

Abstract
BRAF mutation testing to determine eligibility for treatment with vemurafenib was performed on archival skin lesions of a 54-year-old patient diagnosed with Erdheim–Chester disease (ECD) in 1999. Sanger sequencing of DNA extracted from a 2008 skin lesion identified two non-contiguous base substitutions in BRAF, which were shown by next-generation sequencing (NGS) to be located in the same allele. Due to its long-standing duration, molecular evolution of disease was possible; however, both Sanger and NGS of a 2000 skin lesion were unsuccessful due to the poor quality of DNA. Finally, droplet digital PCR using a probe specific for this novel mutation detected the complex BRAF mutation in both the 2000 and 2008 lesions, indicating this case to be ECD with a novel underlying BRAF p.Thr599_Val600delinsArgGlu mutation. Although well at present, molecular modelling of the mutant BRAF suggests suboptimal binding of vemurafenib and hence reduced therapeutic effectiveness.
Keywords: Haematology (incl blood transfusion), Oncology
Background
Erdheim–Chester disease (ECD), a rare form of non-Langerhans cell histiocytosis, is characterised by xanthogranulomatous inflammation and the infiltration of tissues with lipid-laden cells expressing macrophage lineage markers.1 Affecting multiple organs including bone, skin, soft tissue, lungs, cardiovascular system, kidneys and the central nervous system, disease-associated morbidity and prognosis are influenced by the sites of organ involvement.1 2 It was recently shown that the disease cells of 40–60% of ECD cases contained somatic BRAF V600E (p.Val600Glu) mutations and clinical responses to the BRAF inhibitor, vemurafenib have been documented.3–5 Additional BRAF point mutations or small insertion/deletions have not yet been reported. Here we describe ECD associated with a unique complex BRAF mutation, p.Thr599_Val600delinsArgGlu (BRAF p.T599_V600>RE), in a patient with long-standing ECD. Although in vitro or in vivo models of ECD are not available, molecular modelling of the mutant BRAF indicates that conformational changes induced by substitutions of the two amino acids would reduce the efficacy of vemurafenib binding. The case illustrates rational usage of currently available sequencing and bioinformatics technologies to overcome the technical difficulties associated with poor tissue quality and limited proportions of disease cells that are typical of this type of pathology and to characterise novel and complex disease-associated mutations.
Case presentation
The patient first presented in 1999 when 37 years of age for evaluation of a left adrenal mass. A diagnosis of ECD was suspected initially on the radiological features and subsequently confirmed on skin biopsy in 2000 (see below). In 2008, he presented with symptomatic pericardial effusion and required surgery with a pericardial window for management of his symptoms. A biopsy at this time was consistent with involvement of the pericardium and skin with ECD. He was referred for haematological review given the development of significant complications of his disease although no specific therapy was offered as his pericardial effusion was satisfactorily managed with pericardial window. He has subsequently undergone regular haematological review with intermittent serial CT scanning to evaluate disease burden. There has been no progressive increase in the disease burden. When last reviewed in 2016, there was diffuse paravertebral infiltration within the thorax and retroperitoneum, but no other parenchymal disease obvious on imaging (figure 1).
Figure 1.
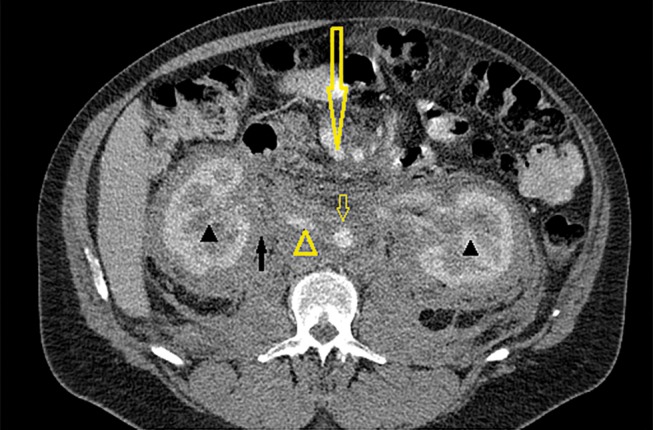
Axial CT image obtained with intravenous contrast in the portal venous phase at the level of the renal hilum. Image shows mildly enhancing soft tissue density infiltrative material in the retroperitoneum (solid arrow), encasing both kidneys (solid arrow heads), inferior vena cava (unfilled arrow head), abdominal aorta (unfilled short arrow) and superior mesenteric vessels (unfilled long arrow).
Investigations
Due to recent reports of the successful treatment of ECD patients with BRAF inhibitors,4 6–8 BRAF mutation analysis was requested for the patient following a routine appointment and two formalin-fixed paraffin-embedded specimens were located in the archives of the Anatomical Pathology department of Royal Perth Hospital, skin lesions from 2000 and 2008. No recent lesional biopsies were available. Review of the pathology from 2000 and 2008 showed superficial dermal lesions each consisting of a localised accumulation of predominantly plump spindled cells with scattered Touton-type giant cells. Both the spindled cells and giant cells were positive for CD68, a macrophage marker, and negative for S100 protein, a marker for neural, melanocytic and Langerhans cells (figure 2).
Figure 2.
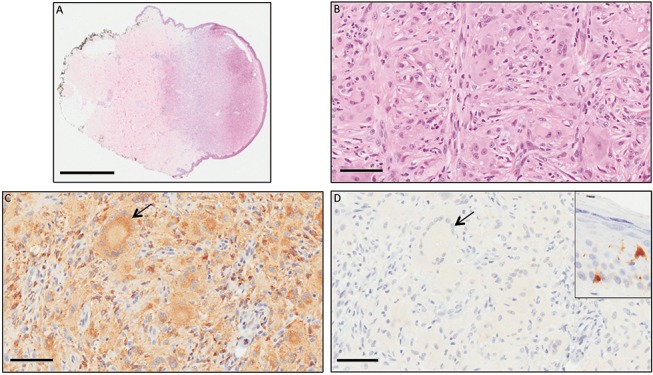
(A) Skin with subcutis showing a nodular infiltrate of cells in superficial and mid-dermis. (B) The infiltrate was composed of abundant plump mononuclear histiocytes and scattered multinucleated histiocytes in a background of dermal fibrosis. (C) The histiocytes, including Touton-type giant cells (arrow), expressed CD68 (C) but did not express S100 (D), which did label Langerhans cells and melanocytes in the epidermis (inset). The features were consistent with a cutaneous deposit of Erdheim–Chester disease. (A) Bar=2 mm; (B–D) bar=70 µm.
DNA extraction from the 2008 specimen was successful; however, DNA yield from the 2000 specimen was low, DNA quality was poor and it was noted that the proportion of disease cells in this specimen was also low. Bidirectional Sanger sequencing for BRAF exon 15 revealed minor levels of sequence variants, c.1796C>G and c.1799T>A, in the 2008 specimen, the appearance of which was consistent with the proportion of disease cells in the lesion (figure 3A). Sequencing quality was not considered suitable to report for the 2000 specimen (not shown). Next-generation sequencing (NGS) was subsequently performed on DNA from both specimens using the Ion AmpliSeq Cancer Hotspot Panel v2, confirming presence of both base substitutions in the 2008 specimen and indicating that the base substitutions always occurred in the same allele (figure 3B). No additional mutations in amplicons targeted in that panel were identified. Thus the BRAF sequence alterations present in this case would result in substitution of two adjacent amino acids, encoding a complex BRAF mutation, c.1796_1799CAGT>GAGA/p.Thr599_Val600delinsArgGlu (p.T599_V600>RE). Sequencing of the 2000 specimen was again unsuccessful (not shown).
Figure 3.

Detection of the complex BRAF c.1796_1799CAGT>GAGA mutation. (A) Sanger sequencing of 2008 skin lesion indicating small mutant peaks in the forward (upper panel) and reverse (lower panel) sequences. (B) Snapshot of Integrative Genomics Viewer analysis of next-generation sequencing from the 2008 skin lesion indicating co-occurrence of the two mutant bases (reverse sequence is depicted, sequencing depth ×1100, mutant allele frequency 9%). (C) Droplet digital PCR showing detection of the complex BRAF mutation in skin lesions from both 2000 (lower left, abundance 4.7%) and 2008 (lower right, abundance 9.2%). Blue = mutant BRAF; green = wild-type BRAF; red = multiple sequences in droplet; black = no probe bound.
In light of the long-standing duration of disease in the patient and because the BRAF mutation included the common base alteration that results in substitution of valine 600 for glutamic acid (BRAF p.Val600Glu/BRAF p.V600E), which normally occurs as the sole mutation, it was considered possible that the second base alteration affecting threonine 599 represented a ‘molecular’ evolution and that the disease originally contained one (either) of the mutations, with the second occurring as the disease progressed. However, this hypothesis required sequencing of DNA from the 2000 specimen, which had not been possible using either Sanger sequencing or NGS. To overcome technical issues associated with the poor quality of DNA in the 2000 specimen, a droplet digital PCR (ddPCR) assay was developed, including design of a TaqMan probe specific for this BRAF mutation (5′-TAGCTAGAGAGAAATC-3′). ddPCR using DNA extracted from the 2000 and 2008 skin lesions identified the complex BRAF mutation in both the 2000 and 2008 specimens (figure 3C). Thus both BRAF base substitutions appear to have been present from shortly following the patient’s diagnosis of ECD.
To further characterise the complex BRAF p.T599_V600>RE mutant, the published crystal structure of BRAF p.V600E in the Protein Data Bank (PDB code: 4MNF) was used to model the structure of BRAF p.T599_V600>RE. Molecular docking and molecular dynamics simulations were carried out to predict the interactions of vemurafenib with dimers of BRAF p.V600E and BRAF p.T599_V600>RE (figure 4). These simulations identified important differences in the stability of vemurafenib binding to BRAF p.T599_V600>RE in comparison to BRAF p.V600E, indicating that vemurafenib is unlikely to bind effectively to BRAF p.T599_V600>RE. Thus at present there is no evidence to support the use of vemurafenib in the management of this patient; however, alternative targeted therapies including MEK inhibitors may be considered.9 Detailed description of the molecular modelling of mutant BRAF will be reported elsewhere.
Figure 4.
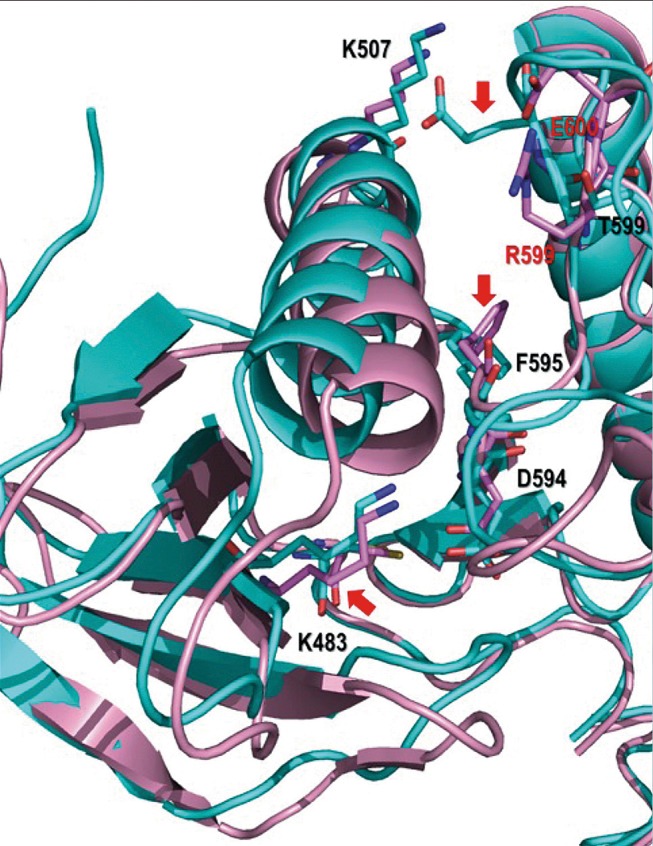
Difference in conformation between BRAF p.T599_V600>RE and BRAF p.V600E. The superimposed structures of the BRAF mutants are excerpts of representative images extracted from the molecular dynamics simulations. Addition of the p.T599R mutation to p.V600E in the BRAF p.T599_V600>RE structure results in the loss of a key interaction between K507 and E600 due to a change in conformation in the latter residue, which now interacts with R599. This results in a number of conformational changes, particularly in the side chains of residues K483, F595 and E600 (highlighted by red arrows). Mutated and key residues in the BRAF p.V600E and BRAF p.T599_V600>RE proteins are shown as sticks in cyan and pink, and labelled in red and black colour, respectively. Hydrogen bonding and π interactions are highlighted with dashed lines in green and black, respectively.
Outcome and follow-up
ddPCR of circulating cell-free DNA has been successfully used to monitor the progression of neoplasms including histiocytoses by sequential detection and quantitation of disease-specific mutations, such as BRAF V600E.7 10–12 Although the patient is well at present, ddPCR using the probe developed against the mutant BRAF c.1796_1799CAGT>GAGA was performed using cell-free DNA extracted from 5 mL plasma;13 however, no evidence of the mutation was identified. The lack of detection of the mutation was consistent with the patient’s current condition. However, in the absence of recent lesional biopsies, it is also possible that his ECD cells now contain a different mutation that is not detected by the probe developed against the BRAF c.1796_1799CAGT>GAGA mutation present in 2008. Now optimised, this test may be used as part of routine monitoring of this patient.
Discussion
The recent demonstration of recurrent BRAF V600E mutations in 40–60% of cases of ECD and Langerhans cell histiocytosis represents a significant advance in knowledge of the aetiology of this family of conditions, providing definitive evidence of the clonal origins of disease.3 In ECD cases in which BRAF V600E is not able to be detected, point mutations in NRAS or MAP2K1 (encoding MEK1) as well as rare BRAF fusions have been identified, indicating similar hyperactivation of RAS-RAF-MEK-ERK signalling.2 9 14 Mutations affecting the PI3K/AKT pathway have also been reported and comparable to other solid tumours, these may co-occur with either BRAF V600E or with alternate mutations activating RAS-RAF-MEK-ERK signalling.9 14 Valine 600, which is substituted for glutamic acid in the BRAF V600E mutation, is located in the activation segment of BRAF. Although ECD is less extensively characterised in comparison to other tumour types, it is interesting to note that until the present case and in contrast to other malignancies, point mutations or small deletion/insertions affecting additional BRAF activation segment residues have not been reported.
The development and implementation of highly sensitive methods for mutation detection including NGS and ddPCR have enabled reliable identification of characteristic disease-associated mutations in a variety of specimens that would not be suitable for conventional Sanger sequencing. These include very small samples with limiting amounts of DNA including DNA extracted from circulating tumour cells, exosomes or cell-free DNA and samples with very low proportions of disease cells, such as histiocytosis lesions. In routine pathology practice, the cost benefit of each of the newer techniques must be balanced with the likely success of the method in relation to both the available specimen and patient outcome. This is particularly relevant for ECD, which can be associated with extended periods of survival, as in the present case.1 2 It was interesting to note in this case that the poor quality of DNA and very low proportion of disease cells in the older skin lesion from 2000 prevented successful NGS in which all DNA in target regions is sequenced, including DNA that is damaged, producing spurious results. In contrast, ddPCR, in which the specific probe binds only to its target DNA sequence and not non-target DNA, was found to be less susceptible to interference from nicked or damaged DNA in the specimen. Consideration of these findings is important for future implementation of newer technologies such as NGS and ddPCR in pathology and oncology practice. This is relevant both in the characterisation of primary lesions for targetable mutations and for ongoing monitoring of disease in circulating DNA, the latter of which could be used to augment routine disease monitoring and provide a semiquantitative measure of response to treatment and disease progression.7
The original purpose of BRAF mutation testing for this patient was to indicate potential suitability for treatment with currently available BRAF inhibitors or other therapies targeting the RAS-MAPK pathway. As such, identification of a novel BRAF mutation that had not been characterised previously was problematical in that there was no precedent to suggest sensitivity or resistance to vemurafenib or dabrafenib. This was pertinent in particular because the complex mutation involved the known sensitive BRAF p.V600E alteration as well as the substitution of an immediately adjacent amino acid. Extensive search of the literature for reports of the BRAF p.Thr599_Val600delinsArgGlu mutation in any malignancy identified a single case of papillary thyroid cancer with the same complex mutation resulting from the identical two base substitutions, which were also confirmed to reside in the same allele.15 Treatment of that patient with a BRAF inhibitor was not reported.
Cancer-associated mutations of BRAF valine 600 are distinct in that they are the only mutant forms of BRAF whose activity in vivo is inhibited by the BRAF inhibitors in current clinical use (eg, vemurafenib, dabrafenib). In contrast, wild-type BRAF and non-V600 activating mutations of BRAF are resistant to the inhibitory effects and may in fact be activated by vemurafenib and dabrafenib.16 Findings from several studies using in vitro models have indicated that the selectivity of current BRAF inhibitors for BRAF p.V600 mutants appears to result from a combination of drug-specific and BRAF mutant-specific characteristics. BRAF typically functions as a dimer with a second BRAF (homodimer) or with the highly related CRAF (heterodimer).17 Although binding of vemurafenib to a BRAF monomer in a homodimer or heterodimer inhibits its activity, the resulting changes in the conformation of the dimer prevent vemurafenib binding to the second (wild-type or mutant) BRAF monomer, which is held in an active conformation and drives MAPK signalling, leading to cell proliferation.18 (Vemurafenib is not active against CRAF.) For reasons that are not yet well-described, BRAF p.V600 mutants are the only forms of BRAF identified thus far able to function as monomers, thereby defining the unique specificity of the inhibitory activity of vemurafenib for BRAF containing a cancer-associated mutation that substitutes valine 600 for glutamic acid (E), asparagine (D), lysine (K) or arginine (R).19 It is notable, however, that as published experimental models have not addressed mutant forms of BRAF which include a p.V600 mutation in addition to other amino acid changes, results could not be extrapolated to the specific mutation identified in our patient with ECD.
Molecular modelling of native protein structures or wild-type and mutant protein complexes is a pivotal tool in the understanding of biological function and drug activity. Although an established technique in research and development, it is not commonly used in medical or pathology practice. Indeed its principal role in basic research has been to explain or support biological observations rather than to predict activity or responses. The routine sequencing of human tumours to detect cancer-specific mutations has resulted in the identification of both common mutations with well-characterised sensitivity or resistance to current targeted therapies and rare uncharacterised mutations of relevant genes with unknown drug interactions and responses. Individually, these latter mutations may be unique or uncommon; however, together they may constitute a significant proportion of tumours with unknown sensitivity or resistance to applicable targeted therapies. Generation of experimental cell or tissue models to test drug sensitivity is not feasible in routine practice; however, the inclusion of molecular modelling techniques may provide information for clinicians that can assist in treatment choice in situations such as the present case for which there is no precedent.
In summary, this case report describes an unusual case of ECD associated with a unique BRAF mutation, its identification broadening the molecular characterisation of this rare disease. The case highlights rational use of modern technologies to both better define disease-associated mutations and to overcome technical issues related to poor quality and limited quantity of specimens, which are frequently encountered in pathology practice. Development of a personalised ddPCR test based on the sequencing data can also be implemented in the ongoing management of this patient, with future experimental data and the introduction of next-generation BRAF inhibitors of broader specificity potentially identifying additional treatment options if required.
Learning points.
Erdheim–Chester disease (ECD) is a rare form of non-Langerhans cell histiocytosis of variable disease severity
BRAF V600E mutations are associated with 40–60% of ECD cases.
Patients with ECD that contains a BRAF V600E mutation may be successfully treated with BRAF inhibitors (vemurafenib, dabrafenib).
A unique case of ECD associated with a previously uncharacterised BRAF mutation, BRAF p.Thr599_Val600delinsArgGlu, is described.
Characterisation of disease-associated mutations enables treatment planning and the development of personalised tests that may be used for ongoing monitoring of disease progression.
Footnotes
Contributors: JMB analysed sequencing results, collated the data and wrote the manuscript. JJR performed the Sanger sequencing and NGS library preparation. MAT performed the Sanger sequencing and designed the ddPCR probe. RA analysed the NGS. EG performed and analysed the ddPCR. RLM and MA performed the protein modelling. SF performed and reviewed the histopathology. PC (clinically) managed the case and provided the case presentation. JP managed the case. All authors were involved in the design of aspects of the analysis of this case and reviewed and approved the final version of the manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Cives M, Simone V, Rizzo FM, et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol 2015;95:1–11. 10.1016/j.critrevonc.2015.02.004 [DOI] [PubMed] [Google Scholar]
- 2. Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood 2014;124:483–92. 10.1182/blood-2014-03-561381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012;120:2700–3. 10.1182/blood-2012-05-430140 [DOI] [PubMed] [Google Scholar]
- 4. Blombery P, Wong SQ, Lade S, et al. Erdheim-Chester disease harboring the BRAF V600E mutation. J Clin Oncol 2012;30:e331–e332. 10.1200/JCO.2012.43.2260 [DOI] [PubMed] [Google Scholar]
- 5. Emile JF, Charlotte F, Amoura Z, et al. BRAF mutations in Erdheim-Chester disease. J Clin Oncol 2013;31:398 10.1200/JCO.2012.46.9676 [DOI] [PubMed] [Google Scholar]
- 6. Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121:1495–500. 10.1182/blood-2012-07-446286 [DOI] [PubMed] [Google Scholar]
- 7. Hyman DM, Diamond EL, Vibat CR, et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2015;5:64–71. 10.1158/2159-8290.CD-14-0742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borys D, Nystrom L, Song A, et al. Erdheim Chester disease with appendicular skeletal, renal and pleural involvement responding to Zelboraf (BRAF inhibitor) treatment: case report. Skeletal Radiol 2016;45:1397–402. 10.1007/s00256-016-2431-6 [DOI] [PubMed] [Google Scholar]
- 9. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154–65. 10.1158/2159-8290.CD-15-0913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janku F, Vibat CR, Kosco K, et al. BRAF V600E mutations in urine and plasma cell-free DNA from patients with Erdheim-Chester disease. Oncotarget 2014;5:3607–10. 10.18632/oncotarget.1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cangi MG, Biavasco R, Cavalli G, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis 2015;74:1596–602. 10.1136/annrheumdis-2013-204924 [DOI] [PubMed] [Google Scholar]
- 12. Cavalli G, Biavasco R, Borgiani B, et al. Oncogene-induced senescence as a new mechanism of disease: the paradigm of Erdheim-Chester disease. Front Immunol 2014;5:281 10.3389/fimmu.2014.00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015;6:42008–18. 10.18632/oncotarget.5788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emile JF, Diamond EL, Hélias-Rodzewicz Z, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood 2014;124:3016–9. 10.1182/blood-2014-04-570937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jung CK, Im SY, Kang YJ, et al. Mutational patterns and novel mutations of the BRAF gene in a large cohort of korean patients with papillary thyroid carcinoma. Thyroid 2012;22:791–7. 10.1089/thy.2011.0123 [DOI] [PubMed] [Google Scholar]
- 16. Joseph EW, Pratilas CA, Poulikakos PI, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci USA 2010;107:14903–8. 10.1073/pnas.1008990107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 2015;16:281–98. 10.1038/nrm3979 [DOI] [PubMed] [Google Scholar]
- 18. Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30. 10.1038/nature08902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao Z, Torres NM, Tao A, et al. BRAF mutants evade ERK-Dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 2015;28:370–83. 10.1016/j.ccell.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]