

Abstract
We describe a 52-year-old woman presenting with acute onset of severe burning paraesthesia in the hands and feet associated with allodynia and antalgic gait. At the time of admission to hospital no motor weakness was present. A diagnosis of Guillain-Barré syndrome (GBS) was considered when neurophysiological studies were completed showing convincing evidence of demyelination on motor conduction studies and sural sparing on sensory nerve studies.1 We describe this case as a sensory variant of GBS. Clinical improvement followed treatment with a single course of intravenous immunoglobulin (IVIG). The patient made a complete clinical recovery within 6 months of onset and repeat neurophysiological studies showed marked improvement. We encourage clinicians to consider an atypical variant of GBS in patients presenting with acute sensory complaints.
Keywords: General Practice / Family Medicine, Clinical Neurophysiology, Pain (neurology), Peripheral Nerve Disease
Background
Typical Guillain-Barré syndrome (GBS) manifests with progressive ascending symmetric motor weakness. However, many phenotypic variants of GBS are now recognised. Sensory variants have been described in the literature, but their place on the GBS spectrum remains controversial.2 Based on the size of sensory nerve fibres involved and the site of primary nerve damage, sensory GBS has been classified into one of three subtypes: acute demyelinating polyneuropathy, acute large fibre sensory neuronopathy and acute small fibre sensory neuropathy.3 This case is instructive as our patient presented with small fibre sensory symptoms and GBS was not initially considered as an aetiology. However, neurophysiological studies identified demyelinating features consistent with a sensory variant of GBS and findings on cerebrospinal fluid (CSF) analysis also supported the diagnosis. This case adds to the growing body of evidence to support including sensory GBS in the differential diagnosis for a patient presenting with sensory complaints in the absence of significant motor weakness.
Case presentation
Our patient is a 52-year-old woman of Filipino origin who presented to the emergency department with an 8-day history of intense burning pain associated with a ‘pins and needles’ sensation. This started in the hands and forearms and 4 days later spread to involve the soles of the feet making it painful to walk. She experienced such severe pain in her hands on contact with any surface that she was unable to do any personal or household tasks including dressing and holding eating utensils. She denied any weakness but reported fatigue and hypersensitivity of the right side of her face and scalp to touch, with discomfort combing her hair or placing that side of her face down on a pillow.
There was no history of antecedent illness, immunisation or viral infection. She had not been started on any new medications. She had a past history of untreated hypertension but was otherwise healthy. She had no history of diabetes or malignancy. She had no history of recent travel. There was no family history of neuropathy or any neurological diseases.
In the emergency department, she was found to have a blood pressure of 228/120 mmHg and a mildly elevated troponin. Although her neurological symptoms were her chief complaint, she was admitted to hospital primarily for investigation and treatment of her hypertension.
Neurological examination on admission was notable for antalgic gait with difficulty with toe and heel walking primarily related to pain. Pain made the objective assessment of strength difficult but her motor examination was felt to be normal. Deep tendon reflexes were present at 1+ in the upper extremities and at the knees, with absent ankle reflexes. On sensory examination light touch was intact throughout. However, there was stocking glove distribution hyperaesthesia to pain and temperature in the upper extremities to the level of the wrist and in the lower extremities to the mid-calf. Vibration sense was absent at the toes, present at the ankles and vibration threshold was impaired in the distal fingertips. Proprioception was intact to small amplitude toe and finger movements.
Investigations for her hypertension included a CT angiogram, which was negative for aortic dissection, renovascular stenosis and arteritis. Cardiac workup was negative for arrhythmia. Her blood pressure normalised in hospital with standard medical treatment but the neurological symptoms persisted without a clear cause on initial workup. Therefore she underwent neurophysiological testing to attempt to elucidate a cause for her symptoms.
Neurological examination 5 days after admission, at the time of her nerve conduction studies, had changed slightly in that she had mild weakness of finger extension and of the intrinsic muscles graded 4+/5 and of toe extension and flexion graded 4+/5. Her neurological examination was otherwise unchanged.
Investigations
Vitamin B12 level, fasting glucose, haemoglobin A1C, serum protein electrophoresis and autoimmune workup were all unremarkable. Workup for porphyria was not completed.
Neurophysiological studies showed markedly prolonged median (9 ms; normal: <4.4 ms) and peroneal (10.5 ms; normal: <6.5 ms) distal motor latencies with prolonged median and ulnar F waves (36.5 ms) (table 1). Sensory studies were notable for normal sural sensory amplitude with absent median and very small amplitude ulnar sensory response (figure 1)(table 2).
Table 1.
Motor nerve conduction studies at the time of initial diagnosis showing prolongation of the median & fibular distal motor latencies (bold)
| Nerve | Lat (ms) | Dist (mm) | Amp P-P (mV) | Amp O-P (mV) | Amp% (%) | Area (ms*mV) | Stimulus intensity (mA) | CV (m/s) |
| Median motor right | ||||||||
| Wrist—APB | 9.04 | 70 | 7.3 | 5.4 | – | 31.4 | 37.2 | – |
| Elbow—Wrist | 13.4 | 196 | 7.4 | 5.5 | 1.85 | 32.2 | 32.0 | 45.0 |
| Ulnar motor right | ||||||||
| Wrist—ADM | 3.96 | 70 | 7.2 | 5.1 | – | 31.5 | 30.2 | |
| Bl. elbow—Wrist | 7.13 | 176 | 6.6 | 4.7 | −7.8 | 29.0 | 37.0 | 55.5 |
| Ab. elbow—Bl. elbow | 9.04 | 120 | 6.5 | 4.8 | 2.1 | 29.4 | 9.8 | 62.8 |
| Fibular motor right | ||||||||
| Ankle—EDB | 10.5 | 90 | 7.4 | 5.3 | – | 32.3 | 15.3 | – |
| Bl. FibHd—Ankle | 16.8 | 255 | 6.9 | 5.0 | −5.7 | 30.7 | 52.6 | 40.5 |
| Ab. FibHd—Bl. FibHd | 18.5 | 67 | 7.3 | 5.4 | 8.0 | 32.7 | 24.4 | 39.4 |
Ab., above; ADM, abductor digiti minimi; Amp, amplitude; APB, abductor pollicis brevis; Bl., below; CV, conduction velocity; dis, distal; EDB, extensor digitorum brevis; FibHd, fibular head; lat, latency; O-P, onset to peak; P-P, peak to peak.
Figure 1.
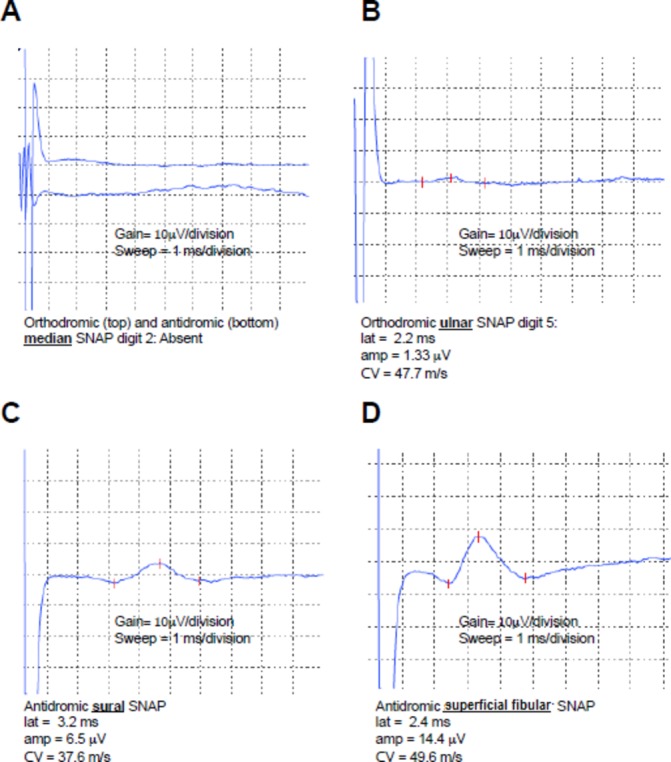
Sensory nerve action potentials of the median (A), ulnar (B) sural (C) and superficial fibular (D) nerves at the time of initial presentation. Gain and sweep speed are highlighted below the individual waveforms for reference. Parameters of the sensory nerve action potentials, including latency, peak amplitude and conduction velocity are listed below the respective waveforms. Amp, peak amplitude; CV, conduction velocity; lat, latency; SNAP, sensory nerve action potential.
Table 2.
Sensory nerve conduction studies at the time of initial diagnosis
| Nerve | Lat (ms) | Dist (mm) | Amp (µV) | Stimulus intensity (mA) | Temperature (˚C) | CV (m/s) |
| Median sensory right | ||||||
| Orthodromic—F2 | – | 126 | – | 8.1 | – | – |
| Antidromic—F2 | – | 126 | – | 7.5 | – | – |
| Ulnar sensory right | ||||||
| Orthodromic—F5 | 2.20 | 105 | 1.33 | 7.9 | – | 47.7 |
| Sural sensory right | ||||||
| Antidromic—Lat. Mal | 3.19 | 120 | 6.5 | 11.4 | 28.6 | 37.6 |
| Superficial fibular sensory right | ||||||
| Antidromic—Lat. Foot | 2.42 | 120 | 14.4 | 11.8 | – | 49.6 |
Amp, amplitude; CV, conduction velocity; dis, distal; lat, latency; Lat., lateral; Mal., malleolus.
Lumbar puncture was completed and CSF analysis revealed elevated CSF protein at 1100 mg/L (normal: <450 mg/L) and a leucocyte count of 0.001×109/L. These findings were consistent with albuminocytological dissociation. Neurological antibodies including GM1, GM2, GD1a, GD1b, GQ1b and GT1b were all within normal limits.
Workup for secondary causes of hypertension was unremarkable.
Differential diagnosis
The neurophysiological finding of sural sparing is a characteristic finding of GBS helping distinguish it from other length dependent neuropathies.1 The striking prolongation of the distal motor latencies and F waves were consistent with an acute demyelinating neuropathy, and with the finding of albuminocytological dissociation on the CSF a presumed diagnosis of GBS was made. No other diagnosis was felt likely given unremarkable findings on the rest of the completed investigations.
Treatment
Our patient was treated with intravenous immunoglobulin (IVIG) at a total dose of 2 g/kg given over 4 days. Symptoms of disabling neuropathic pain were treated with increasing dosages of pregabalin and nortriptyline. Her blood pressure was well controlled on amlodipine 10 mg daily, ramipril 10 mg twice daily and hydrochlorothiazide 12.5 mg daily.
Outcome and follow-up
The patient was seen in neurological follow-up at 3, 4 and 6 months after symptom onset. Neurophysiological studies were repeated at 6 months. At 3 months after onset her neurological symptoms were markedly improved and she had returned to full-time work as a healthcare aide. At 4 months, she no longer required neuropathic pain medications. At 6 months, her neurological examination had normalised and neurophysiological studies were markedly improved. Distal motor latencies were still mildly prolonged (figure 2)(table 3). The median and ulnar sensory responses were now present but mildly reduced in amplitude. At 6-month follow-up, her blood pressure was well controlled at 130/80 mmHg on ramipril and amlodipine. Consideration was given to whether the severe hypertension on admission was secondary to autonomic dysfunction related to GBS. However, given the response to usual antihypertensive medications, previously documented untreated hypertension and persistent need for antihypertensives even after the GBS had resolved this was felt to be unlikely. Our clinical impression remains that the patient had underlying untreated essential hypertension exacerbated by pain on presentation.
Figure 2.
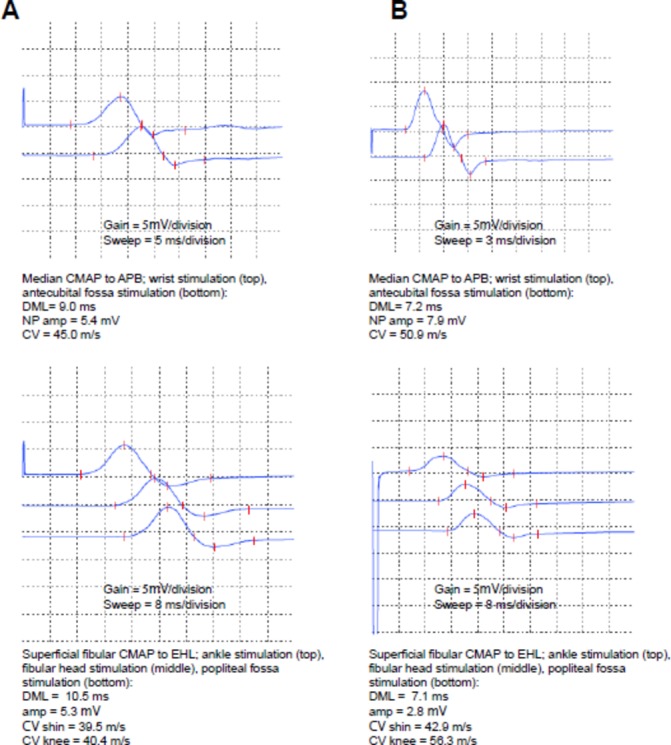
Compound muscle action potentials for the median (top) and superficial fibular (bottom) nerves at the time of initial presentation (A) and at follow-up (B). Gain and sweep speed are highlighted below the individual waveforms for reference. Note the difference in sweep speed for the median study (top panels), to explain the difference in waveform morphology between A and B. Parameters of the compound muscle action potentials are listed below the respective waveforms for reference. APB, abductor pollicis brevis; CMAP, compound muscle action potential; CV, conduction velocity; DML, distal motor latency; EDB, extensor digitorum brevis; NP amp, negative peak amplitude.
Table 3.
Motor nerve conduction studies 6 months after initial diagnosis showing partial improvement in the median and fibular distal motor latencies (bold)
| Nerve | Lat (ms) | Dist (mm) | Amp P-P (mV) | Amp O-P (mV) | Amp% (%) | Area (ms*mV) | Stimulus intensity (mA) | Temperature (˚C) | CV (m/s) |
| Median motor right | |||||||||
| Wrist—APB | 7.17 | 70 | 11.5 | 7.9 | – | 29.7 | 15.0 | 33.6 | – |
| Elbow—Wrist | 11.1 | 200 | 10.2 | 6.8 | −13.9 | 23.8 | 37.0 | 50.9 | |
| Ulnar motor right | |||||||||
| Wrist—ADM | 4.38 | 70 | 9.6 | 6.8 | – | 22.8 | 20.4 | ||
| Bl. Elbow—Wrist | 8.42 | 195 | 8.3 | 6.0 | −11.8 | 20.8 | 13.0 | 48.3 | |
| Ab. Elbow—Bl. Elbow | 11.1 | 110 | 7.7 | 5.7 | −5.0 | 22.7 | 18.7 | 41.0 | |
| Fibular motor right | |||||||||
| Ankle—EDB | 7.08 | 90 | 3.9 | 2.8 | – | 16.3 | 95.2 | – | |
| Bl. FibHd—Ankle | 12.6 | 237 | 4.5 | 3.3 | −17.9 | 17.5 | 95.2 | 42.9 | |
| Ab. FibHd—Bl. FibHd | 14.2 | 90 | 4.6 | 3.4 | 3.0 | 17.9 | 95.2 | 56.3 | |
Ab., above; ADM, abductor digiti minimi; Amp, amplitude; APB, abductor pollicis brevis; Bl., below; CV, conduction velocity; dis, distal; EDB, extensor digitorum brevis; FibHd, fibular head; lat, latency; O-P, onset to peak : P-P, peak to peak.
Discussion
Most reported cases of ‘sensory GBS’ in the literature rely on criteria set forth by Asbury in 1981 for a sensory and areflexic variant of GBS.4 At that time, he stated that ‘the precise diagnostic limits of GBS remain uncertain’ and this is still true today. Although several variants of GBS are widely recognised, the question of whether patients presenting with predominantly sensory findings can truly be considered to have GBS is still unanswered, with some supporting its inclusion as a subtype and others arguing that it should be considered a separate clinical entity.2 5 6 There are ongoing efforts to clarify whether there is a place on the GBS spectrum for a sensory variant and to characterise this clinical entity using clinical and pathological findings.7
The criteria put forward by Asbury include rapid onset of sensory symptoms, symmetric and widespread distribution, complete or nearly complete recovery, characteristic electrodiagnostic results showing demyelination and elevated CSF protein. Despite this, a review of diagnostic criteria for GBS, which is still widely used today, does not formally include the sensory variant.8 An updated set of eight criteria for sensory GBS was proposed by Oh et al in 2001 and supported by a series of eight cases; however, they have not been widely adopted. The proposed criteria include: (1) acute onset of sensory symptoms, (2) peak deficit achieved within 4 weeks, (3) diminished or absent reflexes, (4) normal motor strength, (5) nerve conduction evidence of demyelination in at least two nerves, (6) monophasic course, (7) no other known cause for neuropathy and (8) no family history of neuropathy.9
Our case fulfils five of the five criteria proposed by Asbury and seven of the eight diagnostic criteria proposed by Oh et al for sensory GBS.4 9 Our case had mild distal weakness and thus did not fulfil the criteria of normal motor strength.
In 2012, Uncini and Yuki continued to advocate for the recognition of a sensory variant of GBS, identifying 22 suspected cases from 1980 to early 2011. Based on the size of sensory nerve fibres involved and the site of primary nerve damage Uncini and Yuki subclassified sensory GBS into one of three subtypes: an acute demyelinating polyneuropathy, an acute large fibre neuronopathy and an acute small fibre sensory neuropathy.3 Our case would fall into the acute demyelinating polyneuropathy subtype on the basis of the nerve conduction studies showing evidence of demyelination.
More recently, several case reports of ‘sensory GBS’ have emerged in venues including conference proceedings. The lack of formal diagnostic criteria is problematic because authors characterise the cases as sensory GBS, but the clinical presentations are heterogeneous, including descriptions of trigeminal nerve involvement,10 a case with normal reflexes11 and a number of cases with normal nerve conduction studies that were characterised as possible small fibre variants.12 13
The lack of a clear consensus in the literature has contributed to uncertainty regarding whether a distinct clinical entity such as sensory GBS exists. Patients presenting with purely sensory symptoms and signs may have fewer objective abnormalities on neurological examination and the diagnosis of GBS in this situation may be more challenging. This case report is important, as GBS was not initially considered at the time of our patient’s admission to hospital. It was the subsequent neurophysiological studies which led to a diagnosis of GBS and optimal treatment.
In conclusion, GBS is a remarkably diverse condition with varied clinical presentations including predominantly sensory variants. Further research into the relationships of acute peripheral neuropathy with specific anti-ganglioside antibodies may prove helpful in better understanding the varied phenotypes of GBS.14
Learning points.
This case adds to the body of evidence supporting a sensory variant of Guillain-Barré syndrome (GBS).
A sensory variant of GBS should be considered in the differential diagnosis of acutely evolving sensory symptoms even in absence of weakness.
Referral to a neurologist for nerve conduction studies and cerebrospinal fluid analysis should be undertaken in suspected cases of sensory GBS.
Diagnosis of GBS in this setting facilitates specific treatment such as intravenous immunoglobulin and may prevent unnecessary investigations such as MRI or peripheral nerve biopsy.
Acknowledgments
The authors wish to acknowledge the contributions of the clinical staff of the Clinical Teaching Unit and Neurology services at Vancouver General Hospital who contributed to the care of this patient. We would also like to acknowledge and thank the patient and her family who have graciously allowed us to share the details of her case.
Footnotes
Contributors: MS, GG, PT and LK were all involved in the clinical care of the patient. Each author contributed to the content and writing of the initial draft of the manuscript and to revisions. GG prepared the accompanying tables and figures. All approved the final draft of the manuscript to be submitted for publication and agree to take public responsibility for the content of the manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Derksen A, Ritter C, Athar P, et al. . Sural sparing pattern discriminates Guillain-Barré syndrome from its mimics. Muscle Nerve 2014;50:780–4. 10.1002/mus.24226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barré syndrome. The Lancet 2016;388:717–27. 10.1016/S0140-6736(16)00339-1 [DOI] [PubMed] [Google Scholar]
- 3.Uncini A, Yuki N. Sensory Guillain-Barré syndrome and related disorders: an attempt at systematization. Muscle Nerve 2012;45:464–70. 10.1002/mus.22298 [DOI] [PubMed] [Google Scholar]
- 4.Asbury AK. Diagnostic considerations in Guillain-Barré syndrome. Ann Neurol 1981;9(Suppl):1–5. 10.1002/ana.410090703 [DOI] [PubMed] [Google Scholar]
- 5.Hughes RAC. Sensory form of Guillain-Barré syndrome. The Lancet 2001;357:1465 10.1016/S0140-6736(00)04672-9 [DOI] [PubMed] [Google Scholar]
- 6.Callaghan BC, Price RS, Chen KS, et al. . The importance of rare subtypes in diagnosis and treatment of peripheral neuropathy: a review. JAMA Neurol 2015;72:1510–8. 10.1001/jamaneurol.2015.2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang J, Huan M, Jiang H, et al. . Pure sensory Guillain-Barré syndrome: a case report and review of the literature. Exp Ther Med 2014;8:1397–401. 10.3892/etm.2014.1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol 1990;27:S21–4. 10.1002/ana.410270707 [DOI] [PubMed] [Google Scholar]
- 9.Oh SJ, LaGanke C, Claussen GC. Sensory Guillain-Barré syndrome. Neurology 2001;56:82–6. 10.1212/WNL.56.1.82 [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Liu N, Zhang ZC, et al. . Sensory Guillain-Barré syndrome: a case report. Exp Ther Med 2014;8:1713–6. 10.3892/etm.2014.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aggarwal S, Malik D, Amaraneni A, et al. . Pure sensory variant of Guillain-Barré syndrome with normal reflexes - a very rare phenomenon. J Gen Intern Med 2015;30:S443. [Google Scholar]
- 12.Seneviratne U, Gunasekera S. Acute small fibre sensory neuropathy: another variant of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2002;72:540–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang H, Trivedi A, Adoni S, et al. . A case of pure sensory variant of Guillain-Barré syndrome. Neurology 2016;86:P3.373. [Google Scholar]
- 14.Kogawa S, Nakajima A, Kobashi S, et al. . [A case of pure-sensory-type Guillain-Barré syndrome with galactocerebroside antibody]. Rinsho Shinkeigaku 2015;55:55:171–173. 10.5692/clinicalneurol.55.171 [DOI] [PubMed] [Google Scholar]