

Abstract
Type 2 diabetes is increasingly recognized as a risk factor for Alzheimer’s disease (AD), but the underlying mechanisms remain poorly understood. Hyperphosphorylation of the microtubule-associated protein tau has been reported in rodent models of diabetes, including db/db mice, which exhibit insulin resistance and chronically elevated glucocorticoids due to leptin receptor insufficiency. In this report, we investigated endocrine mechanisms for hippocampal tau phosphorylation in db/db and wildtype (Wt) mice. By separately manipulating peripheral and intrahippocampal corticosterone levels, we determined that hippocampal corticosteroid exposure promotes tau phosphorylation and activates glycogen synthase kinase 3β (GSK3β). Subsequent experiments in hippocampal slice preparations revealed evidence for a nongenomic interaction between glucocorticoids and GSK3β. To examine whether GSK3β activation mediates tau phosphorylation and impairs memory in diabetes, db/db and Wt mice received intrahippocampal infusions of TDZD-8, a non-ATP competitive thiadiazolidinone inhibitor of GSK3β. Intrahippocampal TDZD-8 blocked tau hyperphosphorylation and normalized hippocampus-dependent memory in db/db mice, suggesting that pathological synergy between diabetes and AD may involve glucocorticoid-mediated activation of GSK3β.
Keywords: Insulin resistance, diabetes, hippocampus, tau phosphorylation, learning and memory, corticosterone
Graphical abstract
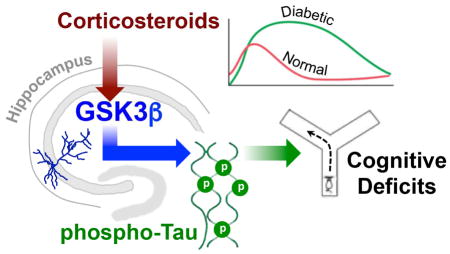
1. Introduction
Insulin resistant diabetes is characterized by increases in circulating glucose and insulin levels, leading to reduced sensitivity to insulin among multiple somatic tissues including the brain. This condition arises from obesity, sedentary lifestyle, and genetic predisposition (Lazar, 2005). Age is also a risk factor for insulin resistance, with older adults developing type 2 diabetes at a higher rate than middle-aged or younger populations (Defronzo, 1981). Aging is often accompanied by cognitive decline, specifically on tasks that recruit the hippocampus and associated cortical structures (Burke and Barnes, 2010). Susceptibility to cognitive decline is greater among insulin resistant individuals (Velayudhan et al., 2010, Ott et al., 1996). Insulin resistance is associated with cognitive impairment among middle aged (Sandeep et al., 2004) and aged individuals without dementia (Convit et al., 2003; Reynolds et al., 2010). Changes in brain structure in insulin resistant diabetes occur within temporal lobe circuits that are also sensitive to aging and Alzheimer’s disease (AD; Bishop et al., 2010). Individuals with insulin resistant diabetes exhibit hippocampal atrophy (Rasgon et al., 2011; Bruehl et al., 2009; Wu et al., 2008), and impairment of resting-state connectivity between the hippocampus and neocortical regions implicated in cognitive function (Zhou et al., 2010). Taken together with behavioral evidence that diabetes has a negative impact on cognition, the neuroimaging data suggest that similar negative outcomes occur during diabetes and brain aging.
Tangles composed of hyperphosphorylated tau protein are a major clinical hallmark of Alzheimer’s disease (AD), the most common form of age-related dementia (Wang and Mandelkow, 2016). Individuals with type 2 diabetes are at increased risk for developing dementia, including AD (Biessels and Reagan, 2015), but the mechanisms underlying this increased susceptibility are not yet fully understood. In two separate studies by independent groups, analysis of tau phosphorylation in human brain samples from individuals with AD, diabetes, or both conditions revealed that diabetes exacerbates tau pathology (Liu et al., 2011; Valente et al., 2010). Changes in glucocorticoid levels and rhythmicity are a shared feature in rodent models of AD (Green et al., 2006; Baglietto-Vargas et al., 2013), and rodent models of type 2 diabetes (Stranahan et al., 2008). Lowering corticosterone levels reduces plaque burden and tau pathology in a triple-transgenic mouse model of AD (Green et al., 2006). Similar commonalities are evident in humans, where inhibition of cortisol synthesis or bioactivity attenuates memory deficits in aged individuals and middle-aged diabetics (Lupien et al., 2005; Sandeep et al., 2004). These findings point to a possible mechanistic relationship between glucocorticoids and AD pathology in diabetes.
Leptin receptor deficient (db/db) mice exhibit insulin resistance and hyperglycemia (Hummel et al., 1966), as well as increases in hippocampal tau phosphorylation and activation of glycogen synthase kinase 3β, a prominent tau kinase (GSK3β; Kim et al., 2009). db/db mice also exhibit elevated levels of corticosterone (Takeshita et al., 2000; Stranahan et al., 2008), but the potential relationship between elevated glucocorticoids and tau phosphorylation has yet to be explored in this model. To investigate possible associations between hippocampal corticosteroid exposure, GSK3β activation, and tau phosphorylation, we selectively manipulated hippocampal and peripheral corticosterone levels using intrahippocampal corticosterone infusions and pharmacological inhibition of adrenal steroidogenesis in db/db and Wt mice. After observing that increases in hippocampal corticosterone were necessary and sufficient for tau phosphorylation and GSK3β activation in these experiments, we investigated whether hippocampal glucocorticoids activate GSK3β via nongenomic mechanisms. To examine the role of GSK3β in tau phosphorylation and memory impairment in vivo, db/db and Wt mice received chronic intrahippocampal infusions of TDZD-8, a selective inhibitor of GSK3β. These experiments revealed that GSK3β-mediated tau phosphorylation underlies memory deficits and hippocampal tau phosphorylation in db/db mice. Additional work will be required to determine whether corticosteroids similarly promote tangle formation in htau mutant mice with diabetes, but the current findings provide a compelling rationale for further investigation into pathological synergy between diabetes and AD pathology.
2. Materials and Methods
2.1. Animals and systemic drug treatment
Male C57Bl6J and B6.Leprdb/J (db/db) homozygous mice were obtained from Jackson Laboratories at 5 weeks of age. Upon arrival in the animal facility, mice were housed three per cage at 28°C and 55% humidity. Following one week of acclimation, mice were treated with metyrapone (100 mg/kg, IP; Tocris Bioscience, Bristol, UK) or vehicle (saline with 20% polyethylene glycol) for two weeks to lower and normalize corticosterone levels, as described (Wosiski-Kuhn et al., 2014; Dey et al., 2014). Metyrapone was administered daily between 08:00 and 10:00 (lights-on at 06:00) and mice were euthanized by decapitation under Isoflurane anesthetic two hours after the final injection. For initial experiments, (n=6) mice were generated in each of the following conditions: vehicle-treated Wt, vehicle-treated db/db, metyrapone-treated Wt, metyrapone-treated db/db. Mice were weighed daily and body temperatures were monitored via rectal probe (World Precision Instruments). In some experiments, peripheral injections of metyrapone or vehicle were administered to mice receiving intrahippocampal infusions of corticosterone, as described below. For these experiments, (n=21) Wt and (n=22) db/db mice were randomly assigned to one of the following conditions: intrahippocampal artificial cerebrospinal fluid (ACSF)/systemic vehicle; intrahippocampal corticosterone/systemic vehicle; intrahippocampal ACSF/systemic metyrapone; intrahippocampal corticosterone/systemic metyrapone. All procedures were approved by the Augusta University Animal Care and Use Committee and followed NIH guidelines.
2.2. Stereotaxic surgery and osmotic pump implantation
For stereotaxic surgery, mice were anesthetized with Isoflurane and implanted with bilateral cannulae (Plastics One) at the following coordinates (in mm): anteroposterior, −2.1; mediolateral, ±1.5; dorsoventral, 2.1 (Paxinos and Franklin, 2001). Cannulae were connected to Alzet minipumps (Durect Corporation, Cupertino, CA, USA) that had been primed for 24hr at 37°C in sterile saline. For intrahippocampal corticosterone infusions, a water-soluble complex of corticosterone and 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich, St. Louis, MO, USA) was delivered at 60ng/24hr for two weeks, as described (Wosiski-Kuhn et al., 2014). For intrahippocampal GSK3β inhibition, Alzet pumps were filled with TDZD-8, a non-ATP-competitive selective inhibitor of GSK3β (Martinez et al., 2002). We generated (n=8) mice in each of the following conditions: Wt-Vehicle, db/db-Vehicle, Wt-TDZD8, db/db-TDZD-8. TDZD-8 was delivered at a rate of 2μM/24hr for two weeks, with behavioral testing three days prior to euthanasia as described below.
2.3. Spatial and object recognition memory
Behavioral testing in the Y-maze and novel object preference task was conducted at the onset of the dark cycle (18:00–20:00) under red light illumination, as described (Erion et al., 2014; Wosiski-Kuhn et al., 2014). For the Y-maze, testing was initiated after a ten-minute habituation session where the mouse was confined to the start arm of the maze. Test sessions began when the guillotine door at the end of the start arm was lifted to allow free exploration, and individual sessions did not end until the mouse made a complete arm entry, defined by entry past the base of the tail. After a complete entry, the mouse was again confined to the start arm for a 1min inter-trial interval before the next choice. This sequence was carried out five times and the number of correct alternations was expressed relative to the number of trials for statistical comparison. For novel object preference testing, mice received a 10min acclimation trial in the test arena with no objects present. After the acclimation period, two identical objects were introduced and the mouse was videotaped during free exploration. After the initial training trial., the mouse was returned to the home cage for 30min before being reintroduced to the arena with one familiar and one novel object. Exploration of the familiar and novel objects was videotaped for 5min, with additional trials at 2hr and 24hr after training. Videos were analyzed offline by an experimenter blind to treatment condition using Tracker, a freely available video analysis tool (physlets.org/tracker).
2.4. Protein extraction and western blotting
Hippocampi were homogenized in 10x (wt/vol) modified RIPA buffer containing protease and phosphatase inhibitors, as described (Erion et al., 2014; Wosiski-Kuhn et al., 2014). In brief, lysates were centrifuged at 14,000xg for 15min at 4C and protein content in supernatants was determined using Bradford assay. For gel electrophoresis, samples denatured at 90C for 5min, separated on 10–20% Bis-Tris gels, and transferred to nitrocellulose membranes, as previously reported (Erion et al., 2014; Wosiski-Kuhn et al., 2014). For detection of total and phosphorylated tau, membranes were blocked in 20% SuperBlock (Pierce) in TTBS before overnight incubation with rabbit anti-total tau (Dako Cytomation) and mouse monoclonal antibody CP13, which detects tau phosphorylation at Ser202, or PHF-1, which detects tau phosphorylation at serine 396/404 (both antibodies were a kind gift from Dr. Peter Davies, Yeshiva University). The next day, membranes were washed and incubated with near-infrared fluorescent secondary antibodies directed against mouse (IRDye 680RD) or rabbit (IRDye 800CW; LiCor, Lincoln, NE USA) for 1hr at room temperature. For GSK3β, membranes were blocked in 5% bovine serum albumin (BSA) in Tris-buffered saline containing 0.5%Tween-20 (TTBS) before overnight incubation at 4C with mouse monoclonal anti-GSK3β and a rabbit monoclonal antibody directed against Ser9-phosphorylated GSK3β (1:1,000; Cell Signaling Technology, Danvers, MA USA). Bound primary antibodies were detected using near-infrared fluorescent secondary antibodies before imaging and analysis as described above. After the secondary antibody incubation, membranes were imaged on a LiCor Odyssey Fc imager and band intensities were determined using Image Studio software in a blinded manner. Phospho-tau and pS9GSK3β band intensities were expressed relative to total tau or total GSK3β for subsequent statistical comparisons.
2.5. Enzyme-linked immunosorbent assay
Quantification of total tau by enzyme-linked immunosorbent assay (ELISA) was carried out using a commercially available kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Analysis of tau phosphorylation by ELISA followed previously published protocols (Acker et al., 2013), with modifications. Specifically, high-binding 96-well plates (Immulon 4HBX, Thermo Scientific) were coated with CP13 or PHF-1 antibodies diluted at 6μg/mL in coating buffer (25mM NaHCO3, 25mM Na2CO3; pH=9.7). After 48hr at 4C, wells were washed and blocked using a commercially available blocking buffer (DY995, R&D Systems). Hippocampal homogenates were diluted in blocking buffer, loaded into duplicate wells coated with capture antibody, and incubated overnight at 4C. The next day, wells were washed and incubated with a monoclonal antibody against total tau (DA9; 2μg/mL) for detection of phospho-tau bound to CP13 or PHF-1. Following 2hr incubation with DA9, wells were washed, and the bound antibody complex was amplified with HRP-conjugated goat anti-mouse (Vector Labs; 2μg/mL) for 30min. After additional washes, wells were chromated in TMB One Substrate Solution (R&D Systems) for 10min. The color reaction was stopped with 2M H2SO4 and plates were read at 450nm on a SpectraMax plate reader (Molecular Probes). Phosphorylated tau in homogenates were determined by linear regression using standard curves generated from wells with increasing concentrations of recombinant PHF-tau (Millipore).
2.6. Immunofluorescence
To generate brain sections for immunofluorescence, a subset of db/db and Wt mice from the intrahippocampal TDZD-8 experiments were transcardially perfused with 4% paraformaldehyde, as previously reported (Erion et al., 2014; Wosiski-Kuhn et al., 2014). Brains were cryoprotected and sectioned coronally at 40 micron thickness on a freezing microtome (Leica). Sections were stored at −20 in cryoprotectant before being washed and processed for phospho-tau and total tau immunofluorescence, as reported previously (Stranahan et al., 2011). In brief, sections were quenched in sodium borohydride, blocked in 5% milk, and reacted overnight with mouse monoclonal antibody CP13 (1:200; kind gift of Dr. Peter Davies) and rabbit polyclonal anti-total tau (1:500, Dako Cytomation). The following day, sections were washed and reacted with Alexa fluor-conjugated secondary antibodies (Invitrogen, Molecular Probes, Carlsbad, CA) before being mounted on slides, dried, and counterstained with DAPI. Slides were assigned a numeric code after completion of the immunofluorescence reaction and the code was not broken until completion of the study. Confocal micrographs were acquired through a 25X oil immersion objective on a Zeiss LSM510Meta upright microscope. Micrographs were qualitatively scored, with 0 indicating no phospho-tau labeling, 1 indicating labeling in the CA3 stratum radiatum only, and 2 indicating labeling in the CA3 stratum radiatum and stratum pyramidale.
2.7. Statistics
For experiments involving systemic treatment with metyrapone or vehicle, measures of hippocampal protein expression and serum glucocorticoids were analyzed using 2 × 2 ANOVA with genotype and drug treatment as fixed factors. For experiments that combined intrahippocampal corticosteroids with systemic metyrapone treatment, data were analyzed using within-genotype 2 × 2 ANOVA (systemic drug treatment and intrahippocampal corticosteroid treatment as fixed factors). The effect of glucocorticoids and transcriptional repression on activation of GSK3β in acute slices was also analyzed using 2 × 2 ANOVA. Two-way ANOVA was also applied for statistical comparison of protein expression and spatial alternation following intrahippocampal GSK3β inhibition. In the novel object recognition paradigm, data were analyzed using 2×2 repeated-measures ANOVA. For all endpoints, statistical significance was set at p<0.05 and post hoc tests were conducted using t-tests with Bonferroni’s correction for multiple comparisons. Statistical analyses were conducted in PASW statistics or Graphpad Prism 5.0.
3. Results
3.1. Lowering corticosterone prevents tau phosphorylation and GSK3β activation in diabetic mice
Systemic treatment with the glucocorticoid synthesis inhibitor metyrapone effectively lowered circulating corticosterone levels in db/db mice (Figure 1A; F1,20=7.8, p<0.05). Metyrapone treatment also reduced hippocampal tau phosphorylation at serine 202 (Figure 1B; F1,20 = 16.52, p<0.01) and serine 396/404 (Figure 1C; F1,20 = 15.36, p<0.01). Hippocampal tau phosphorylation has previously been linked with impaired body temperature regulation in leptin receptor mutant mice (El Khoury et al., 2016). The metabolic phenotype of the db/db mutation varies between background strains (Hummel et al., 1972; Mao et al., 2006), and is less severe on the C57Bl6 background used in the current studies, relative to the BKS background used by El Khoury et al (2016). Consistent with this milder physiological phenotype, there was no effect of genotype or corticosteroid inhibition on rectal temperature in the current experiments (°C, mean ± sem: Wt/Veh = 37.1 ± 0.2; db/Veh = 36.7 ± 0.3; Wt/Met = 37.0 ± 0.3, db/Met = 36.9 ± 0.3).
Figure 1. Metyrapone treatment prevents tau phosphorylation and GSKβ activation in the db/db mouse hippocampus.

(A), Systemic metyrapone treatment reduces circulating corticosterone levels in db/db mice. (B), Western blotting demonstrated that lowering corticosterone levels with metyrapone reduces phosphorylation of tau at serine 202 in db/db mice. (C), Glucocorticoid inhibition also blocked tau phosphorylation at S396/404 in db/db mice. (D), Metyrapone prevented dephosphorylation and activation of GSK3β in whole hippocampal homogenates from db/db mice. For all graphs, bar height represents the average from (n=6) mice in each condition, error bars represent the sem, and asterisks (*) denote statistical significance at p<0.05 by 2×2 ANOVA.
Glucocorticoid exposure dephosphorylates and activates the tau kinase GSK3β in non-diabetic animals (Beck et al., 2009), therefore we next examined hippocampal GSK3β activation in db/db mice treated with metyrapone to determine whether corticosterone activates GSK3β in diabetes. Vehicle-treated db/db mice exhibit increased hippocampal GSK3β activation, based on reduced levels of pS9GSK3β in hippocampal homogenates (F1,20 = 11.60, p<0.01). Metyrapone treatment eliminated reductions in hippocampal pS9GSK3β in db/db mice (Figure 1D), without affecting pS9GSK3β in Wt mice, indicating that increases in circulating corticosterone dephosphorylate and activate GSK3β in obesity and diabetes.
3.2. Hippocampal glucocorticoid exposure activates GSK3β and promotes tau phosphorylation
Systemic metyrapone treatment reduces glucocorticoids, but systemic reductions in corticosterone also attenuate hyperglycemia in db/db mice (Dey et al., 2014). To address whether increases in hippocampal corticosterone induce tau phosphorylation, we independently manipulated central and peripheral corticosterone levels in Wt and db/db mice (Figure 2A). Successful intrahippocampal delivery of corticosterone via bilateral cannulae and Alzet minipumps was confirmed by ELISA in solvolytic extracts, as previously reported (Wosiski-Kuhn et al., 2014). In Wt mice, chronic intrahippocampal corticosterone increased tau phosphorylation, as determined by sandwich ELISA (Acker et al., 2013). This effect was comparable in Wt mice treated with metyrapone or vehicle, and was detected at both pS202 (Figure 2B; F1,16=12.97, p<0.01) and pS396/404 (Figure 2C; F1,16=18.40, p<0.01). In metyrapone-treated db/db mice, reductions in tau phosphorylation were blocked by intrahippocampal corticosterone (Figure 2B–C; for pS202, F1,16=5.39, p<0.05; for pS396/404, F1,16=6.27, p<0.05).
Figure 2. Intrahippocampal exposure to elevated corticosterone levels promotes tau phosphorylation and activates GSKβ.

(A), Schematic represents experimental design for independent manipulation of central and peripheral corticosterone levels. (B), Analysis of pS202-tau by ELISA revealed that local increases in corticosterone promote hippocampal tau phosphorylation in both genotypes. (C), Intrahippocampal corticosterone infusions also promote accumulation of pS396/404-tau, independently of systemic glucocorticoid inhibition. (D), Hippocampal glucocorticoid exposure dephosphorylates and activates GSK3β in whole hippocampal homogenates. For all graphs, bar height represents the average from (n= 5–6) mice in each condition, error bars represent the sem, and asterisks (*) denote statistical significance at p<0.05 by 2×2 ANOVA with Bonferroni-corrected planned post hoc comparisons.
To examine the effects of hippocampal glucocorticoids on GSK3β activation, we quantified total and pS9GSK3β by western blotting. In vehicle-treated Wt mice, hippocampal corticosterone infusion significantly reduced pS9GSK3β, indicative of dephosphorylation and activation following intrahippocampal corticosterone (Figure 2D; F1,16=12.07, p<0.01). Corticosterone-mediated activation of GSK3β was also detected in Wt mice treated systemically with metyrapone (Figure 2D). In metyrapone-treated db/db mice, intrahippocampal corticosterone blocked increases in pS9GSK3β (Figure 2E, F1,16=16.91, p<0.01), suggesting that inactivation of GSK3β by systemic glucocorticoid inhibition requires local reductions in hippocampal corticosterone.
3.3. Nongenomic activation of GSK3β by glucocorticoids in hippocampal slices
To determine whether corticosteroids regulate GSK3β via genomic or nongenomic mechanisms, we applied the synthetic glucocorticoid dexamethasone and the transcriptional inhibitor actinomycin D (ActD) to hippocampal slice preparations from Wt mice (Figure 3A). Acute slices were prepared from adrenally intact Wt mice, and serum corticosterone levels were within the expected range for the circadian trough (ng/mL, mean ± sem: 107.1±11.8). After incubation with dexamethasone, ActD, or dexamethasone plus ActD, hippocampi were excised and frozen for protein extraction and quantification of pS9GSK3β (Figure 3B). These experiments revealed that dexamethasone dephosphorylates and activates GSK3β (Figure 3C; F1,16=10.26, p<0.01). Because GSK3β activation was unaffected by inhibition of gene transcription with ActD (Figure 3C), it is likely that hippocampal glucocorticoid exposure activates GSK3β via nongenomic mechanisms.
Figure 3. Nongenomic activation of GSK3β by hippocampal glucocorticoid exposure.

(A), Experimental design for glucocorticoid exposure and transcriptional inhibition in acute slices. (B), Representative western blot membrane probed with antibodies to inactive (pS9) and total GSK3β. (C), Dephosphorylation and activation of GSK3β by the synthetic glucocorticoid dexamethasone (Dex) is unaffected by inhibiting transcription with actinomycin D (ActD), indicative of a nongenomic interaction. For all graphs, bar height represents the average of (n= 6) mice in each condition, error bars represent the sem, and asterisks (*) denote statistical significance at p<0.05 by 2×2 ANOVA with Bonferroni-corrected planned post hoc comparisons.
3.4. Hippocampal GSK3β inhibition rescues learning and memory in db/db mice
To examine the role of hippocampal GSK3β in cognitive dysfunction, groups of db/db and Wt mice received intrahippocampal infusions of TDZD-8, a thiadiazolidinone (TDZD) inhibitor with high selectivity for GSK3β (Martinez et al., 2002). Unlike previous generations of ATP-competitive GSK3β inhibitors, members of the TDZD family directly interfere with dephosphorylation and activation of GSK3β (Serenó et al., 2009) and this was evident in hippocampal lysates from the current experiments (Figure 4A). TDZD-8 blocked activation of GSK3β in db/db mice, based on increased pS9GSK3β in hippocampal protein extracts (Figure 4B; F1,22=14.76, p<0.01). Intrahippocampal treatment with TDZD-8 had no effect on body weight, food intake, or serum glucose levels (Supplementary Figure 1A–C), indicating that hippocampal GSK3β inhibition did not attenuate obesity and diabetes in db/db mice.
Figure 4. Hippocampal GSK3β inhibition rescues spatial memory in db/db mice.

(A), Top panel shows experimental design for chronic intrahippocampal delivery of TDZD-8, a non-ATP-competitive thiadiazolidinone inhibitor with high selectivity for GSK3β (Martinez et al., 2002). Bottom panel shows representative western blot membrane probed with antibodies against active (pS9) and total GSK3β. (B), Quantification of band intensities revealed that intrahippocampal delivery of TDZD-8 blocked dephosphorylation and activation of GSK3β in db/db mice. (C), Hippocampal GSK3β inhibition normalizes spatial recognition memory in db/db mice, based on increased alternation in the Y-maze. (D), Deficits in novel object exploration time at early post-training intervals in db/db mice were blocked by intrahippocampal TDZD-8. (E), Persistent, GSK3β-mediated reductions in novel object preference were evident at longer retention intervals in db/db mice based on comparison of exploration bout number. For all graphs, bar height represents the mean of (n=7–8) mice in each condition, error bars represent the sem, and asterisks (*) denote statistical significance at p<0.05 by 2×2 ANOVA (B–C) or 2×2 repeated measures ANOVA (D–E).
Spatial alternation behavior in the Y-maze is impaired by hippocampal lesions (Conrad et al, 1996), and we previously demonstrated that db/db mice exhibit deficits in spatial working memory using this paradigm (Erion et al, 2014; Wosiski-Kuhn et al, 2014). Consistent with these previous reports, db/db mice exhibit reduced alternation in the Y-maze (Figure 4C; effect of genotype, F1,22=7.63, p<0.01). Intrahippocampal treatment with TDZD-8 normalized alternation behavior in db/db mice, without influencing performance in Wt mice (genotype × drug interaction, F1,22=9.37, p<0.01). Although locomotor speed was reduced in both vehicle- and TDZD-8-treated db/db mice (effect of genotype, F1,22=9.65, p<0.01; cm/sec, mean±sem: Wt-Veh = 1.2±0.03; db/db-Veh = 0.78±0.03; Wt-TDZD8 = 1.18±0.03; db/db-TDZD8 = 0.72±0.04), the test paradigm used in these experiments requires a complete arm entry in every trial, with no maximum trial duration. db/db mice took more time to complete each trial than Wt mice (effect of genotype, F1,22=6.21, p<0.05), and there was no effect of intrahippocampal TDZD-8 on trial duration (mean±sem: Wt-Veh = 1.3 ± 0.4; db/db-Veh = 3.7 ± 0.5; Wt-TDZD8 = 1.4±0.4; db/db-TDZD8 = 3.8 ± 0.5). Given that successful alternation was independent of reduced locomotion in TDZD-8-treated db/db mice, it is likely that dissociable systems underlie cognitive impairment and locomotor suppression in this model.
db/db mice exhibit reductions in object recognition memory, in addition to deficits in spatial working memory (Stranahan et al, 2008). To determine whether hippocampal GSK3β activation impairs object recognition memory in db/db mice, we measured novel object preference at increasing intervals after training with two identical objects. Analysis of novel object exploration relative to total object exploration revealed that hippocampal GSK3β inhibition rescued preference for the novel object thirty minutes after the initial training session in db/db mice (Figure 4D; F1,22=3.26, p<0.05). There were no differences in total time spent exploring both objects in any of the trials (Supplementary Figure 2A), and latency to approach the objects was similar in all groups of mice (Supplementary Figure 2B). However, db/db and Wt mice exhibited different patterns of object exploration during each trial. Consistent with their hypoactive phenotype (Stranahan et al, 2009), db/db mice explored the stimulus objects less frequently (effect of genotype, F1,22=26.51, p<0.01; Supplementary Figure 2C), with commensurate increases in the average duration of exploratory bouts, relative to Wt mice (F1,22=8.51, p<0.01; Supplementary Figure 2D). To control for differences in the temporal pattern of object exploration, we quantified the number of novel object exploration bouts relative to total exploratory bout number during each trial. This measure revealed that reductions in novel object preference in vehicle-treated db/db mice persist at longer retention intervals (F1,22=11.95, p<0.01), and were eliminated by hippocampal GSK3β inhibition (Figure 4E). Taken together, these patterns demonstrate that activation of GSK3β impairs hippocampus-dependent spatial and object recognition memory in db/db mice.
3.5. GSK3β-mediated phosphorylation of tau in db/db mice
To determine whether GSK3β activation is required for tau phosphorylation in db/db mice, we quantified pS202- and pS396/404-Tau after intrahippocampal infusions of TDZD-8. Western blotting revealed that intrahippocampal GSK3β inhibition blocked accumulation of pS202-Tau in db/db mice, without affecting Wt mice (Figure 5A; F1,22=5.58, p<0.01). GSK3β phosphorylates tau at multiple sites, including at the pS396/404 epitope detected by antibody PHF-1 (Cavallini et al., 2013). Quantification of pS396/404-Tau with antibody PHF-1 revealed that hippocampal GSK3β inhibition prevents tau phosphorylation at multiple epitopes in db/db mice (Figure 5B; F1,22=7.22, p<0.01).
Figure 5. GSK3β activation is required for hippocampal tau phosphorylation in db/db mice.

(A), Top panel shows representative western blot membrane probed with antibodies against pS202Tau and total tau. Analysis of band intensities (bottom graph) revealed that intrahippocampal delivery of TDZD-8 prevented accumulation of pS202Tau in db/db mice. (B), Top panel shows western blotting for pS396/404Tau in hippocampal protein extracts. Quantification of band intensities revealed that blocking GSK3β prevented hyperphosphorylation of pS396/404Tau in db/db mice. For all graphs, bar height represents the mean of (n=7–8) mice in each condition, error bars represent the sem, and asterisks (*) denote statistical significance at p<0.05 by 2×2 ANOVA.
GSK3β-mediated phosphorylation of tau leads to misfolding and aggregation in the somatodendritic compartment (Wang and Mandelkow, 2016). To examine the cellular localization of phosphorylated tau in these experiments, we performed immunofluorescence labeling for pS202Tau and total tau in hippocampal sections (Figure 6). These experiments revealed prominent accumulation of pS202Tau in the stratum radiatum of hippocampal area CA3 in db/db mice (Figure 6B). pS202 and total tau immunofluorescence were also detected in the CA3 pyramidal cell layer in db/db mice, suggestive of somatodendritic accumulation (Figure 6B). Changes in the amount and the anatomical distribution of pS202Tau immunoreactivity were observed in db/db mice receiving intrahippocampal vehicle, but not in db/db mice that received TDZD-8 (Figure 6D). These results indicate that GSK3β-mediated phosphorylation of tau may be a shared pathological mechanism in diabetes and AD.
Figure 6. Hippocampal inhibition of GSK3β activation reduces somatodendritic tau phosphorylation in db/db mice.

Immunofluorescence labeling of pS202Tau, total tau, and DAPI in hippocampal area CA3 of Wt mice receiving intrahippocampal vehicle (A), db/db mice infused with vehicle (B), Wt mice receiving intrahippocampal TDZD-8 (C), or db/db mice that received TDZD-8 (D). Scalebar in (A) = 50 microns and applies to (A–D).
4. Discussion
These studies demonstrate that hippocampal GSK3β activation mediates tau phosphorylation and memory impairment in mice with type 2 diabetes. The data also revealed that increases in peripheral corticosterone result in local., glucocorticoid-induced tau phosphorylation in the hippocampus of diabetic and normoglycemic mice. This effect is likely mediated by nongenomic interactions between hippocampal glucocorticoid receptors and GSK3β, as dephosphorylation and activation of GSK3β was observed in acute slices when gene transcription was inhibited pharmacologically. Because hippocampal GSK3β inhibition rescued spatial memory and reduced tau phosphorylation in db/db mice, we conclude that glucocorticoid-mediated activation of GSK3β underlies tau phosphorylation and cognitive deficits in diabetes.
GSK3β is a multifunctional kinase that regulates other pathways known to influence memory without altering tau phosphorylation. For example, improvements in learning with hippocampal insulin-like growth factor II (IGF-II) administration are dependent on GSK3β inactivation in rats (Chen et al., 2011), consistent with the role of GSK3β as a negative regulator of insulin and insulin-like growth factor signaling (Doble and Woodgett, 2003). Hippocampal Wnt signaling also promotes memory formation by inhibiting GSK3β activation (Fortress et al., 2013; Vargas et al., 2014), indicating that GSK3β constrains learning and memory via multiple mechanisms. However, the cognitive rescue observed following hippocampal GSK3β inhibition in the current study was likely attributable to attenuation of tau phosphorylation in db/db mice, as there was no effect of intrahippocampal TDZD-8 on spatial recognition in Wt mice. Although other groups have reported enhanced memory in pharmacological models of GSK3β inactivation (Vargas et al., 2014), we did not observe improved memory in Wt mice treated with TDZD-8 in these experiments. However, the behavioral paradigms used in these experiments were optimized for successful completion by db/db mice, resulting in maximal performance in Wt mice. It is likely that more complex paradigms would have revealed memory enhancement in Wt mice, similar to previous reports (Vargas et al., 2014).
Studies in rodent models of AD indicate that hippocampal GSK3β inhibition attenuates cognitive impairment induced by intracerebroventricular amyloid beta in rats (Hu et al., 2009), and viral vector-mediated knockdown of GSK3β reduced neuropathology in PDAPP/PS19 transgenic mice with plaques and tangles (Hurtado et al., 2012). Although db/db mice do not develop neurofibrillary tangles, viral vector-mediated expression of human tauP301 in db/db mice generates more extensive pathology, relative to Wt mice (Platt et al., 2016). While effects in db/db mice have primarily been interpreted as a consequence of leptin receptor insufficiency, db/db mice exhibit widespread physiological disruption that impacts other known regulators of tau phosphorylation, including glucocorticoids and insulin receptor signaling (Doble and Woodgett, 2003; Beck et al., 2009). There is evidence for crosstalk between leptin receptor activation and glucocorticoid receptor signaling in primary cells, where co-application of leptin prevented dephosphorylation and activation of GSK3β in neurospheres treated with dexamethasone (Garza et al., 2012). Cell type-specific manipulation of leptin receptor expression and sensitivity will be required to fully elucidate the consequences of leptin for tau phosphorylation, relative to other metabolic hormones.
The current study represents additional evidence that leptin receptor-independent normalization of hippocampal function occurs in db/db mice, as hippocampal GSK3β inhibition is unlikely to reverse the point mutation in the long-form leptin receptor that generates obesity and diabetes in this model (Hummel et al., 1966). We have previously demonstrated that glucocorticoid inhibition with adrenalectomy and physiological corticosterone replacement or pharmacological inhibition of adrenal steroidogenesis also rescues hippocampal function at the behavioral and synaptic level in db/db mice (Stranahan et al., 2008; Wosiski-Kuhn et al., 2014). Normalization of hippocampal function by reducing corticosterone was not associated with reinstatement of hippocampal insulin sensitivity in db/db mice, based on analysis of insulin-stimulated phosphorylation of insulin receptor subunit in acute slices from db/db mice treated with metyrapone (Dey et al., 2014). However, there is unquestionably more work to be done to fully characterize changes in neuronal hormone sensitivity in db/db mice and other models of obesity and diabetes.
The relative contributions of leptin, insulin, and corticosteroids to tau phosphorylation have yet to be resolved in models of diabetes, but multiple groups have shown that administration of exogenous leptin reduces cognitive impairment and neuropathology in AD mouse models (Greco et al., 2010; Irving and Harvey, 2013). The results of the current experiments could be considered leptin receptor-independent, but humans and rodents express multiple leptin receptor isoforms. The long-form leptin receptor (ObRb) plays the most prominent role in activating downstream signaling cascades, including JAK2 and STAT3 (Banks et al., 2000), but the short-form membrane leptin receptors (ObRa, ObRc, ObRd) possess a single JAK2 binding site on the intracellular tail that has limited signaling capacity (Bjørbaek et al., 1997; Li et al., 2013). On the C57Bl6J genetic background, db/db mutant mice retain expression of the short-form leptin receptor variants, while db/db mice on the original mixed BKS background lack both long- and short-form leptin receptor expression (Hummel et al., 1972; Mao et al., 2006). The short-form leptin receptor variants are physiologically relevant, as B6.db/db mice respond to exogenous leptin with reductions in body weight, adiposity, and fasting glucose levels, unlike BKS.db/db mice (Harris et al., 2001). Metabolic responses to leptin are diminished in B6.db/db mice, relative to Wt (Harris et al., 2001), but the persistence of short-form leptin receptor isoforms in the B6.db/db line used in the current report prevents us from ruling out contributions of other isoforms to the observed results.
There have been numerous reports of increases in hippocampal tau phosphorylation in rodents treated with the pancreatic beta-cell toxin streptozotocin (STZ), a non-obese model of insulin deficient diabetes and hyperglycemia (Planel et al., 2007a; King et al., 2013). Hippocampal tau hyperphosphorylation has also been demonstrated previously in rodents with obesity and insulin resistance, including models of leptin or leptin receptor deficiency (Kim et al., 2009; El-Khoury et al., 2016). Reductions in body temperature have been proposed as a physiological mechanism for hyperphosphorylation of tau in diabetes (Planel et al., 2007a; El-Khoury et al., 2016), but db/db mice in the current studies did not exhibit impaired thermoregulation. It is therefore possible that the effects of hypothermia-induced tau phosphorylation may differ from those of tau phosphorylation in normothermic models of diabetes. Consistent with this interpretation, widespread and reversible phosphorylation of tau occurs in hibernating mammals (Arendt et al., 2003). Deficits in hippocampal function are dissociable from hypothermia-induced tau phosphorylation, as hibernating mammals awakened from torpor exhibit intact learning and memory despite widespread hippocampal tau phosphorylation (Bullman et al., 2016). Anesthesia-induced hypothermia also promotes hippocampal tau phosphorylation in normal mice (Planel et al., 2007b), but hippocampal long-term potentiation persists in anesthetized rodents despite a drop in core body temperature (Pavlides et al., 2002), and LTP magnitude is independent of brain temperature in awake rodents (Davis et al., 2004). LTP is not the only measure of synaptic plasticity related to cognition, but the lack of clear associations between hypothermia and neurocognitive impairment suggest that hypothermia-induced tau phosphorylation may differ from pathological tau phosphorylation in AD. Additional studies will be required to distinguish between reversible modifications of tau under physiological conditions and pathological tau phosphorylation that leads to aggregation and neurofibrillary tangles, but addressing these questions may identify cellular mechanisms that can be targeted to block neuropathology in AD and other dementias.
Supplementary Material
Research Highlights.
Diabetes promotes hippocampal tau phosphorylation by increasing glucocorticoids
Hippocampal glucocorticoid exposure activates the tau kinase GSK3β
Hippocampal GSK3β antagonism prevents tau hyperphosphorylation in diabetic mice
Blocking hippocampal GSK3β activation rescues memory in diabetic mice
Acknowledgments
We are grateful to Dr. Peter Davies of Yeshiva University for the antibodies against total and phosphorylated tau. This work was supported by the National Institutes of Health [grant numbers K01DK100616 and R03DK101817 to AMS].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acker CM, Forest SK, Zinkowski R, Davies P, d’Abramo C. Sensitive quantitative assays for tau and phospho-tau in transgenic mouse models. Neurobiol Aging. 2013;34:338–50. doi: 10.1016/j.neurobiolaging.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Stieler J, Strijkstra AM, Hut RA, Rüdiger J, Van der Zee EA, Harkany T, Holzer M, Härtig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–81. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglietto-Vargas D, Medeiros R, Martinez-Coria H, LaFerla FM, Green KN. Mifepristone alters amyloid precursor protein processing to preclude amyloid beta and also reduces tau pathology. Biol Psychiatry. 2013;74:357–66. doi: 10.1016/j.biopsych.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–72. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- Beck IM, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev. 2009;30:830–82. doi: 10.1210/er.2009-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessels GJ, Reagan LP. Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci. 2015;16:660–71. doi: 10.1038/nrn4019. [DOI] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–35. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–95. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- Bullmann T, Seeger G, Stieler J, Hanics J, Reimann K, Kretzschmann TP, Hilbrich I, Holzer M, Alpár A, Arendt T. Tau phosphorylation-associated spine regression does not impair hippocampal-dependent memory in hibernating golden hamsters. Hippocampus. 2016;26:301–18. doi: 10.1002/hipo.22522. [DOI] [PubMed] [Google Scholar]
- Bruehl H, Wolf OT, Convit A. A blunted cortisol awakening response and hippocampal atrophy in type 2 diabetes mellitus. Psychoneuroendocrinology. 2009;34:815–21. doi: 10.1016/j.psyneuen.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SN, Barnes CA. Senescent synapses and hippocampal circuit dynamics. Trends Neurosci. 2010;33:153–61. doi: 10.1016/j.tins.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallini A, Brewerton S, Bell A, Sargent S, Glover S, Hardy C, Moore R, Calley J, Ramachandran D, Poidinger M, Karran E, Davies P, Hutton M, Szekeres P, Bose S. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J Biol Chem. 2013;288:23331–47. doi: 10.1074/jbc.M113.463984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–7. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad CD, Galea LA, Kuroda Y, McEwen BS. Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav Neurosci. 1996;110:1321–34. doi: 10.1037//0735-7044.110.6.1321. [DOI] [PubMed] [Google Scholar]
- Convit A, Wolf OT, Tarshish C, de Leon MJ. Reduced glucose tolerance is associated with poor memory performance and hippocampal atrophy among normal elderly. Proc Natl Acad Sci U S A. 2003;100:2019–22. doi: 10.1073/pnas.0336073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CD, Jones FL, Derrick BE. Novel environments enhance the induction and maintenance of long-term potentiation in the dentate gyrus. J Neurosci. 2004;24:6497–506. doi: 10.1523/JNEUROSCI.4970-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA. Glucose intolerance and aging. Diabetes Care. 1981;4:493–501. doi: 10.2337/diacare.4.4.493. [DOI] [PubMed] [Google Scholar]
- Dey A, Hao S, Erion JR, Wosiski-Kuhn M, Stranahan AM. Glucocorticoid sensitization of microglia in a genetic mouse model of obesity and diabetes. J Neuroimmunol. 2014;269:20–7. doi: 10.1016/j.jneuroim.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury NB, Gratuze M, Petry F, Papon MA, Julien C, Marcouiller F, Morin F, Nicholls SB, Calon F, Hébert SS, Marette A, Planel E. Hypothermia mediates age-dependent increase of tau phosphorylation in db/db mice. Neurobiol Dis. 2016;88:55–65. doi: 10.1016/j.nbd.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Erion JR, Wosiski-Kuhn M, Dey A, Hao S, Davis CL, Pollock NK, Stranahan AM. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J Neurosci. 2014;34:2618–31. doi: 10.1523/JNEUROSCI.4200-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Schram SL, Tuscher JJ, Frick KM. Canonical Wnt signaling is necessary for object recognition memory consolidation. J Neurosci. 2013;33:12619–26. doi: 10.1523/JNEUROSCI.0659-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza JC, Guo M, Zhang W, Lu XY. Leptin restores adult hippocampal neurogenesis in a chronic unpredictable stress model of depression and reverses glucocorticoid-induced inhibition of GSK-3β/β-catenin signaling. Mol Psychiatry. 2012;17:790–808. doi: 10.1038/mp.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco SJ, Bryan KJ, Sarkar S, Zhu X, Smith MA, Ashford JW, Johnston JM, Tezapsidis N, Casadesus G. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1155–67. doi: 10.3233/JAD-2010-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–56. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RB, Mitchell TD, Yan X, Simpson JS, Redmann SM., Jr Metabolic responses to leptin in obese db/db mice are strain dependent. Am J Physiol Regul Integr Comp Physiol. 2001;281:R115–32. doi: 10.1152/ajpregu.2001.281.1.R115. [DOI] [PubMed] [Google Scholar]
- Hummel KP, Coleman DL, Lane PW. The influence of genetic background on expression of mutations at the diabetes locus in the mouse. I. C57BL-KsJ and C57BL-6J strains. Biochem Genet. 1972;7(1):1–13. doi: 10.1007/BF00487005. [DOI] [PubMed] [Google Scholar]
- Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153:1127–8. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Carroll JC, Macdonald C, Aboagye AK, Trojanowski JQ, Lee VM. Selectively Silencing GSK-3 Isoforms Reduces Plaques and Tangles in Mouse Models of Alzheimer’s Disease. J Neurosci. 2012;32:7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving AJ, Harvey J. Leptin regulation of hippocampal synaptic function in health and disease. Philos Trans R Soc Lond B Biol Sci. 2013;369:20130155. doi: 10.1098/rstb.2013.0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. 2009;150:5294–301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MR, Anderson NJ, Guernsey LS, Jolivalt CG. Glycogen synthase kinase-3 inhibition prevents learning deficits in diabetic mice. J Neurosci Res. 2013;91:506–14. doi: 10.1002/jnr.23192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–5. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- Li Z, Ceccarini G, Eisenstein M, Tan K, Friedman JM. Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Mol Metab. 2013;2:364–75. doi: 10.1016/j.molmet.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol. 2011;225:54–62. doi: 10.1002/path.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Xu GH, Wang K, Cao JL, Gu EW, Li YH, Liu XS. Involvement of GSK3β/β-catenin signaling in the impairment effect of ketamine on spatial memory consolidation in rats. Neurobiol Learn Mem. 2014;111:26–34. doi: 10.1016/j.nlm.2014.02.012. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, Fiocco A, Wan N, Maheu F, Lord C, Schramek T, Tu MT. Stress hormones and human memory function across the lifespan. Psychoneuroendocrinology. 2005;30:225–42. doi: 10.1016/j.psyneuen.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Mao HZ, Roussos ET, Peterfy M. Genetic analysis of the diabetes-prone C57BLKS/J mouse strain reveals genetic contribution from multiple strains. Biochim Biophys Acta. 2006;1762:440–6. doi: 10.1016/j.bbadis.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alonso M, Castro A, Pérez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J Med Chem. 2002;45:1292–9. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Nivón LG, McEwen BS. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 2002;12:245–57. doi: 10.1002/hipo.1116. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2. San Diego: Academic Press; 2001. [Google Scholar]
- Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, Yu WH, Luchsinger JA, Wadzinski B, Duff KE, Takashima A. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci. 2007a;27:13635–48. doi: 10.1523/JNEUROSCI.3949-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007b;27:3090–7. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt TL, Beckett TL, Kohler K, Niedowicz DM, Murphy MP. Obesity, diabetes, and leptin resistance promote tau pathology in a mouse model of disease. Neuroscience. 2016;315:162–74. doi: 10.1016/j.neuroscience.2015.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasgon NL, Kenna HA, Wroolie TE, Kelley R, Silverman D, Brooks J, Williams KE, Powers BN, Hallmayer J, Reiss A. Insulin resistance and hippocampal volume in women at risk for Alzheimer’s disease. Neurobiol Aging. 2011;32:1942–8. doi: 10.1016/j.neurobiolaging.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds RM, Strachan MW, Labad J, Lee AJ, Frier BM, Fowkes FG, Mitchell R, Seckl JR, Deary IJ, Walker BR, Price JF Edinburgh Type 2 Diabetes Study Investigators. Morning cortisol levels and cognitive abilities in people with type 2 diabetes: the Edinburgh type 2 diabetes study. Diabetes Care. 2010;33:714–20. doi: 10.2337/dc09-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandeep TC, Yau JL, MacLullich AM, Noble J, Deary IJ, Walker BR, Seckl JR. 11Beta-hydroxysteroid dehydrogenase inhibition improves cognitive function in healthy elderly men and type 2 diabetics. Proc Natl Acad Sci U S A. 2004;101:6734–9. doi: 10.1073/pnas.0306996101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, Clarimón J, Lleó A, Gómez-Isla T. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–67. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Arumugam TV, Lee K, Cutler RG, Egan JP, Mattson MP. Diabetes impairs hippocampal function via glucocorticoid-mediated effects on new and mature neurons. Nature Neuroscience. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Haberman RP, Gallagher M. Cognitive decline is associated with reduced reelin expression in the entorhinal cortex of aged rats. Cerebral Cortex. 2011;21:392–400. doi: 10.1093/cercor/bhq106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–61. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita N, Yoshino T, Mutoh S. Possible involvement of corticosterone in bone loss of genetically diabetic db/db mice. Horm Metab Res. 2000;32:147–51. doi: 10.1055/s-2007-978610. [DOI] [PubMed] [Google Scholar]
- Valente T, Gella A, Fernàndez-Busquets X, Unzeta M, Durany N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol Dis. 2010;37:67–76. doi: 10.1016/j.nbd.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Vargas JY, Fuenzalida M, Inestrosa NC. In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J Neurosci. 2014;34:2191–202. doi: 10.1523/JNEUROSCI.0862-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayudhan L, Poppe M, Archer N, Proitsi P, Brown RG, Lovestone S. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br J Psychiatry. 2010;196:36–40. doi: 10.1192/bjp.bp.109.067942. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- Wosiski-Kuhn M, Erion JR, Gomez-Sanchez EP, Gomez-Sanchez CE, Stranahan AM. Glucocorticoid receptor activation impairs hippocampal plasticity by suppressing BDNF expression in obese mice. Psychoneuroendocrinology. 2014;42:165–77. doi: 10.1016/j.psyneuen.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Brickman AM, Luchsinger J, Ferrazzano P, Pichiule P, Yoshita M, Brown T, DeCarli C, Barnes CA, Mayeux R, Vannucci SJ, Small SA. The brain in the age of old: the hippocampal formation is targeted differentially by diseases of late life. Ann Neurol. 2008;64:698–706. doi: 10.1002/ana.21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lu W, Shi Y, Bai F, Chang J, Yuan Y, Teng G, Zhang Z. Impairments in cognition and resting-state connectivity of the hippocampus in elderly subjects with type 2 diabetes. Neurosci Lett. 2010;473:5–10. doi: 10.1016/j.neulet.2009.12.057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.