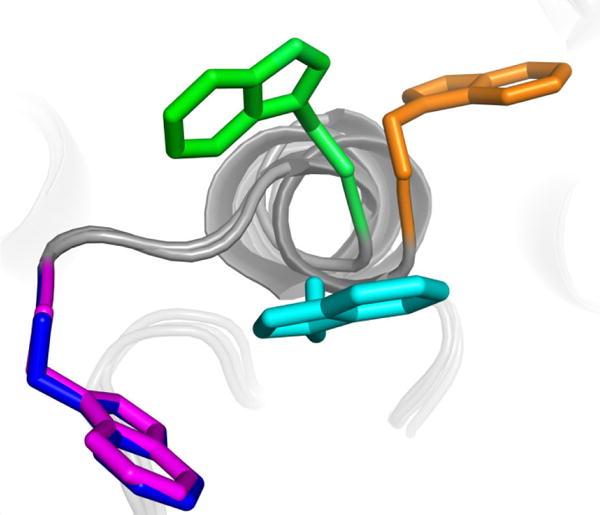
Superimposed structures of W519 in TcdA-GTD with UDP-glucose (blue), apo TcdA-GTD (green), TcdA-GTD with UDP (cyan), and apo TcdA 1–1830 (orange). TcdA-GTD bound to U2F (magenta) or UDP-glucose have nearly identical W519 conformations.
As previously discussed, earlier structures of TcdA have indicated that the conformation of the W519 loop depends on the presence or absence of UDP or UDP-glucose within the active site. TcdB-GTD had only been crystallized in a single conformation, in complex with UDP + glucose. In an effort to better characterize the conformations and flexibility of the TcdB W520 loop, we sought to crystallize TcdB-GTD in an apo form. Crystals of TcdB-GTD alone diffracted poorly (>7 Å) and were difficult to reproduce as single, non-twinned crystals. The inclusion of apigenin in crystal screens with TcdB-GTD yielded several new conditions and a different space group. We collected a 2.8 Å dataset from TcdB-GTD crystals grown without UDP-glucose or U2F, which depended on the presence of apigenin (Table 1). The data were indexed as space group C2221, with a relatively large unit cell (dimensions 92 Å, 154 Å, 271 Å, 90°, 90°, 90°). Molecular replacement led to a solution where two copies of TcdB-GTD were joined along alpha helices 15 and 16 within the asymmetric unit (ASU) (Figure 6A). Overall, both chains within the ASU were highly similar (RMSD of 0.293 Å2) with minor differences at the C-terminus and an unstructured loop bridging two helices (residues 156–171). Both chains within the TcdB-GTD ASU demonstrated apo-like conformations in the W520 loop, though the precise position of W520 differed (Figure 6B).
Figure 6.
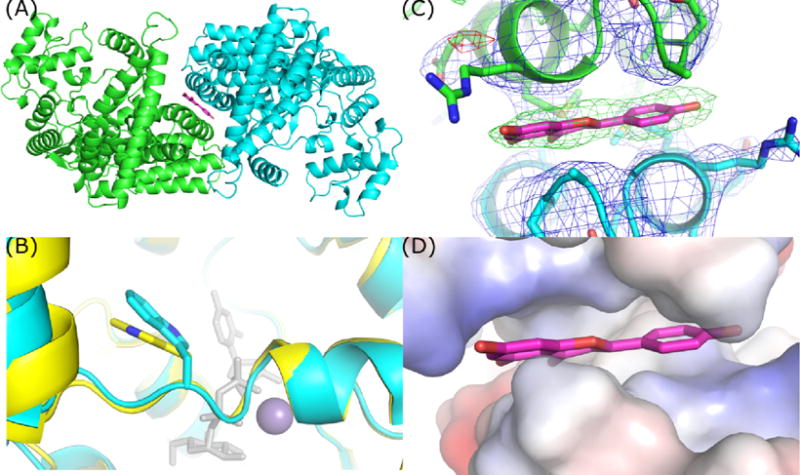
TcdB-GTD crystallized in an apo-like conformation contains two chains within the ASU. (A) Top-down view demonstrating the position of apigenin and non-crystallographic symmetry of the TcdB-GTD chains. (B) Overlay of apo TcdA-GTD (yellow) and TcdB-GTD (cyan) in an apo-like conformation. (C) 2mFo-DFc and Fo-Fc maps of apigenin site contoured at 1 and 4 sigma respectively. (D) Vacuum electrostatic model of the hydrophobic patch occupied by apigenin.
After some iterative refinement, we observed a consistent area of positive density in our difference maps at the junction of the two ASU chains. These differences were coincident with a planar ellipse of density in 2mFo-DFc maps at the same location that looked similar in shape and size to apigenin (Figure 6C). The inclusion of apigenin at this chain-chain interface explains the new crystal form and the dependence of this form on apigenin. In this instance, we propose that apigenin merely fills a hydrophobic void between the GTD chains in the ASU of our TcdB-GTD structure (Figure 6D).
Discussion
The growing number of deposited LCT-GTD structures allows for a more comprehensive analysis of GTD function. The primary difference between apo and UDP-glucose bound states across TcdB, TcsL, TcdA, and Tcnα-GTDs are the conformations of the active site “lid” that includes the conserved 519/520 tryptophan. (Figure 1, magenta). Crystal structures of multiple LCT-GTDs suggest that the position of the tryptophan is related to the presence or absence of substrate.
The relationship between this tryptophan loop and UDP-glucose is clear when comparing structures of TcdA-GTD in apo and UDP-glucose bound forms. In these cases, TcdA-GTD was crystallized in the same space group and the W519 loop was shifted into the active site in the presence of UDP-glucose (Pruitt et al., 2012). TcdA-GTD bound to UDP-glucose adopted a conformation nearly identical to that of TcdB-GTD, though it contains an intact substrate (Figure 5, blue). TcsL-GTD was crystallized in the presence of UDP-glucose and shares the substrate bound conformation with TcdA and TcdB-GTD (Ziegler et al., 2008). Tcnα-GTD, however, was determined in an apo state. The apo form of TcdA-GTD is similar to that of Tcnα-GTD, where the loop tryptophan was rotated outside of the active site (Figure 5, green). Two additional TcdA-GTD structures show the conserved W519 in identical conformations under apo and UDP-bound conditions (Figure 5, cyan) (D’Orzo et al., 2012). This conformation could represent an intermediate state between the apo and substrate-bound forms. Importantly, the absence of glucose in the UDP-bound structure suggests that UDP is insufficient to stabilize the loop and tryptophan within the active site. This correlates well with the conformation of W520 in TcdB-GTD in complex with hydrolyzed UDP-glucose. In this case, the glucose was retained in the active site and likely stabilizes W520 in its inward, substrate-bound conformation.
Conclusion
In this study, we report new crystal structures of C. difficile toxin glucosyltransferase domains obtained using a non-hydrolyzable UDP-glucose analog, U2F, some in the presence of apigenin—a reported inhibitor of TcdA and TcdB-GTDs. We found that despite the hydroxyl to fluorine substitution in the synthetic compound, the active site and sugar selection residues remained unchanged in both proteins. While the inclusion of apigenin was required for crystallizing an apo-like form of TcdB-GTD, the molecule was not observed in the other crystals, and the mechanism for this inhibitor remains unclear. In determining these new structures of TcdA and TcdB-GTD we observed disparate conformations of the active site tryptophan. These conformations add to the existing group of structures where a conserved tryptophan samples a broad conformational space in the context of a comparatively static active site.
Acknowledgments
Funding: This work was supported by the National Institutes of Health grants AI095755, T32 GM008320, and Department of Veterans Affairs grant BX002943.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H, Pfeifer G, Hofmann F, Maier E, Benz R, Aktories K. Low ph-induced Formation of Ion Channels by Clostridium difficile Toxin B in Target Cells. J Biol Chem. 2001;276:10670–10676. doi: 10.1074/jbc.M009445200. [DOI] [PubMed] [Google Scholar]
- Breton C, Šnajdrová L, Jeanneau C, Koča J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006;16:29–37. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- Busch C, Hofmann F, Gerhard R, Aktories K. Involvement of a conserved tryptophan residue in the UDP-glucose binding of large clostridial cytotoxin glycosyltransferases. J Biol Chem. 2000;275:13228–13234. doi: 10.1074/jbc.275.18.13228. [DOI] [PubMed] [Google Scholar]
- Busch C, Hofmann F, Selzer J, Munro S, Jeckel D, Aktories K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J Biol Chem. 1998;273:19566–19572. doi: 10.1074/jbc.273.31.19566. [DOI] [PubMed] [Google Scholar]
- Chaves-Olarte E, Weidmann M, Von Eichel-Streiber C, Thelestam M. Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J Clin Invest. 1997;100:1734–1741. doi: 10.1172/JCI119698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumbler NM, Rutherford SA, Zhang Z, Farrow MA, Lisher JP, Farquhar E, Giedroc DP, Spiller BW, Melnyk RA, Lacy DB. Crystal structure of Clostridium difficile toxin. A Nat Microbiol. 2016;1:1–6. doi: 10.1038/nmicrobiol.2015.2. [DOI] [PubMed] [Google Scholar]
- Ciesla WP, Bobak DA. Clostridium difficile toxins A and B are cation-dependent UDP-glucose hydrolases with differing catalytic activities. J Biol Chem. 1998;273:16021–16026. doi: 10.1074/jbc.273.26.16021. [DOI] [PubMed] [Google Scholar]
- D’Orzo N, Malito E, Biancucci M, Bottomley MJ, Maione D, Scarselli M, Martinelli M. The structure of Clostridium difficile toxin A glucosyltransferase domain bound to Mn 2+ and UDP provides insights into glucosyltransferase activity and product release. FEBS J. 2012;279:3085–3097. doi: 10.1111/j.1742-4658.2012.08688.x. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson RP, Tarling CA, Roberts S, Withers SG, Davies GJ. The donor subsite of trehalose-6-phosphate synthase: Binary complexes with udp-glucose and udp-2-deoxy-2-fluoro-glucose at 2 Å resolution. J Biol Chem. 2004;279:1950–1955. doi: 10.1074/jbc.M307643200. [DOI] [PubMed] [Google Scholar]
- Hiromoto T, Honjo E, Tamada T, Noda N, Kazuma K, Suzuki M, Kuroki R. Crystal structure of UDP-glucose:anthocyanidin 3-O-glucosyltransferase from Clitoria ternatea. J Synchrotron Radiat. 2013;20:894–898. doi: 10.1107/S0909049513020712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Herrmann A, Habermann E, Von Eichel-Streiber C. Sequencing and analysis of the gene encoding the α-toxin of Clostridium novyi proves its homology to toxins A and B of Clostridium difficile. Mol Gen Genet. 1995;247:670–679. doi: 10.1007/BF00290398. [DOI] [PubMed] [Google Scholar]
- Jank T, Aktories K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008;16:222–229. doi: 10.1016/j.tim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Jank T, Giesemann T, Aktories K. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J Biol Chem. 2007;282:35222–35231. doi: 10.1074/jbc.M703138200. [DOI] [PubMed] [Google Scholar]
- Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995 doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS Acta Crystallogr. Sect D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahama M, Oda M, Kobayashi K. Glycosylating Toxin of Clostridium perfringens. Glycosylation. 2012 doi: 10.5772/48112. [DOI] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997 doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, Strynadka NC. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat Struct Biol. 2001;8:166–175. doi: 10.1038/84168. [DOI] [PubMed] [Google Scholar]
- Pruitt RN, Chumbler NM, Rutherford SA, Farrow MA, Friedman DB, Spiller B, Lacy DB. Structural determinants of Clostridium difficile toxin A glucosyltransferase activity. J Biol Chem. 2012;287:8013–8020. doi: 10.1074/jbc.M111.298414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke J, Tenzer S, Rupnik M, Koschinski A, Hasselmayer O, Schrattenholz A, Schild H, von Eichel-Streiber C. Autocatalytic cleavage of Clostridium difficile toxin B. Nature. 2007;446:415–419. doi: 10.1038/nature05622. [DOI] [PubMed] [Google Scholar]
- Reinert DJ, Jank T, Aktories K, Schulz GE. Structural basis for the function of Clostridium difficile toxin. B J Mol Biol. 2005;351:973–981. doi: 10.1016/J.Jmb.2005.06.071. [DOI] [PubMed] [Google Scholar]
- Schrödinger L. The PyMOL Molecular Graphics System. Version. 2015;1:8. [Google Scholar]
- Spyres LM, Daniel J, Hensley A, Qa’Dan M, Ortiz-Leduc W, Ballard JD. Mutational analysis of the enzymatic domain of Clostridium difficile toxin B reveals novel inhibitors of the wild-type toxin. Infect Immun. 2003;71:3294–3301. doi: 10.1128/IAI.71.6.3294-3301.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Beilhartz GL, Auger A, Gupta P, Therien AG, Melnyk RA. Small molecule inhibitors of Clostridium difficile toxin B-induced cellular damage. Chem Biol. 2015;22:175–85. doi: 10.1016/j.chembiol.2014.12.010. [DOI] [PubMed] [Google Scholar]
- Teichert M, Tatge H, Schoentaube J, Just I, Gerhard R. Application of mutated Clostridium difficile toxin A for determination of glucosyltransferase-dependent effects. Infect Immun. 2006;74:6006–6010. doi: 10.1128/IAI.00545-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman AW, Dias MVB, Wu S, Blundell TL, Huang F, Spencer JB. Chimeric Glycosyltransferases for the Generation of Hybrid Glycopeptides. Chem Biol. 2009;16:676–685. doi: 10.1016/j.chembiol.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Ziegler MOP, Jank T, Aktories K, Schulz GE. Conformational Changes and Reaction of Clostridial Glycosylating Toxins. J Mol Biol. 2008;377:1346–1356. doi: 10.1016/j.jmb.2007.12.065. [DOI] [PubMed] [Google Scholar]