

Abstract
The retinoic acid receptor-related orphan receptor-α (RORα) is an important regulator of various biological processes, including cerebellum development, circadian rhythm and cancer. Here, we show that hepatic RORα controls lipid homeostasis by negatively regulating transcriptional activity of peroxisome proliferators-activated receptor-γ (PPARγ) that mediates hepatic lipid metabolism. Liver-specific Rorα-deficient mice develop hepatic steatosis, obesity and insulin resistance when challenged with a high-fat diet (HFD). Global transcriptome analysis reveals that liver-specific deletion of Rorα leads to the dysregulation of PPARγ signaling and increases hepatic glucose and lipid metabolism. RORα specifically binds and recruits histone deacetylase 3 (HDAC3) to PPARγ target promoters for the transcriptional repression of PPARγ. PPARγ antagonism restores metabolic homeostasis in HFD-fed liver-specific Rorα deficient mice. Our data indicate that RORα has a pivotal role in the regulation of hepatic lipid homeostasis. Therapeutic strategies designed to modulate RORα activity may be beneficial for the treatment of metabolic disorders.
Hepatic steatosis development may result from dysregulation of lipid metabolism, which is finely tuned by several transcription factors including the PPAR family. Here Kim et al. show that the nuclear receptor RORα inhibits PPARγ-mediated transcriptional activity by interacting with HDAC3 and competing for the promoters of lipogenic genes.
Introduction
Obesity is a high-risk metabolic disorder, leading to various complications, including cardiovascular disease, hyperlipidemia and type II diabetes1–3. Ectopic accumulation of fat in various tissues activates numerous cellular stress and inflammatory signaling pathways, resulting in insulin resistance, pancreatic β-cell dysfunction, and hepatic steatosis4. The liver is the central metabolic organ to regulate key aspects of glucose and lipid metabolism including gluconeogenesis, fatty acid β-oxidation, lipoprotein uptake and secretion and lipogenesis5. Given that portal vein is a critical path to convey insulin signaling from pancreas during fed state, the hepatic glucose and lipid metabolism are directly under control of nutrient signaling.
Dysregulation of hepatic lipid metabolism results in the development of hepatic steatosis, contributing to the chronic insulin resistance and steatotic hepatitis6, 7. The hepatic metabolic pathways are governed by highly dynamic transcriptional networks of orphan nuclear receptors (ONRs), including proliferators-activated receptor-γ (PPARγ), farnesoid X receptor, and liver X receptor8. ONRs are ligand-activated transcription factors with no defined ligands9, 10. Many ONRs are expressed in tissues involved in metabolism, such as skeletal muscle, adipose tissue and liver11, 12, and play critical roles in the regulation of metabolism13. Genetic studies have shown that many ONRs regulate nutrient metabolism and physiology of obesity and type II diabetes14–16. Given that numerous synthesized ligands for ONRs are used for developing putative drugs for human metabolic diseases17–19, ONRs are emerging as therapeutic targets for the treatment of metabolic diseases.
Previously, we have reported that receptor-related orphan receptor-α (RORα), a member of ONRs, possesses tumor suppressive function by transrepressing canonical Wnt/β-catenin signaling leading to inhibition of colon cancer growth and by increasing p53 stability upon DNA damage response20, 21. RORα is known to regulate cerebellum development22. The staggerer (sg) mice, natural Rorα spontaneous mutant mice, display ataxia and severe cerebellar atrophy23. Moreover, RORα functions to regulate circadian rhythm as a key regulator of the cyclic expression of BMAL1 together with REV-ERBα24. The RORα/REV-ERBα feedback loop controls the circadian expression pattern of BMAL1, indicating that RORα plays a key role in the core circadian clock25. In addition, sg mice show lower expression levels of genes involved in lipid metabolism, including apolipoprotein A-1 (apoA1) and apolipoprotein C-III (apoCIII)26, 27. Thus, sg mice exhibit less body weight gain compared with wild-type (WT) mice28. Given that sg mice have huge cerebellar defects, it is still possible that physiological changes observed in sg mice are indirect effects. Thus, the physiological roles of RORα to control transcriptional networks to modulate lipogenesis and gluconeogenesis still remain unclear.
Here, we report that RORα plays a key role to control hepatic lipid metabolism to protect against diet-induced obesity and hepatic steatosis, using liver-specific Rorα-deficient mouse model. High-fat diet (HFD)-fed liver-specific Rorα deficient mice (RORαLKO mice) show severe metabolic defects, including hepatic steatosis, obesity, and insulin resistance, although no physiological changes have been observed with control diet (CD). Genome-wide transcriptome analysis reveals that PPARγ signaling is remarkably elevated in RORαLKO mice. RORα specifically recruits HDAC3 to the PPARγ target promoters to suppress PPARγ transcriptional activity. Finally, PPARγ antagonism by using PPARγ antagonist GW9662, largely ameliorates body weight gain and hepatic steatosis in HFD-fed RORαLKO mice, indicating that dysregulated PPARγ signaling is a critical metabolic cue, leading to metabolic defects in HFD-fed RORαLKO mice. Together, our data demonstrate that RORα controls PPARγ signaling to protect against hepatic metabolic homeostasis and obesity in response to HFD.
Results
HFD induces obesity in liver-specific Rorα-deficient mice
To determine the physiological roles of RORα in the liver, we generated RORα-floxed mice (RORαf/f) by gene targeting in ES cells and crossed RORα floxed mice with Albumin-Cre (Alb-Cre) mice to selectively create liver-specific Rorα conditional knockout (KO) mice (hereafter named RORαLKO) (Fig. 1a, b). The mRNA and protein levels of endogenous hepatic RORα were remarkably depleted in RORαLKO mice compared with littermate controls (hereafter named RORαf/f) (Fig. 1c–f). We measured the growth rate of RORαLKO mice and observed that they attained body weights similar to RORαf/f mice fed CD during 10 weeks from 8 weeks old (Fig. 1g). Body composition analysis revealed that RORαLKO mice exhibited similar fat/lean mass, free body fluid and adipocytes size with those of RORαf/f (Supplementary Fig. 1a, b). However, when placed on a HFD, RORαLKO mice exhibited a significant increase of the weight gain (20 vs. 25 g) compared with their RORαf/f littermates, resulting in extraordinary obesity (Fig. 1g). Body composition analysis and macroscopic view revealed that RORαLKO mice had more fat mass (Fig. 1h, i). All white and brown fat depots from RORαLKO mice were significantly increased in mass relative to RORαf/f (Fig. 1j). During obesity, adipose tissue expands by hyperplastic and/or hypertrophic growth. The cross-sectional area of adipocytes in visceral fat tissue was markedly increased in RORαLKO mice compared with RORαf/f mice (Fig. 1k). Induction of pro-inflammatory genes, including Mcp1, Ifnγ, and F4/80 in visceral fat depot were potentiated in RORαLKO mice (Fig. 1l). Consistent with a significant weight gain in HFD-fed RORαLKO mice, gene expression analysis revealed reduction of Pgc1α, as well as a number of genes involved in thermogenesis, mitochondrial biogenesis, and fatty acid oxidation in brown adipose tissue of RORαLKO mice compared with that of RORαf/f littermates (Fig. 1m). The observation that energy expenditure (EE) in brown fat has been impaired in HFD-fed RORαLKO mice led us to examine whether they have global metabolic defects. Although no obvious defects were observed in mice on CD (Supplementary Fig. 1c), HFD-fed RORαLKO mice were found to produce far less CO2, consume less O2 and expend less energy than RORαf/f littermates, indicating that oxidative phosphorylation is impaired by the hepatic deletion of RORα (Fig. 1n and Supplementary Fig. 1d). Previously, bile acids have been reported to increase EE by promoting intracellular thyroid hormone activation in brown adipose tissue29. We observed that expression of key genes involved in hepatic bile acid synthesis was remarkably reduced in HFD-fed RORαLKO mice (Supplementary Fig. 1e). Consistently, serum bile acid pool sizes in HFD-fed RORαLKO mice were markedly less than RORαf/f littermates (Supplementary Fig. 1f), implicating that reduction of bile acid synthesis and bile acid pool size led to reduced EE in brown adipose tissue of HFD-fed RORαLKO mice.
Fig. 1.
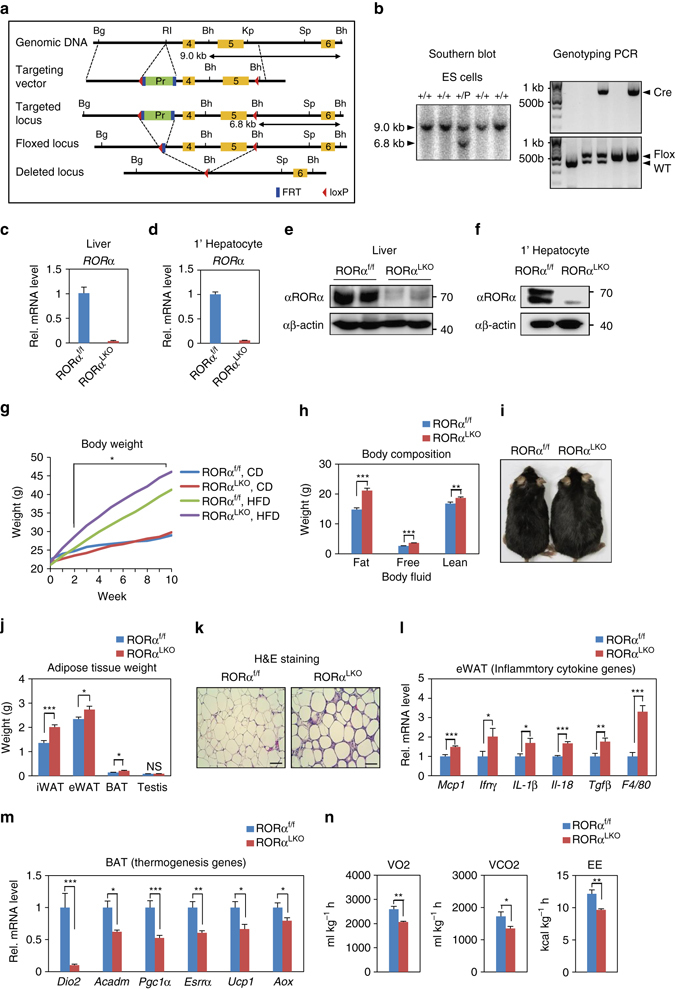
Liver-specific Rorα deleted mice are susceptible to diet-induced obesity. a Schematic representation of the Rorα gene-targeting strategy, including a map of the RORα exon 4 and 5 allele (yellow box) and the targeting vector with loxP sites (red arrowhead), FRT sites (blue box), and puromycin selection gene (green box). Bg: BglII, RI: EcoRI, Bh: BamHI, Kp: KpnI, Sp: SpeI. b Southern blot analysis to screen correctly targeted Rorα + /puro ES cell clones. For BamHI digestion, the bands representing WT and mutant alleles were 9.0 kb and 6.8 kb, respectively. PCR analyses with genomic DNA extracted from tail of WT, RORαf/+, Alb; RORαf/+, RORαf/f and Alb; RORαf/f mice are shown. PCR were performed to amplify the cre (top), floxed and deleted allele (bottom). c, d mRNA expression level of RORα in liver extract c and primary hepatocyte d from RORαf/f and RORαLKO mice. Expression was normalized to 18 s rRNA expression. e, f Protein expression level of RORα in liver extract e and primary hepatocyte f. g Body weight change in RORαf/f and RORαLKO mice fed CD or HFD for 10 weeks (n = 9–12/group). Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, RORαf/f vs. RORαLKO, HFD. h, i RORαf/f and RORαLKO mice were fed with HFD for 10 weeks. h Body composition analysis of RORαf/f and RORαLKO mice (n = 6/group). i Macroscopic views of RORαf/f and RORαLKO mice. j Adipose tissues weight of RORαf/f and RORαLKO mice (n = 6–7/group). k Representative image of epidydimal white adipose tissue (eWAT) from RORαf/f and RORαLKO mice stained with hematoxylin and eosin. Scale bar, 100 μm. l, m Expression levels of inflammatory cytokine genes in eWAT extract l or thermogenesis genes in BAT extract m from RORαf/f and RORαLKO mice (n = 4–5 per group) as determined by qRT-PCR. Expression was normalized to L32 expression. n Metabolic cage studies were performed in RORαf/f and RORαLKO mice (n = 5–6 mice/group). O2 consumption (VO2), CO2 production (VCO2) and energy expenditure were represented (left to right). Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001, NS, Non-Significant. Data expressed as mean ± s.e.m
Hepatic steatosis impairs insulin sensitivity in RORαLKO mice
Obesity is largely associated with hepatic steatosis in humans as well as in rodents. Consistent with obese phenotype in RORαLKO mice, large lipid vesicles with increased amounts were observed in the hepatocytes of HFD-fed RORαLKO mice (Fig. 2a). Macroscopically, liver from HFD-fed RORαLKO mice was markedly enlarged and paler compared with HFD-fed RORαf/f liver (Fig. 2b). Consistently, HFD-fed RORαLKO mice exhibited a remarkable increase of liver weight compared with HFD-fed RORαf/f mice (Fig. 2c). In accordance with hematoxylin and eosin staining, oil red O staining and hepatic triglyceride (TG) analysis showed a dramatic increase in lipid level in the HFD-fed RORαLKO liver compared with the HFD-fed RORαf/f liver, whereas no difference was observed in CD-fed RORαf/f and RORαLKO mice (Fig. 2d, e and Supplementary Fig. 1g). While hepatic gene expression profiles were similar among CD-fed genotypes (Supplementary Fig. 1h), hepatic gene expression profiles of lipogenesis, gluconeogenesis, and lipid sequestration in the HFD-fed RORαLKO were largely increased, indicating that RORα protects against HFD-induced hepatic steatosis (Fig. 2f).
Fig. 2.
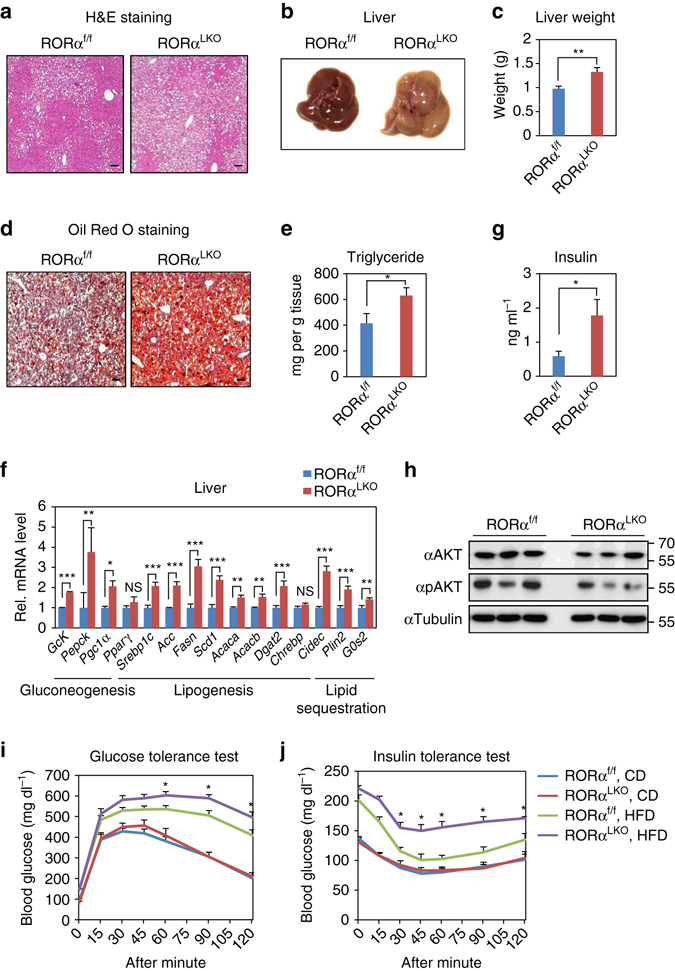
Liver-specific Rorα deleted mice are susceptible to diet-induced hepatic steatosis and insulin resistance. a–j RORαf/f and RORαLKO mice were fed with HFD for 10 weeks. a Representative liver histological section images of RORαf/f and RORαLKO mice stained with hematoxylin and eosin. Scale bar, 100 μm. b Macroscopic view of livers from RORαf/f and RORαLKO mice. c Liver weights of RORαf/f and RORαLKO mice (n = 10–11 per group). d Representative liver histological section images of RORαf/f and RORαLKO mice stained with Oil Red O. Scale bar, 100 μm. e Triglyceride content of livers from RORαf/f and RORαLKO mice (n = 8 per group). f Hepatic gene expression profile involved in metabolism from the livers of RORαf/f and RORαLKO mice (n = 4 per group) as determined by quantitative PCR with reverese transcription (qRT-PCR). Expression was normalized to 36B4 expression. g Fasting insulin levels in RORαf/f and RORαLKO mice (n = 6–7 per group). Data expressed as mean ± s.e.m. Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001, NS = non-significant. h Immunoblot analysis was performed from liver extracts of RORαf/f and RORαLKO mice. i, j Glucose tolerance test i and insulin tolerance test j on RORαf/f and RORαLKO mice fed on CD or HFD for 10 weeks. (n = 4–9/group). Data expressed as mean ± s.e.m. Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, RORαf/f vs RORαLKO, HFD. Data expressed as mean ± s.e.m
Obesity and hepatic steatosis often predispose rodents and humans to impaired glucose homeostasis and insulin resistance30–32. Hepatic deficiency of RORα resulted in elevated fasting insulin levels in RORαLKO mice (Fig. 2g). As elevated fasting insulin level is an indication of insulin resistance, RORαLKO mice predisposed to severe insulin resistance than RORαf/f mice. Consistent with elevated fasting insulin level, an investigation of insulin signaling pathways confirmed reduction of phosphorylated AKT, indicating that insulin signaling was impaired in HFD-fed RORαLKO mice (Fig. 2h). As insulin signaling was impaired in the liver, we performed glucose tolerance tests (GTTs) and insulin tolerance tests (ITTs) to determine if glucose homeostasis was impaired in HFD-fed RORαLKO mice. Glucose intolerance and insulin resistance were observed in HFD-fed RORαLKO mice, although CD-fed RORαLKO mice exhibited little or no difference in glucose homeostasis compared with CD-fed RORαf/f mice (Fig. 2i, j). Altogether, our data strongly demonstrate that hepatic RORα is required for prevention against insulin resistance.
RORαLKO mice exhibit enhanced PPARγ transcriptional activity
To explore molecular mechanism by which hepatic deletion of RORα induces obesity and insulin resistance, we performed mRNA-sequencing analysis of liver tissues obtained from HFD-fed RORαf/f, HFD-fed RORαLKO, CD-fed RORαf/f and CD-fed RORαLKO mice (Supplementary Data Table 1). Using the resulting mRNA expression profiles, we first identified 343 differentially expressed genes (DEGs; see Methods) between HFD-fed RORαLKO and HFD-fed RORαf/f mice (RORαLKO/RORαf/f HFD in Fig. 3a) and also 395 DEGs between CD-fed RORαLKO and CD-fed RORαf/f mice (RORαLKO/RORαf/f CD in Fig. 3a and Supplementary Data 2). Moreover, we further compared log2-fold changes of the DEGs in the two comparisons above ((RORαLKO/RORαf/f HFD)/(RORαLKO/RORαf/f CD) in Fig. 3a) and identified the genes specifically affected by RORα under HFD condition as the DEGs showing significant (P < 0.05) differences in the log2-fold changes (Supplementary Data 2). We categorized these DEGs into eight groups (Groups 1–8) based on differential expression patterns in the three comparisons above. Our data above showed that we only found significant weight gain of RORαLKO mice under HFD condition. Of Groups 1–8, thus, we first focused on Groups 1–4 showing significant changes under HFD condition (Fig. 3a).
Fig. 3.
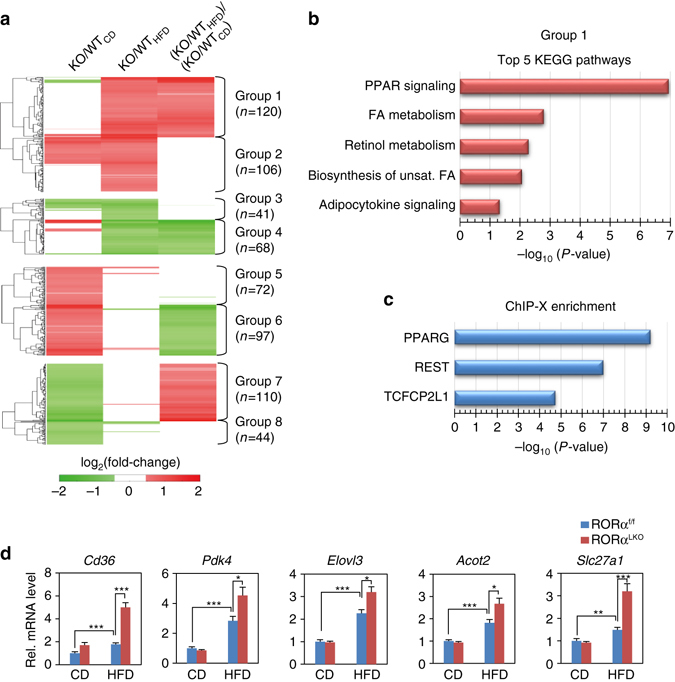
Transcriptome analysis of hepatic gene expression profile in RORαLKO mice. a Up- and down-regulated genes in RORαLKO compared to RORαf/f mice. These genes were categorized into four groups of the up- (Groups 1, 2) and down-regulated genes (Groups 3, 4) in HFD-fed RORαLKO. Besides Groups 1–4, remain genes were also categorized into four groups of the up- (Groups 5, 6) and down-regulated genes (Groups 7, 8) in CD-fed RORαLKO. Groups 1, 2 (or Groups 3, 4) were further divided by the specificity of the RORα effect under HFD condition. Log2-fold changes in the following comparisons were displayed: RORαLKO/RORαf/f HFD, RORαLKO/RORαf/f CD and (RORαLKO/RORαf/f HFD)/(RORαLKO/RORαf/f CD). Hierarchical clustering of the DEGs in Groups 1–8 (Euclidian distance as a dissimilarity measure and average linkage) were used to display the log2-fold changes. b KEGG pathways enrichment analysis for the genes in Group 1. The bars represent the enrichment scores, -log10 (P value). c TF enrichment analysis for the genes in Group 1 using ChEA2 software. Top 3 TFs are shown. The bars represent the enrichment scores, -log10 (P value). d Expression levels of group 1 genes (upregulated genes in RORαLKO mice fed HFD compared with RORαf/f mice) in liver extract from RORαf/f and RORαLKO mice fed CD or HFD for 10 weeks (n = 5–9/group) as determined by qRT-PCR. Expression was normalized to 36B4 expression. Statistical analysis was performed using two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001. Data expressed as mean ± s.e.m
To understand cellular processes represented by Groups 1–4, we performed enrichment analysis of gene ontology biological processes (GOBPs) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for the genes in Groups 1–4 using DAVID software 33, 34 (Supplementary Data 3). Group 1 is mainly involved in the processes related to PPAR and adipocytokine signaling pathways and fatty acid/retinol metabolism (Fig. 3b). Group 4 is mainly involved in the processes related to circadian rhythm (Supplementary Data 3). Since Group 1 is highly associated with the weight gain of HFD-fed RORαLKO, we next examined which transcription factors (TFs) account for up-regulation of the genes in Group 1 under HFD condition. By performing TF enrichment analysis of the genes in Group 1 using ChEA2 software35, PPARγ turned out to be the most enriched TF in Group 1 (Fig. 3c and Supplementary Data 4). Quantitative PCR with reverese transcription analysis confirmed that the genes in Group 1 including PPARγ target genes are largely elevated in HFD-fed RORαLKO mice (Fig. 3d), indicating that PPARγ transcriptional activity is enhanced in the absence of RORα.
PPARα is a transcriptional factor that conducts a key role in hepatic lipid metabolism and shares similar response elements with PPARγ on the target promoters36, 37. To determine whether RORα also mediates PPARα transcriptional network in the liver, we examined the expression of well-known hepatic PPARα target genes, including Acox1 and Fgf21. The hepatic gene expressions of Acox1 and Fgf21 in HFD-fed RORαLKO mice were similar to those of HFD-fed RORαf/f mice, suggesting that RORα deficiency would not further enhance hepatic PPARα transcriptional network (Supplementary Fig. 2a) under HFD condition. We next examined the expression of PPARα target genes in the physiological setting of PPARα activation. It has been widely accepted that PPARα is activated under conditions of energy deprivation38. The induction of PPARα target genes in RORαLKO mice was quite similar to that of RORαf/f mice (Supplementary Fig. 2b). Chromatin immunoprecipitation (ChIP) assay clearly revealed little or no difference of PPARα recruitment to PPAR-response element (PPRE) on the promoters of PPARα target genes (Supplementary Fig. 2c). Recently, PPARα has been reported to bind autophagic gene promoters to coordinate autophagy in response to nutrient deprivation37. We observed that the induction of autophagic genes including LC3a and Sesn2 of RORαLKO mice were similar to those of RORαf/f mice (Supplementary Fig. 2d). Taken together, these data indicate that RORα mainly controls PPARγ transcriptional network rather than PPARα in the liver in response to environmental stress such as HFD.
RORα represses PPARγ transcriptional activity via HDAC3
Since PGC1α is a well-known coactivator for PPARγ39, we examined whether introduction of RORα inhibits PPARγ/PGC1α-dependent transcriptional activation using PPRE-containing luciferase reporter. Expression of PGC1α dramatically increased PPARγ transcriptional activity, and increased expression of RORα progressively attenuated the PPARγ/PGC1α-dependent transcriptional activation (Fig. 4a). In addition, we examined whether RORα inhibits NCOA1 and NCOA2-mediated PPARγ transcriptional activation. NCOA1 and NCOA2, as p160 family members, are also coactivators for PPARγ40. Consistently, RORα significantly reduced NCOA1 and NCOA2-mediated transcriptional activation (Fig. 4b and Supplementary Fig. 3a).
Fig. 4.
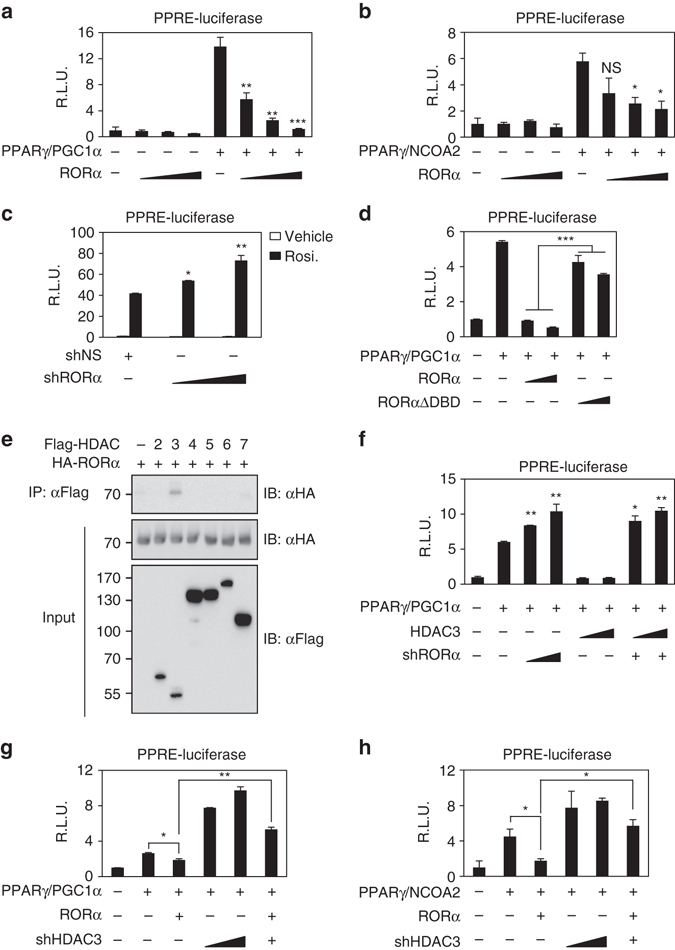
RORα interacts with HDAC3 to repress PPARγ transcriptional activity. a, b Effect of overexpression of RORα on PPRE-luciferase reporter activity with coactivator PGC1α a or NCOA2 b. *P < 0.05, **P < 0.01, ***P < 0.001, NS, non-significant, compared to PPARγ/coactivator group. c Effect of knockdown of RORα on PPRE-luciferase reporter activity. Cells were treated with DMSO (vehicle), rosiglitazone (20 μM) for 24 h. *P < 0.05, **P < 0.01, compared to shNS group. d Effect of RORα ΔDBD mutant on PPRE-luciferase reporter activity. ***P < 0.001. e Co-immunoprecipitation assay was performed to detect the interaction between RORα and HDACs of HEK293T cells. f Effect of RORα on PPRE-luciferase reporter activity by HDAC3 overexpression. *P < 0.05, **P < 0.01, compared to PPARγ/PGC1α group. g, h Effect of knockdown of HDAC3 with coactivator PGC1α g and NCOA2 h on PPRE-luciferase reporter activity. Data expressed as mean ± s.e.m. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc analysis. *P < 0.05, **P < 0.01. Data expressed as mean ± s.e.m
To evaluate the role of RORα in attenuation of the PPARγ-dependent transcriptional activation, we treated Hep3B cells with rosiglitazone, a PPARγ synthetic agonist, and then measured PPRE-luciferase activity. Knockdown of RORα markedly enhanced PPRE-luciferase activity, indicating that RORα functions to repress PPARγ transcriptional activity (Fig. 4c). To determine if DNA-binding domain (DBD) of RORα is required for inhibiting PPARγ transcriptional activation, we introduced DBD-deleted RORα mutant (RORα ΔDBD). We observed that RORα WT markedly suppressed PPARγ transcriptional activation, whereas the RORα-mediated repression was remarkably relieved by introduction of RORα ΔDBD (Fig. 4d). As RORα failed to interact with PPARγ, our data proposed that RORα suppresses PPARγ transcriptional activation through DBD and possibly competes with PPARγ for the binding to PPRE. Consistently, the recruitment of RORα was markedly reduced in PPRE-deleted synthetic promoter region (Supplementary Fig. 3b, c).
Since histone acetylation promotes transcriptional activation, we next examined whether RORα interacts with specific histone deacetylases for the repression of PPARγ transcriptional activity. Co-immunoprecipitation assay revealed that RORα specifically interacts with HDAC3 (Fig. 4e and Supplementary Fig. 3d). To determine if HDAC3 is required for RORα-mediated repression of PPARγ transcriptional activity, we further examined repressive function of HDAC3 for PPRE-luciferase activity in the presence or absence of RORα. Intriguingly, knockdown of RORα relieved the HDAC3-dependent repressive function indicating that HDAC3 exerted repressive function on PPARγ transcriptional activity in the presence of RORα (Fig. 4f). Consistently, knockdown of HDAC3 largely reversed RORα-mediated repression of PPARγ transcriptional activity (Fig. 4g, h and Supplementary Fig. 3e). These results indicate that RORα recruits HDAC3 to potentiate repression of PPARγ transcriptional activity.
RORα/HDAC3 dynamically regulate PPARγ target gene expression
PPRE consists of a direct repeat (DR) sequence of (A/G)GGTCA spaced by one nucleotide, whereas consensus RORα response element (RORE) consists of core motif (A/G)GGTCA preceded by a 6-bp A/T-rich sequence. Thus, given that RORE and PPRE share core motif, we hypothesized that RORα directly binds the PPRE of PPARγ target promoters for transcriptional repression. To examine whether RORα and HDAC3 are co-recruited to the PPARγ target promoters for the repression, we performed ChIP assay with anti-RORα, PPARγ, PPARα, RNA polymerase II (Pol II), acetylated H3 (H3Ac) and HDAC3 antibodies from the mouse liver extracts of CD or HFD-fed RORαf/f and RORαLKO mice. ChIP assays revealed that RORα and HDAC3 were co-recruited to the Cd36, Scd1 and Plin2 promoters in the liver of HFD-fed RORαf/f mice, although no changes were observed with CD-fed RORαf/f mice (Fig. 5a and Supplementary Fig. 4a, b). In the absence of RORα, PPARγ recruitment was markedly increased, whereas HDAC3 recruitment was largely diminished along with elevated acetylated H3 levels on the Cd36, Scd1 and Plin2 promoters in the liver of HFD-fed RORαLKO mice (Fig. 5a and Supplementary Fig. 4a, b). Unlike PPARγ, PPARα recruitment was barely detected from the PPARγ target promoters containing PPRE as assessed by ChIP assay (Fig. 5a and Supplementary Fig. 4b, c).
Fig. 5.
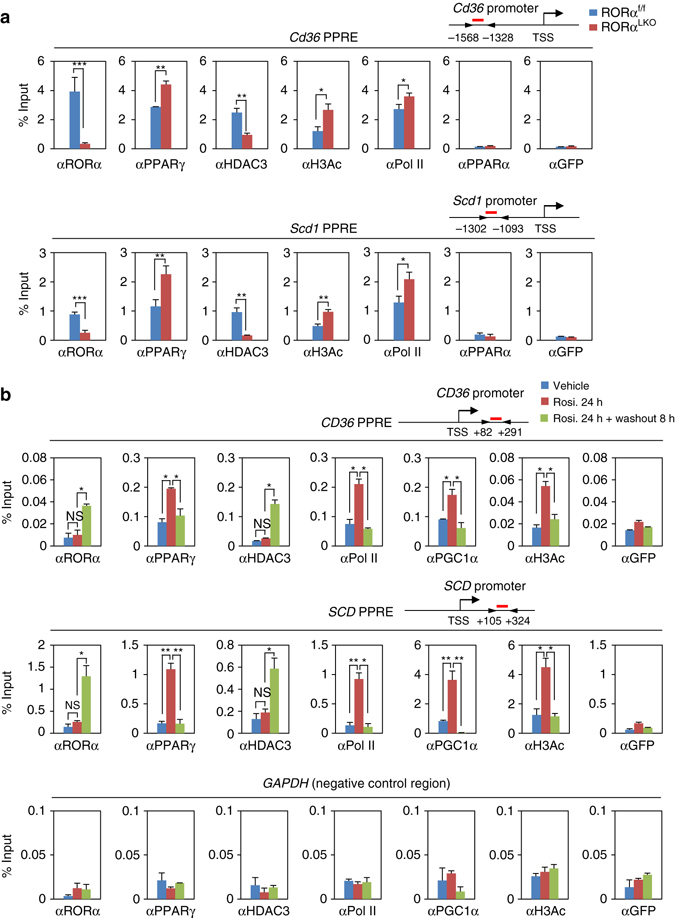
RORα recruits to the PPARγ target gene promoters with HDAC3. a ChIP assays were performed on the Cd36 and Scd1 promoters in liver extract form RORαf/f and RORαLKO mice fed HFD for 10 weeks (n = 3 per group). Promoter occupancy by RORα, PPARγ, HDAC3, H3Ac, Pol II, PPARα and GFP was analyzed. Schematic of promoter region was represented with gene name. Red bar depicts locations of PPRE. Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001. b ChIP assays were performed on the CD36, SCD promoters and GAPDH-negative region in Hep3B cells with or without Rosiglitazone (20 μM) treatment for 24 h and washout 8 h. Promoter occupancy of RORα, PPARγ, HDAC3, Pol II, PGC1α, H3Ac and GFP was analyzed. Schematic of promoter region was represented with gene name. Red bar depicts locations of PPRE. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc analysis. *P < 0.05, **P < 0.01. Data expressed as mean ± s.e.m
We next determined if both RORα and HDAC3 are recruited to the PPRE in response to PPARγ agonist in Hep3B cells (Supplementary Fig. 5a). Treatment of rosiglitazone largely induced the expression of PPARγ target genes (Supplementary Fig. 5b). Interestingly, 8 h washout after rosiglitazone treatment dramatically reduced PPARγ target gene expressions (Supplementary Fig. 5a, b). Consistent with gene expressions, treatment of rosiglitazone increased recruitment of PPARγ, PGC1α and Pol II with elevated histone H3 acetylation level on PPARγ target promoters as well as induction of PPARγ target genes (Fig. 5b and Supplementary Fig. 5c). Strikingly, further increased recruitment of RORα to PPRE was observed along with enhanced HDAC3 recruitment in the setting of washout of rosiglitazone for 8 h (Fig. 5b and Supplementary Fig. 5c). Increased recruitment of RORα and HDAC3 substantially diminished PGC1α and Pol II recruitment on PPRE with decreased histone H3 acetylation level on PPRE (Fig. 5b and Supplementary Fig. 5c).
Next, we further determined whether PPARγ antagonist GW9662 also resulted in the increased recruitment of RORα and HDAC3 to the PPARγ target promoters. Consistent with the results from 8 h washout, GW9662 treatment significantly reduced the expression levels of PPARγ target genes (Supplementary Fig. 6a). ChIP assay revealed that recruitment of RORα and HDAC3 to the PPARγ target promoters were markedly increased, while PPARγ and Pol II recruitments were markedly reduced in response to GW9662 treatment (Supplementary Fig. 6b).
RORα competes with PPARγ for binding to PPARγ target promoters
To address HDAC3 recruitment to PPARγ target gene promoters requires RORα, we first examined the PPARγ target gene induction in the presence or absence of RORα. The induction of PPARγ target genes by rosiglitazone was further enhanced by RORα siRNA, indicating that RORα is a critical transcriptional repressor for PPARγ target gene expression (Supplementary Fig. 7a). RORα knockdown largely increased the recruitment of PPARγ for transcriptional activation with increased levels of H3 acetylation to the PPARγ target gene promoters (Fig. 6a and Supplementary Fig. 7b). While remarkably increased by rosiglitazone washout, the HDAC3 recruitment was substantially reduced by RORα knockdown even with setting of rosiglitazone washout (Fig. 6a and Supplementary Fig. 7b). Taken together, these data clearly indicate that RORα is required for recruitment of HDAC3 to PPARγ target gene promoters.
Fig. 6.
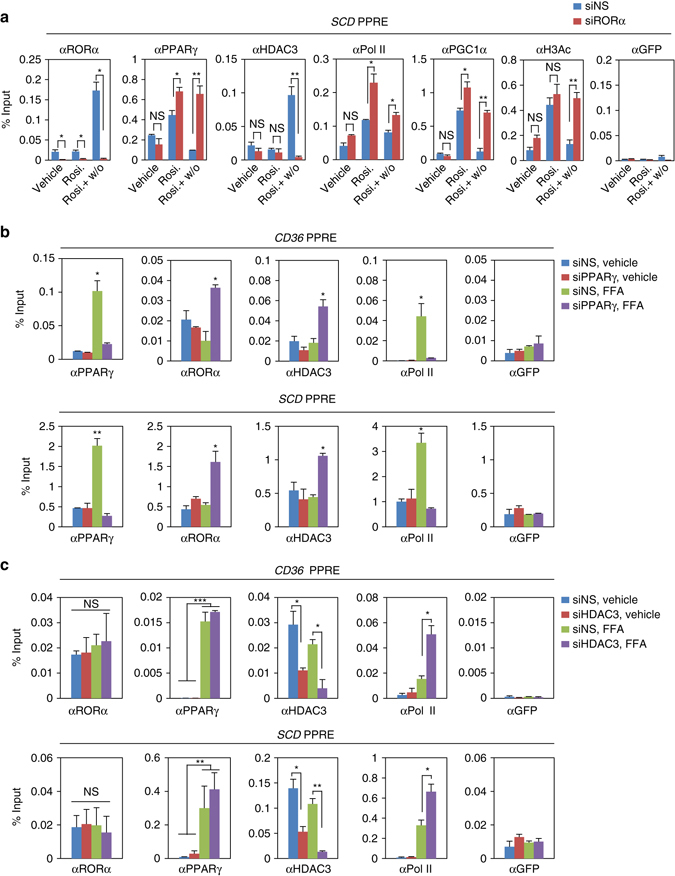
Recruitment of RORα and PPARγ to the PPARγ target gene promoters are mutually exclusive. a ChIP assays were performed in the absence or presence of RORα on SCD promoters in Hep3B cells with or without Rosiglitazone (20 μM) treatment for 24 h and washout 8 h. Promoter occupancy of RORα, PPARγ, HDAC3, Pol II, PGC1α, H3Ac and GFP was analyzed. Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, **P < 0.01, NS, non-significant. b, c ChIP assays were performed in the absence or presence of PPARγ b/HDAC3 c on the CD36 and SCD promoters in Hep3B cells with or without free fatty acid (free fatty acid: Oleic acid 200 μM and Palmitic acid 100 μM) treatment for 24 h. Promoter occupancy of PPARγ, RORα, HDAC3, Pol II and GFP was analyzed. Data expressed as mean ± s.e.m. Statistical analysis was performed using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001. Data expressed as mean ± s.e.m
To determine if RORα competes with PPARγ for the binding to the PPARγ target promoters, we transiently knocked down PPARγ in Hep3B cells. To mimic HFD feeding conditions in vitro, we treated cells with free fatty acid (FFA) and examined expression of PPARγ target genes. We observed that FFA treatment markedly increased PPARγ target gene expressions in both WT and Pparα-null mouse primary hepatocytes, indicating that PPARα failed to influence on PPARγ transcriptional network in the setting of HFD (Supplementary Fig. 7c). Next, we tested PPARγ target gene expressions in the presence of PPARγ siRNA or HDAC3 siRNA. While repressed by PPARγ siRNA, expression of PPARγ target genes was largely enhanced by HDAC3 siRNA in response to FFA treatment (Supplementary Fig. 7d). Consistent with gene expression, increased PPARγ recruitment by FFA was substantially diminished by PPARγ siRNA (Fig. 6b). Interestingly, recruitment of RORα was dramatically increased to the PPARγ target gene promoters, and HDAC3 recruitment was accompanied by the RORα recruitment to PPARγ target promoters by knockdown of PPARγ (Fig. 6b). Furthermore, Re-ChIP assay clearly indicated that PPARγ and RORα are able to bind to the same genomic region and their recruitments to promoter of target genes are mutually exclusive (Supplementary Fig. 7e). These data strongly indicate that RORα and HDAC3 compete with PPARγ for the binding to the target gene promoters for regulation of gene expressions with opposite transcriptional outputs. Altogether, our data demonstrate that RORα functions as a corepressor along with HDAC3 and is co-recruited to the PPARγ target promoters for the repression of PPARγ-mediated transcriptional activity.
To determine whether RORα recruitment is accompanied by the presence of HDAC3, we examined recruitment of RORα to the PPARγ target gene promoters in the presence of HDAC3 siRNA. Although little or no difference of the recruitment of RORα and PPARγ was observed, Pol II recruitment to the PPARγ target gene promoters was markedly increased by HDAC3 knockdown (Fig. 6c), indicating that the presence of HDAC3 affected the recruitment of RNA polymerase II to the PPARγ target gene promoters. Taken together, these data indicate that both RORα and HDAC3 serve as transcriptional corepressors on the PPARγ target gene promoters for the repression of PPARγ target gene expressions.
PPARγ antagonism restores metabolic homeostasis in RORαLKO mice
Since RORα turned out to play a key role to repress PPARγ transcriptional activity in vitro and in vivo, we next examined if inhibition of PPARγ transcriptional activities restores impaired metabolic homeostasis. For this, PPARγ antagonist GW9662 were treated to RORαf/f and RORαLKO mice for 5 weeks with HFD. Intriguingly, the body weight gain of both RORαf/f and RORαLKO mice were markedly reduced by GW9662 treatment compared with vehicle-treated control mice (Fig. 7a). The reduction of body weight gain by GW9662 in RORαLKO mice was much greater, leading to similar body weight to GW9662-treated RORαf/f mice, indicating that inhibition of PPARγ activity remarkably reduces body weight gain in RORαLKO mice (Fig. 7a). Similar to reduced body weight gain, tissue weights of the liver and epidydimal white adipose tissue were markedly reduced by GW9662 treatment in both RORαf/f and RORαLKO mice (Fig. 7b, c). Consistently, cross-sectional area of adipocytes was significantly reduced by GW9662 treatment (Fig. 7d). In accordance with body weight reduction, PPARγ antagonism markedly reduced hepatic steatosis in both RORαf/f and RORαLKO mice (Fig. 7e). Consistently, gene expression profile analysis exhibited that target gene expression levels involved in hepatic gluconeogenesis and lipogenesis are largely reduced by GW9662 treatment (Fig. 7f, g). Together, we demonstrate that enhanced PPARγ transcriptional activity by RORα deficiency is de-activated by PPARγ antagonism to restore metabolic homeostasis, including body weight gain, hepatic steatosis and glucose and lipid metabolism.
Fig. 7.
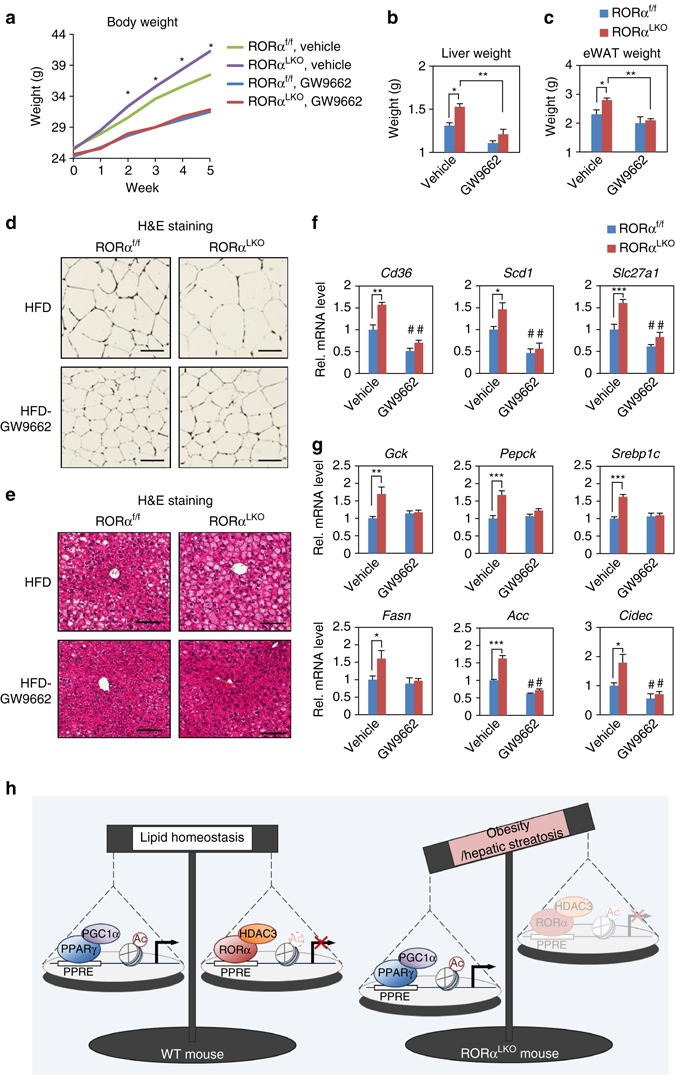
PPARγ antagonism restores metabolic homeostasis in RORαLKO mice. a–e RORαf/f and RORαLKO mice were fed HFD with or without GW9662 for 5 weeks (n = 4–5 per group). a Body weight curves. Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, RORαf/f vs RORαLKO, vehicle. b, c Liver b and epidydimal white adipose tissue (eWAT) c weight of RORαf/f and RORαLKO mice were fed HFD with or without GW9662 for 5 weeks. Statistical analysis was performed using two-way ANOVA. *P < 0.05, **P < 0.01. d, e Representative histological section images from eWAT d and liver e of RORαf/f and RORαLKO mice fed HFD with or without GW9662 for 5 weeks. Scale bar, 100 μm. f, g Expression levels of PPARγ target genes f or gluconeogenesis/lipogenesis/lipid sequestration genes g in liver from RORαf/f and RORαLKO mice fed HFD with or without GW9662 for 5 weeks as determined by quantitative PCR with reverese transcription. Expression was normalized to 36B4 expression. Statistical analysis was performed using Student’s unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001, # P < 0.05 compared to each vehicle group. Data expressed as mean ± s.e.m. h Proposed model for the role of RORα in hepatocyte. RORα regulates PPARγ signaling via HDAC3 recruitment to the PPARγ target promoters for transcriptional repression
Discussion
Hepatic nuclear receptors play critical roles in the regulation of lipid and glucose metabolism in response to environmental stress, including nutrient and hormonal cues41. Dysfunction of hepatic nuclear receptors is largely linked to metabolic diseases including obesity and type II diabetes. We found that PPARγ signaling is a critical pathway affected by hepatic deletion of RORα. Dysregulated PPARγ signaling in RORαLKO mice results in uncontrolled lipogenesis, contributing to the development of hepatic steatosis and diet-induced obesity on a HFD. Furthermore, treatment of PPARγ antagonist GW9662 decreased the susceptibility to obesity42, 43. Consistent with previous reports, we also observed that elevated PPARγ transcriptional activity in RORαLKO mice are downregulated after treatment of GW9662, resulting in decrease of diet-induced hepatic steatosis and obesity. Our data confirm that RORα is a key factor for the repression of PPARγ signaling to protect against diet-induced hepatic steatosis and obesity in vivo.
Together, PPARγ signaling turns out to be significantly activated in HFD-fed RORαLKO mice while increased RORα reduces PPARγ transcriptional activity, providing a direct molecular link between RORα and PPARγ. Furthermore, our data indicate that RORα regulates PPARγ signaling through RORα-mediated HDAC3 recruitment to the PPARγ target promoters. Thus, RORα plays a crucial role in maintaining homeostasis of lipid metabolism in liver by negatively regulating PPARγ signaling via HDAC3 recruitment to the PPARγ target promoters for transcriptional repression (Fig. 7h).
Thiazolidinedione (TZD) is a synthetic PPARγ agonist and has been clinically approved to improve glucose homeostasis and fatty liver in human patients. Although the molecular mechanisms still remain unclear, the ‘lipid steal’ hypothesis has been widely accepted to explain of how TZD treatment improves insulin resistance in type II diabetes patients44–46. However, though PPARγ activation has shown to reduce blood glucose level and hepatic gluconeogenesis, and improve glucose tolerance47, 48, several reports have shown that PPARγ activation leads to hepatic steatosis49, 50. In general, the expression of PPARγ is very low in human and mouse liver. Interestingly, the expression level of hepatic PPARγ is significantly upregulated in obese rodent model51 and high level of PPARγ in mouse liver is sufficient for the induction of adipogenic transformation of hepatocytes with adipose tissue-specific gene expression and lipid accumulation52. These data indicate that PPARγ plays a key role in development of hepatic steatosis. Accordingly, inhibition of PPARγ signaling and hepatic deficiency of PPARγ in ob/ob mice have shown to ameliorate fatty liver53, 54. A recent study clearly showed that PPARγ antagonism improves insulin sensitivity, promotes the browning of white adipose tissue and reduces lipogenic and glucogenic gene expressions in the liver to prevent against diet-induced obesity55. Intriguingly, liver-specific PPARγ-deficient mice exhibit resistance to HFD-induced hepatic steatosis56. Expression of numerous genes involved in lipid uptake and lipid transport was remarkably decreased in the liver-specific PPARγ-deficient mice, resulting in reduction of hepatic steatosis56. Altogether, these studies suggest that local activation of hepatic PPARγ may promote ectopic fat deposition in the liver whereas systemic activation of PPARγ may promote fat deposition in adipose tissue rather than liver leading to clinical improvements of metabolic syndromes including hepatic steatosis. Thus, negative control of RORα to suppress hepatic PPARγ activation is important to maintain physiological hepatic lipid homeostasis. Therefore, these results strongly indicate that the PPARγ signaling pathway is involved in diet-induced hepatic steatosis, and hepatic lipid accumulation is prevented by suppression of PPARγ transcriptional network in the liver.
It has been widely accepted that PPARα is a major nutrient-sensing PPAR isoform to modulate hepatic gene expressions37. As no substantial activation of PPARα has been observed in the fasted or HFD-fed RORαLKO mice, we believe that RORα mainly controls PPARγ transcriptional network to maintain hepatic homeostasis in response to HFD. However, it has been well established that PPARα is a promising therapeutic target to upregulate beta oxidation gene expressions and inhibit hepatic de novo lipogenesis. Consistently, animal model using PPARα-null mice have been reported to develop remarkable hepatic steatosis38, 57. Thus, it is highly possible that hepatic steatosis phenotype in HFD-fed RORαLKO mice may be resulted from both upregulated PPARγ activation and suppressed PPARα transcriptional activity in the absence of RORα. Understanding of the contribution of PPARα transcriptional network with RORα would be critical to delineate the molecular mechanisms of how PPAR isoforms including PPARα and PPARγ modulate hepatic lipid homeostasis with various transcriptional factors in response to environmental stress such as HFD.
It has been reported that RORα may compete with PPARγ for the binding to PPRE58. It is well established that PPRE contains a DR1 motif consisting of two core DRs of AGGTCA separated by a single nucleotide59. Among nucleotides of DR1 motif, the four nucleotides immediately 5′ of DR1 motif are highly conserved and exhibit a consensus of A(A/T)CT. Previous study has reported that the binding of the DBD of PPARs to the single core binding site requires the AT-rich 5′-extended binding site which is quite similar to the binding site for the monomer of RORα60. Thus, the similarity in the binding sequences for PPARγ and RORα appears to allow RORα to modulate PPAR signaling by competing with PPARγ for binding to PPREs61.
The physiological role of HDAC3 has been reported to repress hepatic steatosis. In liver-specific Hdac3-deficient mice, little or no body weight change was observed. As HDAC3 regulates the expression of lipogenic genes in an enzymatic activity-independent manner62, fasting phase markedly promotes hepatic steatosis in liver-specific Hdac3-deficient mice63. An intriguing observation in this study is that RORα is crucial to recruit HDAC3 to repress hepatic PPARγ-mediated lipogenic genes and protect against diet-induced hepatic steatosis and obesity. Furthermore, repressive role of RORα-mediated HDAC3 on lipid metabolism is coupled with elevated hepatic gluconeogenesis. Though hepatic HDAC3 has been shown to promote gluconeogenesis by repressing lipid synthesis and sequestration63, we observed notable increase of gene expression involved in hepatic gluconeogenesis in HFD-fed RORαLKO mice. Intriguingly, HDAC3 ablation upregulated hepatic expression of perilipin gene which contributes to lipid sequestration to ameliorate glucose tolerance63. We also noticed that perilipin 2, hepatic isoform of perilipin was substantially elevated in HFD-fed RORαLKO mice. Unlike hepatic HDAC3 ablation, impaired RORα-mediated HDAC3 transcriptional repression led to interfere hepatic homeostasis of PPARγ signaling. Therefore, disturbed regulatory mechanism of PPARγ signaling in HFD-fed RORαLKO mice would be the main cause of the insulin resistance and glucose intolerance. Consistent with elevated fasting glucose level in HFD-fed RORαLKO mice, mRNA level of the rate-limiting enzyme, phosphoenol pyruvate carboxykinase (PEPCK), in the hepatic gluconeogenesis pathway, was largely elevated in HFD-fed RORαLKO mice. Together, our data strongly indicate that physiological role of HDAC3 in the liver is to suppress PPARγ transcriptional activity via RORα to control hepatic lipid and glucose metabolism.
Previously, it has been reported that bile acid signaling pathway is critical to modulate EE in brown adipose tissue. Bile acids activates mitogen-activated protein kinase pathways and serve as ligands for the G protein-coupled receptor TGR529. Thus, hepatic bile acid synthesis and bile acid pool size in the serum is critical to control metabolic rate. Bile acids induces cyclic-AMP-dependent thyroid hormone activating enzyme type 2 iodothyronine deiodinase (D2)29. Thus, bile acid-TGR5-cAMP-D2 signaling pathway in the brown adipose tissue has been known as a crucial mechanism to modulate EE29. We observed that several key genes involved in bile acid synthesis were largely downregulated as well as serum bile acid pool size in HFD-fed RORαLKO mice. Though we still do not know the direct mechanism of how hepatic bile acid signaling was impaired in RORαLKO mice, we speculate that impaired hepatic bile acid synthesis would impair TGR5 activation in brown adipose tissue to reduce EE.
Several of the observed metabolic alterations in the RORαLKO mice are indeed different from those observed in sg mice. For example, RORαLKO mice gain significantly more weight than WT mice and develop hepatic steatosis when fed with HFD. However, sg mice are protected from HFD-induced obesity and fatty liver and display improved insulin sensitivity28, 64. A strong difference between these two mouse models is their overall growth condition. RORαLKO mice have no obvious phenotypic abnormalities under normal dietary conditions, whereas sg mice suffer from severe growth retardation that would likely be attributed to a number of developmental defects. In addition, defective RORα function in other tissues including brain is likely to systemically affect energy intake and expenditure in the sg mice, making it difficult to specifically dissect hepatic function of RORα. Therefore, it would be helpful to utilize these two mice and compare their phenotypes in certain conditions together for understanding of RORα function in vivo. Collectively, our data indicate that liver-specific RORα deficient mice were successfully developed and the utilization of the mice allowed us to be able to study in vivo functions of RORα in liver more precisely by excluding the potential secondary effect of sg mice.
In conclusion, our data indicate that RORα requires HDAC3 to regulate PPARγ signaling to maintain lipid homeostasis in response to over-nutrient cue. We demonstrate that major target of RORα in the liver is the PPARγ signaling and lipid/glucose metabolism. Our findings provide a direct link between RORα and hepatic fatty acid and glucose metabolism. Thus, therapeutic strategies designed to modulate RORα activity may be beneficial for the treatment of hepatic disease as well as obesity-associated metabolic diseases.
Methods
Generation of conditional Rorα-deficient mice and animal care
To generate mice with a floxed RORα allele, a 16.5 kb region used to construct the targeting vector was first subcloned from a BAC clone (bMQ-293I20, Source BioScience) into a pBluescript phagemid system. The FRT-flanked puromycin cassette containing a loxP sequence was inserted at the front of exon 4 and the single loxP site was inserted at the back of exon 5. The target region was ~15.2 kb which included exons 4 and 5. Twenty micrograms of the targeting vector was linearized by SalI and then electroporated to E14Tg2A ES cells. Surviving clones after puromycin selection were expanded and analyzed by Southern blot to confirm recombinant ES clones. After BamHI digestion, the bands representing WT and mutant alleles are 9.0 and 6.8 kb, respectively. Targeted ES cells were selected for microinjection into C57BL/6 blastocysts to generate chimeras. The male chimeras were bred with C57BL/6 female mice to select for germline transmission. To remove the puromycin selection cassette, targeted heterozygous F1 was crossed with Flp deleter strain (FLPeR mice, The Jackson Laboratory strain 003946). The mice were backcrossed to C57BL/6 then crossed with Alb-Cre mice (The Jackson Laboratory strain 003574) to generate liver-specific Rorα-deficient mice. Male RORαf/f and RORαLKO mice at 8 weeks of age were fed a CD or a 60% kcal fat HFD (Research diet, D12492) during 10 weeks. The sample sizes for all animal studies were announced in each figure legend. Mice were housed in a specific pathogen-free AAALAC-accredited facility under controlled conditions of temperature (25 °C) and light (12 h light:12 h dark, lights switched on at 7:00 a.m.). Food and water were available ad libitum. All mice used in these experiments were backcrossed to C57BL/6 at least seven generations. Animals for each group of experiments were chosen randomly. The primers used in PCR analysis for genotyping floxed alleles are: forward 5′-GCTTGTGGGTTTCTCCTACA-3′ and reverse 5′-GCAGCAAGTGTTGTGTCCCA-3′. This study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of National Cancer Center Research Institute.
Body composition
Fat and lean body masses were assessed by 1H magnetic resonance spectroscopy (Bruker BioSpin).
Indirect calorimetry
Oxygen consumption (VO2), carbon dioxide production (VCO2), respiratory exchange ratios, EE and food consumption were measured using an indirect calorimetry system PHENOMASTER (TSE System). Mice in each chamber were maintained at a constant environmental temperature of 22 °C.
Isolation and culture of primary mouse hepatocytes
Mouse primary hepatocytes were isolated from the liver of 8-week-old male RORαf/f and RORαLKO mice or WT and PPARα null mice by the collagenase perfusion method65. Dissociation into individual hepatocytes was performed in Dulbecco’s modified Eagles’ medium (DMEM) (Welgene) containing 10% heat-inactivated fetal bovine serum (FBS), 1% antibiotics, 20 mM HEPES, 100 nM insulin, 1 nM dexamethasone. For each hepatocyte preparation, cell viability was estimated by the exclusion of trypan blue.
Total bile acid (TBA) measurement
The quantitative determination of total bile acid of mice serum that was collected after centrifugation of mice blood was measured using the total bile acids assay kit (DZ042A-K, Diazyme Laboratories), according to the manufacturer’s instructions.
Histology
When mice were euthanized by CO2 asphyxiation, livers and white adipose tissues (WATs) were rapidly fixed in 10% formalin (Sigma) at 4 °C overnight. After fixation, tissues were sequentially dehydrated in ethanol with increasing concentrations ranging from 50 to 100%. Dehydrated specimens were subsequently infiltrated with 100% xylene and embedded in paraffin wax. For hematoxylin and eosin (H&E) staining, tissues were sectioned at 5 μm thickness, deparaffinized, rehydrated and stained with hematoxylin for 3 min followed by counterstaining with eosin for 1 min. For Oil red O staining, fresh samples of liver embedded in OCT tissue freezing medium (Sakura Finetek). 0.5% Oil red O solution was prepared by dissolving 0.5 g Oil red O powder (Sigma) in 100 ml propylene glycol (sigma). Fresh frozen specimens were cryosectioned at 8 μm thickness and air dried. Then fix in ice cold 10% formalin for 10 min, air dried again, and rinsed with distilled water. Sections were placed in 100% propylene glycol for 5 min and stained with pre-warmed 0.5% Oil red O solution in propylene glycol for 15 min in 60 °C oven. Then sections were rinsed with distilled water and followed by counterstaining with hematoxylin. Images were acquired using digital microscopes (Leica DMD108, Leica microsystems) equipped with ×10 and ×20 objective lenses.
Quantitative real-time RT-PCR
Total RNAs were extracted using Trizol (Invitrogen) and reverse transcription was performed from 2.5 μg of total RNAs using the M-MLV cDNA Synthesis kit (Enzynomics). The abundance of mRNA was detected by a CFX384 TouchTM Real-Time PCR Detection System (Bio-Rad) with SYBR Green (Enzynomics). The quantity of mRNA was calculated using ΔΔCt method and normalized by using primers indicated in each figure legend. All reactions were performed as triplicates. Primers used for analysis are listed in Supplementary Data 5.
Intraperitoneal glucose or insulin tolerance tests
For GTTs, 2 g of glucose per kg of mice body weight was injected i.p. to overnight fasted mice. For ITTs, 0.75 U of insulin (Humulin R, Eli Lilly) per kg of mice body weight was injected i.p. to 6 h fasted mice. Mice blood was drawn at indicated time intervals from the tail tip puncture, and blood glucose level was measured by accu-check perfoma glucometer (Roche).
Generation of mRNA-sequencing data
Four groups of mice, CD-fed RORαf/f, CD-fed RORαLKO, HFD-fed RORαf/f and HFD-fed RORαLKO mice, were analyzed by RNA-sequencing. Four mice per group were killed, and livers from two mice were pooled to generate two samples per group, that is, duplicate experiments for each group were performed. Total RNA extraction was performed using Trizol (Invitrogen). Poly(A) mRNA isolation from total RNA and fragmentation were performed using the Illumina Truseq RNA Sample Prep Kit with poly-T oligo-attached magnetic beads, according to the manufacturer’s instructions. Reverse transcription of RNA fragments was performed using Superscript II reverse transcriptase (Life Technologies). The adaptor-ligated library was size-selected by band excision after agarose gel electrophoresis and purified using the QIAquick gel extraction kit (Qiagen). The prepared mRNA-sequencing libraries were pair-end sequenced on an Illumina Hi-seq 2500. The accession number for the mRNA-sequencing data in this paper is GSE83338.
Analysis of mRNA-sequencing data
After removing adapter sequences (TrueSeq universal and index adapters), we used cutadapter software66 to trim the reads that PHRED scores lower than 20. Remaining reads were aligned to the mouse reference genome (GRCm38) using TopHat aligner67. After the alignment, we quantified the expression of genes as Fragments Per Kilobase of transcript per Million mapped reads (FPKM) for each gene using Cufflinks68. To identify the DEGs, we first selected the ‘expressed’ genes as the ones with FPKM larger than 1 under at least one of the eight samples. For the expressed genes, log2(FPKM + 1) values were normalized across eight samples using the quantile normalization method. To identify the DEGs, for each gene, we calculated a T-statistic and log2-fold-change in the comparisons of RORαLKO/RORαf/f HFD and RORαLKO/RORαf/f CD. We then estimated empirical distributions of T-statistics and log2-fold changes for the null hypothesis by random permutation of the eight samples (1000 permutations). On the basis of the distributions, for each gene, we computed adjusted P values for the observed T-statistic and log2-fold-change and the combined these P values with Stouffer’s method69. Finally, we identified the DEGs as the ones that have the combined P-value ≤ 0.05 and absolute log2-fold-change ≥ 0.439, which is a cutoff value (the 95th percentile of the empirical distribution for log2-fold changes) for each comparison. We further identified RORα-dependent genes under HFD condition as the ones with significant differences between the log2-fold-change in the two comparisons above (RORαLKO/RORαf/f HFD and RORαLKO/RORαf/f CD) larger than 0.439.
Functional enrichment analysis and TF enrichment analysis
For the genes in Groups 1–8, enrichment analysis of GOBPs and KEGG pathways were performed using a DAVID software34. We selected the GOBPs and KEGG pathways with P-value < 0.05 as the ones represented by the genes analyzed. For the genes in Group 1, TF enrichment analysis was performed using a ChEA2 software33. Among the TF-target gene data, only mouse TF-target gene data were used for the enrichment analysis. We selected the TFs with P-value < 0.01 as the ones significantly regulating the genes in Group 1.
Luciferase reporter assay
HEK293T cells (ATCC) and Hep3B cells (Korean Cell Line Bank) were grown and transiently transfected by using polyethylenimine (PEI) and turbofect (Thermo Scientific, R0531). All cell lines used in the study were regularly tested for mycoplasma contamination. For luciferase reporter assays, 1 × 105 cells were seeded in DMEM supplemented with 10% FBS and 1% antibiotics. Cells were transfected with PPRE-luciferase reporters and β-galactosidase expression constructs along with several expression constructs were indicated in each figure. Using a luciferase assay system (Promega), the luciferase activity was measured with a luminometer (Berthold Technologies) after 48 h of transfection. Transfection efficiency was normalized by β-galactosidase expression. The results were obtained from at least three independent experiments
Co-immunoprecipitation assay
HEK293T cells that transfected with Flag-HDACs and HA-RORα were cultured and lysed with lysis buffer (200 mM NaCl, 50 mM Tris-HCl, pH 8.0 and 0.5% NP40). About 20 mg of cell extracts was immunoprecipitated with each 1 μg of anti-Flag antibody overnight and then incubated with 35 μl (50% slurry) of protein A/G agarose beads for 1 h. The immunoprecipitated materials were washed with 500 μl of washing buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.0 and 0.5% NP40) for four times and bound materials were eluted by boiling in 50 μl of sampling buffer (2% β-mercaptoethanol, 5% glycerol, 1% SDS and 60 mM Tris-HCl, pH 6.8) and subjected to immunoblot analysis. Protein samples were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis Images of the immunoblots were visualized and recorded using the LAS 4000-mini system (Fujifilm). Original uncropped images of immunoblots used in this study can be found in Supplementary Fig. 8.
Chromatin Immunoprecipitation (ChIP) and Re-ChIP assays
The ChIP assays were conducted as described. Cells were crosslinked with 1% formaldehyde for 10 min at room temperature. Mouse livers were harvested and quickly washed with PBS and crosslinked with 1% formaldehyde for 10 min at room temperature, followed by quenching with 0.125 M glycine solution for 5 min. Then, cells or harvested mouse livers were washed with ice-cold PBS two times. Chromatin fragmentation was performed by sonication in ChIP lysis buffer (50 mM Tris-HCl (pH 8.1), 1% SDS, 10 mM EDTA (pH 7.6), and protease inhibitor cocktail) with an average size of approximately 500 bp. Proteins were immunoprecipitated in ChIP dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl (pH 8.1), and protease inhibitor cocktail). Crosslinking was reversed overnight at 65 °C in elution buffer (1% SDS, 0.1 M NaHCO3), and DNA was purified with a QIAquick Gel extraction Kit (QIAGEN). For the Re-ChIP assay, components were eluted from the first immunoprecipitation reaction by incubation with 10 mM DTT at 37 °C for 30 min and diluted 1:50 with ChIP dilution buffer containing 20 mM Tris-HCl (pH 8.1), 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100 followed by reimmunoprecipitation with the secondary antibody. Precipitated DNA was analyzed by quantitative PCR. For real-time quantitative PCR analysis, 2 μl from 60 μl DNA extractions was used. All reactions were performed in triplicates. Primers used for analysis are listed in Supplementary Data 5.
GW9662-treated mice
RORαf/f and RORαLKO male mice at 8 weeks of age were subjected to GW9662 at a dose of 0.35 mg per kg body weight per day or an equivalence volume of vehicle in their drinking water for 5 weeks with feeding HFD. The sample sizes for this study was announced in figure legend.
Antibodies
Commercially available antibodies were used: anti-RORα (sc-28612; 1:1000 dilution for IB analysis, 5 μg for ChIP assay), anti-tubulin (sc-8035, 1:5000 dilution for IB analysis), anti-AKT (sc-8312; 1:1000 dilution for IB analysis), anti-PPARα (sc-9000x, 1 μg for ChIP assay) and anti-GFP (sc-9996, 1 μg for ChIP assay) from Santa Cruz Biotechnology; anti-β-actin (A5441; 1:5000 dilution for IB analysis) and anti-FLAG (F3165, Sigma, 1:5000 dilution for IB analysis, 1 μg for IP assay) from Sigma; anti-HA (MMS-101R; 1:5000 dilution for IB analysis, 1 μg for IP assay) from Covance; anti-H3Ac (#06-599, 1 μg for ChIP assay) from Millipore; anti-phospho-AKT(Ser473) (#4051 S, 1:1000 dilution for IB analysis) from Cell Signaling; anti-PPARγ (ab41928, 1 μg for ChIP assay), anti-PGC1α (ab54481, 1 μg for ChIP assay) and anti-HDAC3 (ab7030, 1 μg for ChIP assay) from Abcam; anti-RNA polymerase II (MMS-126R, 1 μg for ChIP assay) from Berkeley antibody company; anti-V5 (R96025; 1:5000 dilution for IB analysis) from Invitrogen.
Statistical analysis
For animal studies, sample size for experiments were determined empirically based on previous studies to ensure appropriate statistical power. Animals for each group of experiments were chosen randomly. No animals were excluded from statistical analysis, and the investigators were not blinded in the studies. The statistical analysis of different groups is realized using the Student’s unpaired t-test or one-way analysis of variance (ANOVA) followed by Tukey post hoc test or two-way ANOVA. SPSS software (IBM) was used for all analyses.
Data availability
mRNA-sequencing data that support the findings of this study have been deposited in Gene Expression Omnibus (GEO) with the primary accession codes GSE83338.
Electronic supplementary material
Acknowledgements
We thank members of the Chromatin Dynamics Research Center for technical assistance and discussions, J.B.K. for providing PPARγ constructs. We thank the NCC Animal Sciences Branch for excellent guidance and assistance with the performed mouse experiments. This work was supported by Creative Research Initiatives Program (Research Center for Chromatin Dynamics, 2009-0081563) to S.H.B.; Global PH.D Fellowship Program (NRF-2011-0008101) to K.K. and (NRF-2012H1A2A1009905) to Y.S.Y.; Korea Mouse Phenotyping Project (2013M3A9D5072550) to S.G.Y., I.Y.K., J.K.S. and S.F.; Basic Science Research Program (NRF-2015R1D1A1A01058037) to K.B., (NRF-2014R1A6A3A04057910) to H.K.; and (NRF -2015R1C1A1A01052195) to S.F.; Medical Research Center (NRF-2014R1A5A2010008) to S.-S.I.; Institute for Basic Science (IBS-R013-G1) to D.H. from the National Research Foundation (NRF) grant funded by the Korea government (MSIP). H.L. was supported by the National Cancer Center Grant (NCC-1310100).
Author contributions
K.K., K.I.K., H.L., S.F. and S.H.B. designed the experiments. K.K., K.B., S.K.O. Y.J. and H.L. generated and maintained RORαf/f and RORαLKO mice. S.G.Y., I.Y.K. and J.K.S. performed mouse metabolic cage assay. J.-S.L. and S.-S.I. provided Pparα-null primary hepatocyte. K.K., S.K.O. and S.F. analyzed mouse phenotype. K.K. performed histological assays. K.K., K.B., S.K.O. and H.K. performed the cell biology and biochemistry experiments. K.K., Y.S.Y., J.B. and D.H. performed mRNA-sequencing preparation and analysis. K.K., Y.S.Y., D.H., S.F. and S.H.B. wrote the manuscript. All authors contributed to data analysis.
Competing interests
The authors declare no competing financial interests.
Footnotes
Kyeongkyu Kim and Kyungjin Boo contributed equally to this work.
Electronic supplementary material
Supplementary Information accompanies this paper at doi:10.1038/s41467-017-00215-1.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sungsoon Fang, Email: sfang@yuhs.ac.
Sung Hee Baek, Email: sbaek@snu.ac.kr.
References
- 1.Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 2.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 3.Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. N. Engl. J. Med. 2017;376:254–266. doi: 10.1056/NEJMra1514009. [DOI] [PubMed] [Google Scholar]
- 4.Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. J. Cell Biol. 2015;208:501–512. doi: 10.1083/jcb.201409063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Berghe G. The role of the liver in metabolic homeostasis: implications for inborn errors of metabolism. J. Inherit. Metab. Dis. 1991;14:407–420. doi: 10.1007/BF01797914. [DOI] [PubMed] [Google Scholar]
- 6.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 2004;114:147–152. doi: 10.1172/JCI200422422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samuel VT, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 8.Chawla A, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell. 2001;7:161–171. doi: 10.1016/S1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 9.Giguere V. Orphan nuclear receptors: from gene to function. Endocr. Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 10.Baek SH, Kim KI. Emerging roles of orphan nuclear receptors in cancer. Annu. Rev. Physiol. 2014;76:177–195. doi: 10.1146/annurev-physiol-030212-183758. [DOI] [PubMed] [Google Scholar]
- 11.Bookout AL, et al. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullican SE, Dispirito JR, Lazar MA. The orphan nuclear receptors at their 25-year reunion. J. Mol. Endocrinol. 2013;51:T115–140. doi: 10.1530/JME-13-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearen MA, Muscat GE. Orphan nuclear receptors and the regulation of nutrient metabolism: understanding obesity. Physiology (Bethesda). 2012;27:156–166. doi: 10.1152/physiol.00007.2012. [DOI] [PubMed] [Google Scholar]
- 14.Li L, et al. The nuclear orphan receptor COUP-TFII plays an essential role in adipogenesis, glucose homeostasis, and energy metabolism. Cell Metab. 2009;9:77–87. doi: 10.1016/j.cmet.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim DK, et al. Orphan nuclear receptor estrogen-related receptor gamma (ERRgamma) is key regulator of hepatic gluconeogenesis. J. Biol. Chem. 2012;287:21628–21639. doi: 10.1074/jbc.M111.315168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranhotra HS. The orphan nuclear receptors in cancer and diabetes. J. Recept. Signal. Transduct. Res. 2013;33:207–212. doi: 10.3109/10799893.2013.781624. [DOI] [PubMed] [Google Scholar]
- 17.Pellicciari R, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 18.Billin AN. PPAR-beta/delta agonists for Type 2 diabetes and dyslipidemia: an adopted orphan still looking for a home. Expert. Opin. Investig. Drugs. 2008;17:1465–1471. doi: 10.1517/13543784.17.10.1465. [DOI] [PubMed] [Google Scholar]
- 19.Katz A, et al. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J. Clin. Pharmacol. 2009;49:643–649. doi: 10.1177/0091270009335768. [DOI] [PubMed] [Google Scholar]
- 20.Lee JM, et al. RORalpha attenuates Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in colon cancer. Mol. Cell. 2010;37:183–195. doi: 10.1016/j.molcel.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 21.Kim H, et al. DNA damage-induced RORalpha is crucial for p53 stabilization and increased apoptosis. Mol. Cell. 2011;44:797–810. doi: 10.1016/j.molcel.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 22.Vogel MW, Sinclair M, Qiu D, Fan H. Purkinje cell fate in staggerer mutants: agenesis versus cell death. J. Neurobiol. 2000;42:323–337. doi: 10.1002/(SICI)1097-4695(20000215)42:3<323::AID-NEU4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 23.Doulazmi M, et al. Cerebellar Purkinje cell loss during life span of the heterozygous staggerer mouse (Rora(+)/Rora(sg)) is gender-related. J. Comp. Neurol. 1999;411:267–273. doi: 10.1002/(SICI)1096-9861(19990823)411:2<267::AID-CNE7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 24.Guillaumond F, Dardente H, Giguere V, Cermakian N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J. Biol. Rhythms. 2005;20:391–403. doi: 10.1177/0748730405277232. [DOI] [PubMed] [Google Scholar]
- 25.Sato TK, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–537. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Vu-Dac N, et al. Transcriptional regulation of apolipoprotein A-I gene expression by the nuclear receptor RORalpha. J. Biol. Chem. 1997;272:22401–22404. doi: 10.1074/jbc.272.36.22401. [DOI] [PubMed] [Google Scholar]
- 27.Mamontova A, et al. Severe atherosclerosis and hypoalphalipoproteinemia in the staggerer mouse, a mutant of the nuclear receptor RORalpha. Circulation. 1998;98:2738–2743. doi: 10.1161/01.CIR.98.24.2738. [DOI] [PubMed] [Google Scholar]
- 28.Lau P, et al. The orphan nuclear receptor, RORalpha, regulates gene expression that controls lipid metabolism: staggerer (SG/SG) mice are resistant to diet-induced obesity. J. Biol. Chem. 2008;283:18411–18421. doi: 10.1074/jbc.M710526200. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe M, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 30.Kahn BB, Flier JS. Obesity and insulin resistance. J. Clin. Invest. 2000;106:473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004;432:1027–1032. doi: 10.1038/nature03047. [DOI] [PubMed] [Google Scholar]
- 32.Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu. Rev. Med. 2005;56:45–62. doi: 10.1146/annurev.med.56.082103.104751. [DOI] [PubMed] [Google Scholar]
- 33.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 35.Kou Y, et al. ChEA2: gene-set libraries from ChIP-X experiments to decode the transcription regulome. Multidisciplinary research and practice for information systems. CD-ARES 2013. Lect. Notes Comput. 2013;8127:416–430. doi: 10.1007/978-3-642-40511-2_30. [DOI] [Google Scholar]
- 36.Dreyer C, et al. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 37.Lee JM, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kersten S, et al. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 40.Hong H, Kohli K, Trivedi A, Johnson DL, Stallcup MR. GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc. Natl Acad. Sci. USA. 1996;93:4948–4952. doi: 10.1073/pnas.93.10.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53:1023–1034. doi: 10.1002/hep.24148. [DOI] [PubMed] [Google Scholar]
- 42.Nakano R, et al. Antagonism of peroxisome proliferator-activated receptor gamma prevents high-fat diet-induced obesity in vivo. Biochem. Pharmacol. 2006;72:42–52. doi: 10.1016/j.bcp.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 43.Ma S, et al. Thyrotropin and obesity: increased adipose triglyceride content through glycerol-3-phosphate acyltransferase 3. Sci. Rep. 2015;5:7633. doi: 10.1038/srep07633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye JM, et al. Direct demonstration of lipid sequestration as a mechanism by which rosiglitazone prevents fatty-acid-induced insulin resistance in the rat: comparison with metformin. Diabetologia. 2004;47:1306–1313. doi: 10.1007/s00125-004-1436-1. [DOI] [PubMed] [Google Scholar]
- 45.Kim JK, et al. Differential effects of rosiglitazone on skeletal muscle and liver insulin resistance in A-ZIP/F-1 fatless mice. Diabetes. 2003;52:1311–1318. doi: 10.2337/diabetes.52.6.1311. [DOI] [PubMed] [Google Scholar]
- 46.Auwerx J. PPARgamma, the ultimate thrifty gene. Diabetologia. 1999;42:1033–1049. doi: 10.1007/s001250051268. [DOI] [PubMed] [Google Scholar]
- 47.Saltiel AR, Olefsky JM. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45:1661–1669. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- 48.Way JM, et al. Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor gamma activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology. 2001;142:1269–1277. doi: 10.1210/endo.142.3.8037. [DOI] [PubMed] [Google Scholar]
- 49.Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2005;288:E1195–1205. doi: 10.1152/ajpendo.00513.2004. [DOI] [PubMed] [Google Scholar]
- 50.Lee YJ, et al. Nuclear receptor PPARgamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. USA. 2012;109:13656–13661. doi: 10.1073/pnas.1203218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vidal-Puig A, et al. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Invest. 1996;97:2553–2561. doi: 10.1172/JCI118703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu S, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J. Biol. Chem. 2003;278:498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 53.Yamauchi T, et al. Inhibition of RXR and PPARgamma ameliorates diet-induced obesity and type 2 diabetes. J. Clin. Invest. 2001;108:1001–1013. doi: 10.1172/JCI12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsusue K, et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 2003;111:737–747. doi: 10.1172/JCI200317223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi SS, et al. PPARgamma antagonist gleevec improves insulin sensitivity and promotes the browning of white adipose tissue. Diabetes. 2016;65:829–839. doi: 10.2337/db15-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moran-Salvador E, et al. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–2550. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- 57.Hashimoto T, et al. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000;275:28918–28928. doi: 10.1074/jbc.M910350199. [DOI] [PubMed] [Google Scholar]
- 58.Ohoka N, Kato S, Takahashi Y, Hayashi H, Sato R. The orphan nuclear receptor RORalpha restrains adipocyte differentiation through a reduction of C/EBPbeta activity and perilipin gene expression. Mol. Endocrinol. 2009;23:759–771. doi: 10.1210/me.2008-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palmer CN, Hsu MH, Griffin HJ, Johnson EF. Novel sequence determinants in peroxisome proliferator signaling. J. Biol. Chem. 1995;270:16114–16121. doi: 10.1074/jbc.270.27.16114. [DOI] [PubMed] [Google Scholar]
- 61.Harding HP, Lazar MA. The monomer-binding orphan receptor Rev-Erb represses transcription as a dimer on a novel direct repeat. Mol. Cell Biol. 1995;15:4791–4802. doi: 10.1128/MCB.15.9.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun Z, et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell. 2013;52:769–782. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Z, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 2012;18:934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lau P, Fitzsimmons RL, Pearen MA, Watt MJ, Muscat GE. Homozygous staggerer (sg/sg) mice display improved insulin sensitivity and enhanced glucose uptake in skeletal muscle. Diabetologia. 2011;54:1169–1180. doi: 10.1007/s00125-011-2046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang HS, et al. Metformin stimulates IGFBP-2 gene expression through PPARalpha in diabetic states. Sci. Rep. 2016;6:23665. doi: 10.1038/srep23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 67.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hwang D, et al. A data integration methodology for systems biology. Proc. Natl Acad. Sci. USA. 2005;102:17296–17301. doi: 10.1073/pnas.0508647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
mRNA-sequencing data that support the findings of this study have been deposited in Gene Expression Omnibus (GEO) with the primary accession codes GSE83338.