

Abstract
Chronic lymphocytic leukaemia (CLL) is characterised by a lymphocytosis of mature-appearing clonal CD5+, CD23+ B lymphocytes. CLL cells arise from the bone marrow and infiltrate lymphoid tissues such as lymph nodes and spleen. Presentation is usually through discovery of lymphocytosis or lymphadenopathy. Unusual presentations, especially paraneoplastic syndromes are rare. Here, we describe a rare case presenting with severe nephrotic syndrome associated with the presence of a monoclonal protein in serum. Workup for suspected plasma cell dyscrasia led instead to the diagnosis of bone marrow infiltration by atypical CLL without lymphocytosis. Renal biopsy showed a glomerulonephritis that turned out to be paraneoplastic as it went into remission after treatment for CLL. Our case shows an unusual presentation of CLL and prompts for increased awareness of lymphoproliferative disorders in the context of seemingly unrelated conditions that may be paraneoplastic in origin.
Keywords: haematology (drugs and medicines), nephrotic syndrome
Background
B-cell chronic lymphocytic leukaemia (B-CLL) is the most common type of leukaemia in Western countries.1 B-CLL cells derive from mature, postgerminal centre, immunologically competent B lymphocytes that proliferate and accumulate within the blood, bone marrow, lymph nodes and spleen.1 CLL cells are usually positive for CD19, CD5 and CD23 surface markers,2 and their main genomic alterations are represented by deletions on the long arm of chromosomes 13 and 11, of the short arm of chromosome 17 and by trisomy of chromosome 12.1 In rare cases, CLL cells show an atypical phenotype, i.e. they are positive for CD5 but negative for CD23, and these cases are referred to as atypical CLL. Especially in atypical CLL, positivity for CD200 expression can help discriminate this entity from mantle cell lymphoma, which is CD5+, CD23- and CD200-.3 About 25% of CLL cases display autoimmune complications during the course of the disease. The most common autoimmune manifestations are autoimmune haemolytic anaemia, which occurs in 10%–35% of patients with CLL and immune thrombocytopaenia (1%–2% of patients).4–6 Non-haematological autoimmune manifestations are very rare, yet at least two syndromes have been clearly identified: pemphigus and various forms of glomerulopathies, such as acute or chronic glomerulonephritis or nephrotic syndrome.6–13 The actual incidence of paraneoplastic glomerulonephropathies in CLL is unknown, as this rare condition has only been described in case reports so far. However, when the two condition coexist, CLL is evident through lymphocytosis and/or lymphadenopathies in about 50% of patients.14
More frequently, CLL can affect kidney function in several other ways, ranging from uric acid nephropathy to leukaemic parenchymal infiltration.8 12 15 16 Renal failure can be diagnosed at the beginning or during progression of CLL.10 15 16 Conversely, plasmacellular disorders are usually characterised by detection of an intact monoclonal immunoglobulin in serum and/or urine, and by a monoclonal plasma-cell infiltration of the bone marrow or extramedullary sites. Some cases may either only secrete light chains or be non-secretory altogether, showing only plasmacellular infiltration in the bone marrow and/or in bone lesions.17 18
A causal relationship between tumour and a nephrotic syndrome is suggested if proteinuria develops either 6 months before or after the diagnosis of malignancy.7 In either scenario, criteria for the diagnosis of a paraneoplastic syndrome also include: remission after chemotherapy-induced complete remission, a pathophysiological link between the two diseases and relapse associated with tumour recurrence.7
Very few cases of paraneoplastic nephrotic syndrome in CLL have been reported.8–12 16 19 The most common glomerulonephropathy associated with CLL is membranoproliferative glomerulonephritis (MPGN).9 MPGN displays a pattern of glomerular injury characterised by mesangial hypercellularity, endocapillary proliferation and double-contour formation along the glomerular capillary walls. It can present with nephritic or nephrotic syndrome.20 Other glomerulopathies associated with CLL include membranous glomerulonephritis, minimal change disease and amyloidosis.16 21 22
Often a glomerulonephropathy can be associated with a positive serum protein immunofixation. The Mayo Clinic reported a series of 28 patients affected by hepatitis-negative MPGN associated with a positive immunofixation. In some of these patients, monoclonal glomerular deposits of the same isotype as the circulating paraprotein were found in the glomeruli.21 Furthermore, the bone marrow trephine performed in these patients revealed a lymphoproliferative neoplasm in six of them.21 Further review of the literature revealed that about 30% of proliferative glomerulonephritis with a monoclonal IgG deposit have detectable monoclonal protein in the serum of the same isotypes as the glomerular deposits.23 The immunotactoid glomerulonephropathy is yet another renal disease with glomerular deposition of precipitates in the form of microtubules that stain positive for immunoglobulins by immunofluorescence: 50% of these cases concur with lymphoma or CLL. Based on the above data, the entity ‘monoclonal gammopathy of renal significance’ has been recently defined.22 In cases where monoclonal deposits are demonstrated in the kidney, the same monoclonal protein should be searched in the peripheral blood. Subsequently, a bone marrow trephine should be performed to assess the origin of the monoclonal component.22
Here, we describe an unusual presentation of CLL whose first clinical manifestation was a paraneoplastic glomerulonephropathy.
Case presentation
A 71-year-old Caucasian male developed progressive malaise and leg oedema over a few weeks. He neither had significant comorbidities nor he took any drug. Family history was negative for both kidney disease and cancer.
Blood analysis showed renal impairment (serum creatinine 2 mg/dL, normal range (nr) 0.1–1.2), mild macrocytic anaemia and a low-level monoclonal IgG kappa monoclonal protein on serum electrophoresis (300 mg/dL), associated with hypogammaglobulinaemia.
An abdominal ultrasound displayed only liver and kidney cysts, kidneys appeared of normal size and shape and presented normal renal vasculature. Meanwhile, the patient was referred to our attention because of the monoclonal gammopathy. He presented an Eastern Cooperative Oncology Group performance status of 2. Physical examination showed only new-onset hypertension and pitting oedema of the lower extremities; neither significant lymphadenopathies nor hepatosplenomegaly were present; pruritus and B symptoms were both absent. Chest X-ray was normal.
Blood count showed mild normocytic anaemia (Hb 11.8 g/dL, nr 14–18, MCV 93 fL), normal leucocyte count (10.8 x109/L) with normal lymphocyte count (3.3x10^9/L). Serum electrophoresis was negative for monoclonal protein but immunofixation was positive for an IgG/kappa component. A free light chain dosage showed elevated serum kappa light chains (86 mg/L, nr 3–19) with normal lambda chains and an altered kappa/lambda ratio of 7.1 (nr 0.26–1.65). Total serum proteins were low (5.2 g/dL, nr 6–8.3), with hypoalbuminaemia (3.3 g/dL, nr 3.9–4.9) and severe hypogammaglobulinaemia (IgG 91, nr 700–1600, IgA 13, nr 70–400 and IgM 8 nr 40–230 mg/dL). Cholesterol and triglycerides levels were normal. Coagulation times were normal, but fibrinogen (510 mg/dL, nr 200–400) and D-dimer (4.15 µg/mL, nr 0–0.5) were slightly elevated. Tests for autoimmunity revealed a weak positivity (1:40) for antinuclear antigen antibody. Complement components C3 and C4 were normal. Serology for hepatitis B and hepatitis C virus was negative.
Microscopic examination of urine revealed more than 50 red blood cells and 30 white blood cells per high power field. The patient suffered from severe non-selective glomerular proteinuria of 8.5 g/24 hours (nr 0.01–0.250). Bence Jones proteinuria was positive for kappa chains (40 mg/24 hours, nr <20).
Our results suggested the hypothesis of hyposecretory multiple myeloma or amyloidosis. Therefore, we performed bone marrow analysis. Bone marrow morphology showed instead a marked infiltration of small lymphocytes representing 70% of the whole cellularity. Flow cytometry revealed a monoclonal B-cell population (CD19+, CD20+, CD5+, CD200+, CD22+, FMC7+, CD23-, kappa-, Lambda-, CD38-, CD43-, CD10-). Karyotyping study demonstrated 14q deletion and extramaterial on chromosome 16p. Fluorescence in situ hybridisation analysis of the bone marrow aspirate was negative for chromosome 12 trisomy, deletions of 17p13, 11q22 and 13q and t(11;14)(q13;q32.3) translocation. PCR for BCL-1 rearrangement was negative. On bone marrow trephine, 90% of marrow cells were small–mild mature lymphocytes with a diffuse-interstitial growing pattern and with a CD5+, CD20+, CD3-, CD23-, CD123-, CD138-, Bcl1- phenotype. Contrary to our initial hypothesis, bone marrow results supported the diagnosis of a CD5-positive chronic lymphoproliferative disorder. The B-cell immunophenotype and the negativity for BCL-1 rearrangement suggested the diagnosis of an atypical CLL that is usually CD200 and CD5-positive but lacks the CD23 antigen.3
Because the cause of renal failure remained elusive at that point, in agreement with the nephrologist, the patient also underwent a renal biopsy. The glomeruli of the patient displayed diffuse and global thickening of basement membrane with occasional double-outline features. There was moderate expansion of mesangial matrix with mesangial and endocapillary cellularity. Tubules appeared focally injured with mild focal expansion of basement membrane and vacuolisation of the tubular epithelium (figure 1, arrowhead). Areas of inflammation were found composed of a lymphocytic infiltrate (figure 1, arrow), along with sclerotic thickening of the vascular wall (figure 1, asterisk) and areas of fibrosis (figure 1, square). The immunohistochemistry was positive for segmental-diffuse C3d in the glomerular capillary and kappa chain in the tubular lumen. Congo red staining was negative, ruling out amyloidosis. Glomerular immunofluorescence was weakly positive only for C3, negative for IgG, IgA, IgM, C1q, kappa and lambda chains. Biopsy report and clinical findings were consistent with pauci-immune membranoproliferative necrotising intraextracapillary glomerulonephritis. In light of this findings, we concluded for a diagnosis of atypical CLL presenting as a paraneoplastic nephrotic syndrome.
Figure 1.
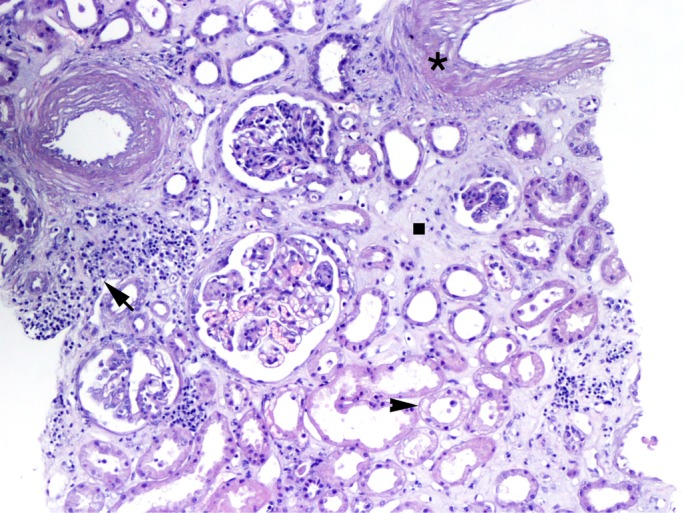
Renal biopsy shows areas of inflammation composed of a lymphocytic infiltrate (arrow), sclerotic thickening of the vascular wall (asterisk), areas of fibrosis (square) and damaged renal tubules with vacuolation (arrowhead).
Treatment
Due to the rapid progressive renal impairment, we decided to start steroid treatment, which was complicated by pneumonia. During pneumonia, his renal function worsened because of both antibiotic therapy and dehydration. After the resolution of pneumonia, we started chemoimmunotherapy with rituximab 375 mg/m2 (day 1), cyclophosphamide 750 mg/m2 (day 1) and prednisone 50 mg/m2 (days 1–5) for a total of six cycles.
Outcome and follow-up
After three cycles, proteinuria decreased to 1.6 g/24 hours, renal function improved (serum creatinine: 1.7 mg/dL) and bone marrow analysis showed a complete disappearance of the lymphoid infiltrate. At the end of six cycles of chemoimmunotherapy, blood analysis showed an increase in the haemoglobin level (Hb 12.4 g/dL) and a negative serum immunofixation with normal serum-free light chain dosage (kappa 8 mg/L, lambda 7 mg/L, ratio 1.14). At the end of treatment, renal function further improved to a proteinuria of 800 mg/24 hours and serum creatinine of 1.3 mg/dL (figure 2). Urine immunofixation was negative. The patient is alive and in complete remission 3 years after the end of treatment.
Figure 2.
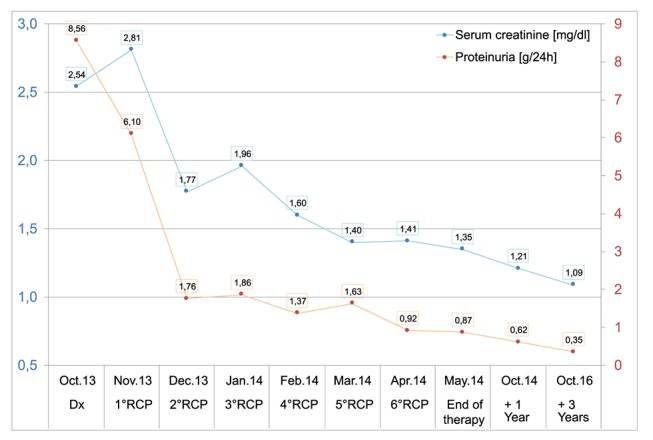
Line chart describing improvement of renal function (serum creatinine, blue, primary Y-axis) and proteinuria (orange, secondary Y-axis). RCP, rituximab, cyclophosphamide and prednisone.
Discussion
Our patient suffered from a nephrotic syndrome due to a pauci-immune MPGN.
In contrast with all the other previously published case reports of paraneoplastic glomerulopathies,8–12 16 19 24–27 no symptoms or laboratory and radiological results were suggestive of CLL at the time of renal failure. Only the bone marrow analysis revealed the underlying lymphoproliferative disorder.
Renal biopsy detected kappa chains in the tubular lumen, but glomerular immunofluorescence was negative for IgG, IgA, IgM, kappa and lambda chains. Most previous reports showed immunoglobulin deposits in the glomeruli while our case showed only light chain deposits in the tubular lumen. Renal biopsy suggested a pauci-immune MPGN, although routine techniques were not able to discriminate between proliferative glomerulonephritis with a monoclonal IgG deposit and immunotactoid glomerulonephropathy.
While electronic microscopy evaluation is important in the diagnostic of glomerulonephropathy, it is not routinely performed in laboratory and it has some limitations since it can result in overlap between types I and III membranoproliferative glomerulonephritis. Immunofluorescence (IF) microscopy-based classification is an alternative system based on the actual pathogenic process, and it is more widespread in pathology laboratories. IF can return valuable information to the practising clinician because it can detect the specific components of the complement pathway and of the immune complexes deposited in the glomerulus, helping directly the correct clinical evaluation and design potential disease-specific treatments.
The case report strongly underlines the utility of performing a bone marrow trephine in all patients presenting an abnormal electrophoresis, immunofixation and/or free light chain ratio, even in the absence of any signs of CLL from physical examination and blood analysis to detect a lymphoproliferative disorder.
The ongoing therapeutic approach for CLL is treating symptomatic patients of Binet B stage and all patients of Binet C stage. In our patient, other than the paraneoplastic syndrome, there was no haematological indication to justify upfront treatment of CLL. Combination therapy with fludarabine, cyclophosphamide and rituximab (FCR) or bendamustine/rituximab are the standard of care, although FCR is not suitable for all patients due to its toxic effects in the elderly and in those with comorbidities.1 28 Ibrutinib and idelalisib have only recently been approved in the first-line treatment of CLL with p53 mutation or 17p deletion.29
Because of our patient age and renal impairment, we decided to start treatment with a steroid pretreatment phase, because of their efficacy both towards lymphoproliferative disorders and immune-mediated glomerulonephritis. Subsequently, after an infectious complication, we opted for a low-intensity cycle aimed primarily at renal manifestations, consisting of prednisone, cyclophosphamide and rituximab. Indeed, cyclophosphamide and rituximab have been shown to be effective both in CLL and glomerulonephritis.30 31 Perry et al reported 14 patients affected by indolent non-Hodgkin lymphomas with monoclonal Ig deposit-related glomerulonephropathy: 63% and 64% of these patients had respectively a complete haematological and renal response following the combination of rituximab, cyclophosphamide and dexamethasone, respectively.32 Consistently, our patient achieved a complete haematological response and a good improvement of renal function, underlying the efficacy of this chemotherapy combination.
Learning points.
Bone marrow trephine is not usually indicated in the diagnostic workup of glomerulonephropathies. In cases associated with monoclonal serum protein and/or hypogammaglobulinaemia and/or altered serum-free light chains, it is important to perform a bone marrow trephine in the suspect of a lymphoproliferative disorder.
Renal biopsy in the context of nephrotic syndromes can show a variety of histological and immmunohistochemical patterns, but may not reveal an underlying lymphoproliferative disorder.
A multidisciplinary approach is needed for the workup and management of unusual presentation of haematological disorders.
Footnotes
Contributors: MS and LF wrote the manuscript. AG contributed to the nephrological data. NB reviewed the case report.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Hallek M. Chronic lymphocytic leukemia: 2013 update on diagnosis, risk stratification and treatment. Am J Hematol 2013;88:803–16. 10.1002/ajh.23491 [DOI] [PubMed] [Google Scholar]
- 2.Rawstron AC, Villamor N, Ritgen M, et al. International standardized approach for flow cytometric residual disease monitoring in chronic lymphocytic leukaemia. Leukemia 2007;21:956–64. 10.1038/sj.leu.2404584 [DOI] [PubMed] [Google Scholar]
- 3.Sandes AF, de Lourdes Chauffaille M, Oliveira CR, et al. CD200 has an important role in the differential diagnosis of mature B-cell neoplasms by multiparameter flow cytometry. Cytometry B Clin Cytom 2014;86:98–105. 10.1002/cyto.b.21128 [DOI] [PubMed] [Google Scholar]
- 4.D'Arena G, Capalbo S, Laurenti L, et al. Chronic lymphocytic leukemia-associated immune thrombocytopenia treated with rituximab: a retrospective study of 21 patients. Eur J Haematol 2010;85:502–7. 10.1111/j.1600-0609.2010.01527.x [DOI] [PubMed] [Google Scholar]
- 5.Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol 2006;33:230–9. 10.1053/j.seminoncol.2006.01.011 [DOI] [PubMed] [Google Scholar]
- 6.Hamblin TJ. Non-hemic autoimmunity in CLL. Leuk Res 2009;33:366–7. 10.1016/j.leukres.2008.09.011 [DOI] [PubMed] [Google Scholar]
- 7.Wągrowska-Danilewicz M, Danilewicz M. Nephrotic syndrome and neoplasia: our experience and review of the literature. Pol J Pathol 2011;62:12–18. [PubMed] [Google Scholar]
- 8.Bartel C, Obermüller N, Rummel MJ, et al. Remission of a B cell CLL-associated membranoproliferative glomerulonephritis Type I with rituximab and bendamustine. Clin Nephrol 2008;69:285–9. 10.5414/CNP69285 [DOI] [PubMed] [Google Scholar]
- 9.Arampatzis S, Giannakoulas N, Liakopoulos V, et al. Simultaneous clinical resolution of focal segmental glomerulosclerosis associated with chronic lymphocytic leukaemia treated with fludarabine, cyclophosphamide and rituximab. BMC Nephrol 2011;12:33 10.1186/1471-2369-12-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moulin B, Ronco PM, Mougenot B, et al. Glomerulonephritis in chronic lymphocytic leukemia and related B-cell lymphomas. Kidney Int 1992;42:127–35. 10.1038/ki.1992.270 [DOI] [PubMed] [Google Scholar]
- 11.Aslam N, Nseir NI, Viverett JF, et al. Nephrotic syndrome in chronic lymphocytic leukemia: a paraneoplastic syndrome? Clin Nephrol 2000;54:492–7. [PubMed] [Google Scholar]
- 12.Barbour SJ, Beaulieu MC, Zalunardo NY, et al. Proliferative glomerulonephritis with monoclonal IgG deposits secondary to chronic lymphocytic leukemia. Report of two cases. Nephrol Dial Transplant 2011;26:2712–4. 10.1093/ndt/gfr251 [DOI] [PubMed] [Google Scholar]
- 13.Strati P, Nasr SH, Leung N, et al. Renal complications in chronic lymphocytic leukemia and monoclonal B-cell lymphocytosis: the Mayo Clinic experience. Haematologica 2015;100:1180–8. 10.3324/haematol.2015.128793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronco PM. Paraneoplastic glomerulopathies: new insights into an old entity. Kidney Int 1999;56:355–77. 10.1046/j.1523-1755.1999.00548.x [DOI] [PubMed] [Google Scholar]
- 15.Da'as N, Polliack A, Cohen Y, et al. Kidney involvement and renal manifestations in non-Hodgkin's lymphoma and lymphocytic leukemia: a retrospective study in 700 patients. Eur J Haematol 2001;67:158–64. 10.1034/j.1600-0609.2001.5790493.x [DOI] [PubMed] [Google Scholar]
- 16.Dou X, Hu H, Ju Y, et al. Concurrent nephrotic syndrome and acute renal failure caused by chronic lymphocytic leukemia (CLL): a case report and literature review. Diagn Pathol 2011;6:99 10.1186/1746-1596-6-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatt ME, Palladini G. 'Light chain amyloidosis 2012: A new era'. Br. J. Haematol 2012;160598:pp. 5822013. [DOI] [PubMed] [Google Scholar]
- 18.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011;364:1046–60. 10.1056/NEJMra1011442 [DOI] [PubMed] [Google Scholar]
- 19.Alzamora MG, Schmidli M, Hess U, et al. Minimal change glomerulonephritis in chronic lymphocytic leukemia: pathophysiological and therapeutic aspects. Onkologie 2006;29:153–6. 10.1159/000091644 [DOI] [PubMed] [Google Scholar]
- 20.Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis--a new look at an old entity. N Engl J Med 2012;366:1119–31. 10.1056/NEJMra1108178 [DOI] [PubMed] [Google Scholar]
- 21.Sethi S, Zand L, Leung N, et al. Membranoproliferative glomerulonephritis secondary to monoclonal gammopathy. Clin J Am Soc Nephrol 2010;5:770–82. 10.2215/CJN.06760909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung N, Bridoux F, Hutchison CA, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 2012;120:4292–5. 10.1182/blood-2012-07-445304 [DOI] [PubMed] [Google Scholar]
- 23.Nasr SH, Satoskar A, Markowitz GS, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol 2009;20:2055–64. 10.1681/ASN.2009010110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hewamana S, Pepper C, Jenkins C, et al. Acute renal failure as the presenting feature of leukaemic infiltration in chronic lymphocytic leukaemia. Clin Exp Nephrol 2009;13:179–81. 10.1007/s10157-009-0129-y [DOI] [PubMed] [Google Scholar]
- 25.Hill PA, Firkin F, Dwyer KM, et al. Membranoproliferative glomerulonephritis in association with chronic lymphocytic leukaemia: a report of three cases. Pathology 2002;34:138–43. 10.1080/003130201201117945 [DOI] [PubMed] [Google Scholar]
- 26.Yahata N, Kawanishi Y, Okabe S, et al. Membranous glomerulonephritis with nephrotic syndrome associated with chronic lymphocytic leukemia. Am J Nephrol;20:402–7. 10.1159/000013626 [DOI] [PubMed] [Google Scholar]
- 27.Rocca AR, Giannakakis C, Serriello I, et al. Fludarabine in chronic lymphocytic leukemia with membranous nephropathy. Ren Fail 2013;35:282–5. 10.3109/0886022X.2012.743912 [DOI] [PubMed] [Google Scholar]
- 28.Fischer K, Cramer P, Busch R, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209–16. 10.1200/JCO.2011.39.2688 [DOI] [PubMed] [Google Scholar]
- 29.Montserrat E, Dreger P. Treatment of chronic lymphocytic leukemia with del(17p)/TP53 mutation: allogeneic hematopoietic stem cell transplantation or BCR-signaling inhibitors? Clin Lymphoma Myeloma Leuk 2016;16:S74–S81. 10.1016/j.clml.2016.02.013 [DOI] [PubMed] [Google Scholar]
- 30.Radhakrishnan J, Cattran DC. The KDIGO practice guideline on glomerulonephritis: reading between the (guide)lines--application to the individual patient. Kidney Int 2012;82:840–56. 10.1038/ki.2012.280 [DOI] [PubMed] [Google Scholar]
- 31.Guiard E, Karras A, Plaisier E, et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol 2011;6:1609–16. 10.2215/CJN.10611110 [DOI] [PubMed] [Google Scholar]
- 32.Perry M, Delarche A, Ribes D, et al. Rituximab–cyclophosphamide–dexamethasone is highly effective in patients with monoclonal Ig deposit-related glomerulopathy and indolent non-Hodgkin lymphomas. Am J Hematol 2014;89:969–73. 10.1002/ajh.23798 [DOI] [PubMed] [Google Scholar]