

Abstract
Cancer cells share several properties, high proliferation potential, reprogramed metabolism, and resistance to apoptotic cues. Acquiring these hallmarks involves changes in key oncogenes and non-oncogenes essential for cancer cell survival and prosperity, and is accompanied by the increased energy requirements of proliferating cells. Mitochondria occupy a central position in cell life and death with mitochondrial bioenergetics, biosynthesis, and signaling are critical for tumorigenesis. Voltage-dependent anion channel 1 (VDAC1) is situated in the outer mitochondrial membrane (OMM) and serving as a mitochondrial gatekeeper. VDAC1 allowing the transfer of metabolites, fatty acid ions, Ca2+, reactive oxygen species, and cholesterol across the OMM and is a key player in mitochondrial-mediate apoptosis. Moreover, VDAC1 serves as a hub protein, interacting with diverse sets of proteins from the cytosol, endoplasmic reticulum, and mitochondria that together regulate metabolic and signaling pathways. The observation that VDAC1 is over-expressed in many cancers suggests that the protein may play a pivotal role in cancer cell survival. However, VDAC1 is also important in mitochondria-mediated apoptosis, mediating release of apoptotic proteins and interacting with anti-apoptotic proteins, such as B-cell lymphoma 2 (Bcl-2), Bcl-xL, and hexokinase (HK), which are also highly expressed in many cancers. Strategically located in a “bottleneck” position, controlling metabolic homeostasis and apoptosis, VDAC1 thus represents an emerging target for anti-cancer drugs. This review presents an overview on the multi-functional mitochondrial protein VDAC1 performing several functions and interacting with distinct sets of partners to regulate both cell life and death, and highlights the importance of the protein for cancer cell survival. We address recent results related to the mechanisms of VDAC1-mediated apoptosis and the potential of associated proteins to modulate of VDAC1 activity, with the aim of developing VDAC1-based approaches. The first strategy involves modification of cell metabolism using VDAC1-specific small interfering RNA leading to inhibition of cancer cell and tumor growth and reversed oncogenic properties. The second strategy involves activation of cancer cell death using VDAC1-based peptides that prevent cell death induction by anti-apoptotic proteins. Finally, we discuss the potential therapeutic benefits of treatments and drugs leading to enhanced VDAC1 expression or targeting VDAC1 to induce apoptosis.
Keywords: apoptosis, cancer, metabolism, mitochondria, voltage-dependent anion channel 1
Voltage-Dependent Anion Channel (VDAC) Isoforms, Structure, and Channel Activity
VDAC Isoforms and Cellular Localization
In mammals, three VDAC isoforms have been identified, VDAC1, VDAC2, and VDAC3, sharing ~70% identity as well as several structural and functional properties (1, 2), although they are significantly different with relation to functionality (1, 3, 4). The three isoforms are expressed in most tissue types, with the expression levels of VDAC2 and VDAC3 being lower in most but not all tissues (1, 2). Here, we only briefly consider the VDAC isoforms, as a full chapter in this issue (De pinto et al.) is devoted to VDAC isoforms.
The expression of hVDAC-2 is associated with neurodegenerative diseases, including amylotropic lateral sclerosis (ALS) (5), epilepsy (6), and Alzheimer’s disease (AD) (7). By contrast, VDAC3−/− mice showed only in heart muscle defects in complex IV activity, a component of the electron transfer chain (8).
While both VDAC1- and VDAC3-deficient mice are viable, which permits the study of the role(s) of VDAC in cellular metabolism in intact animals, the reduced number of Vdac1−/− progeny (according to the Mendelian ratio) suggests partial embryonic lethality (9). Studies using Vdac−/− mice confirmed the importance of this protein as a transporter of metabolites across the outer mitochondrial membrane (OMM). Detergent-“skinned” muscle fibers, which lack VDAC1, displayed reduced ADP-stimulated oxygen consumption, defects in the electron transport chain (ETC) complex activities, reduction of mitochondria-associated hexokinase (HK), and finally, abnormal mitochondrial morphology (9).
A number of regulatory functions involving the generation of reactive oxygen species (ROS), steroidogenesis, and mitochondria-associated endoplasmic reticulum (ER) pathways have been variously ascribed to the different isoforms (10). VDAC1 is involved in apoptosis, interacting with different proteins and factors and mediating the release of Cyto c (1, 11–26). The metabolite transport properties of VDAC1 are also superior to those of VDAC2 and VDAC3 (1). By contrast, VDAC2 is anti-apoptotic (27), is crucial for Bak recruitment (28), and is a critical inhibitor of Bak-mediated apoptosis (29). The anti-tumor agent erastin was found to bind directly to VDAC2 and induce non-apoptotic cell death in some tumor cells that harbored activating mutations in the RAS–RAF–MEK pathway (30).
Immunofluorescence, flow cytometry, and EM immunogold labeling have detected VDAC in other cell compartments in addition to mitochondria (3) [for review, see Ref. (31)]. These compartments include the plasma membrane (3), including location in caveolae and caveolae-like domains (32), the sarcoplasmic reticulum (SR) of skeletal muscles (33), and the ER of rat cerebellum (34). Patch-clamping of intact cells demonstrated channel with properties similar to those of planar-bilayer reconstituted purified VDAC1 (35). VDAC has also been detected in synaptosomes of Torpedo electric organ (36). VDAC2 and VDAC3 have been reported in bovine outer dense fibers and in the cytoskeletal component of sperm flagellum (37).
A possible mechanism for targeting VDAC protein to the plasma membrane proposes that the N-terminal signal peptide of the protein is responsible for this targeting (38). Indeed, plasmalemmal (pl) VDAC1 was found to contain a hydrophobic leader sequence (39). Other targeting mechanisms, such as alternative mRNA untranslated regions, were also suggested (35) for trafficking via ER/mitochondria-associated membranes or plasma membrane/ER associations (40).
Several possible functions of the extra-mitochondrial VDAC were proposed. These include intracellular communication, as mediating calcium signal between the ER and mitochondria (41), being part of the outwardly rectifying depolarization-induced chloride (ORDIC) channel complex (42), regulate cell volume in brain (43), and mediate ATP release (44). Interestingly, silencing VDAC1 expression by specific small interfering RNA (siRNA) was shown to prevent the entry of amyloid beta (Aβ) into the cytosol, as well as Aβ-induced toxicity (45), suggesting the involvement of pl-VDAC1 in Aβ cell entry and in inducing mitochondrial dysfunction and apoptosis (46). These and other proposed functions for plVDAC were recently presented and discussed (31).
VDAC1 Structure, Channel Conductance, Properties, and Regulation
The three-dimensional structure of VDAC1 was solved using X-ray crystallography, NMR, and a combination of both (47–49). The methods propose that VDAC1 is composed of 19 β-strands arranged as a barrel, and with the N-terminal domain located within the pore. The pore diameter of the channel has been estimated to be between 3 and 3.8 nm (47) and decreased to about 1.5 nm when the N-terminal domain is located within the pore (47–49).
This imaging-derived structure is in disagreement with the conclusions of biochemical and biophysical approaches, which argue for the existence of additional extra-membranal VDAC1 regions (50). The discrepancy may be attributed to the fact that all three imaging methods employed refolded recombinant E. coli expressed VDAC1, purified from inclusion bodies and the refolding conditions may be responsible for the appearance of non-native structures.
An important VDAC1 structural element is the stretch of multiple glycine residues (21GlyTyrGlyPheGly25) [1,5] that connects the N-terminal domain to β-strand 1 of the barrel. This glycine-rich 21GYGFG25 sequence, which is highly conserved between mammals, is thought to provide the flexibility required for N-terminal region translocation out of the internal pore of the channel (51). Using site-directed mutagenesis and cysteine substitution, in combination with a thiol-specific cross-linker, it was demonstrated that the N-terminal domain of VDAC1 located within the pore can be translocated (51).
The notion of the mobility of the N-terminal region is further supported by observations that this protein domain moves upon changes in the voltage gradient (51, 52), that antibodies directed against the N-terminal region of the VDAC1 interact with membrane embedded protein (53–55), and that anti-apoptotic and pro-survival factors HK-I, HK-II, and Bcl-2 interact with this domain (16, 51). These results strongly support the suggestion that the N-terminal domain is highly dynamic and can translocate out of the pore (51, 56).
Several different roles have been proposed for the N-terminal segment. These include acting as a voltage sensor (16, 51), as no voltage-dependent conductance was obtained with N-terminal-truncated VDAC1 (16, 57). The N-terminal domain was further proposed to regulate the fluxes of ions and metabolites via VDAC1 (49) and to stabilize the β-barrel (58). The structure of the N-terminal domain of VDAC1, its potential role in regulating barrel shape, and its interaction with HK have been reviewed recently (59). Additional roles of this region in VDAC1 oligomerization and regulation of apoptosis (16, 51) are presented below (see VDAC1 Homo-Oligomer Forming the Cyto c Release Pathway) and as the binding site for HK, Bcl-2, and Bcl-xL (see VDAC1-Based Peptides As Potential Anti-Cancer Therapy). Thus, it seems that the N-terminal domain of VDAC1 regulates a wide variety of VDAC1 functions, such as its channel conductance and apoptosis.
Another property of VDAC1 is the ability to form an oligomeric structure. Chemical cross-linking and fluorescence resonance energy transfer analysis showed dimers, trimers, tetramers, and higher order oligomers (1, 11, 14, 56, 60–62). In addition, the NMR-based structure of recombinant human (h)VDAC1 implies that it forms a dimer of monomers arranged in parallel (47), while analysis of the crystal packing of murine (m)VDAC1 revealed strong anti-parallel dimers that can further assemble into hexamers (63). The modulation and function of VDAC1 oligomerization are presented below (see VDAC1 Homo-Oligomer Forming the Cyto c Release Pathway).
The structural characterization of VDAC2 and VDAC3 is limited, with zebrafish VDAC2 structure was resolved at 2.8A resolution, revealing a dimeric organization (64).
The properties of VDAC1, purified by various procedures and detergents (65), from mitochondria isolated from liver, brain, and other tissues, have been studied when the purified protein was reconstituted into a planar lipid bilayer. Such bilayer-reconstituted VDAC1 assumes a variety of voltage-dependent conformational states, with different selectivities and permeabilities. VDAC1 shows symmetrical bell-shaped voltage-dependent conductance, with the highest conductance (4 nS at 1 M KCl) occurring at low potentials of −20 to +20 mV (1, 66). At low voltages (~10 mV), VDAC1 exists in a high conductive state and shows a preference for transporting anions over cations. VDAC1 is permeable to small ions (e.g., Cl−, K+, Na+), and also to large anions, such as glutamate (66) and ATP (67), and to large cations, such as acetylcholine and dopamine (66). At high positive or negative potentials (>40 mV), VDAC1 switches to lower conductance states with different ionic selectivities and permeabilities (66, 68). In this state, the protein is permeable to small ions but becomes less permeable to ATP and ADP (1, 67, 68).
VDAC1 channel conductance is thought to rely on two separate gating processes, one at positive trans-membrane potentials and the other at negative potentials (1, 68) with the N-terminal α-helical segment of the channel acting as the voltage sensor, gating the pore via conformational changes and/or movements (16, 51). Additional studies are required before the VDAC1 gating mechanism can be fully resolved.
VDAC1 As a Hub Protein
VDAC1 functions in metabolism, Ca2+ homeostasis, apoptosis, and other activities are regulated via the interaction of VDAC1 with many proteins associated with cell survival and cellular death pathways (1, 14, 56, 61). Indeed, VDAC1 in the OMM serves as a hub protein interacting with diverse sets of cytosolic, ER, and mitochondrial proteins that together regulate metabolic and signaling pathways, providing energy for cellular functions or triggering cell death.
As support for this viewpoint, the conserved nature of VDAC1 (1), is in agreement with the finding that hub proteins are more evolutionarily conserved than are non-hub proteins (69). The VDAC1 interactome includes proteins involved in metabolism, apoptosis, signal transduction, anti-oxidation, and DNA- and RNA-associated proteins (Figure 1) (1, 11, 14, 56). Furthermore, these proteins may be located in the OMM, inner mitochondrial membrane (IMM), the IMS, the cytosol, ER, plasma membrane, and nucleus. Thus, VDAC1 functions as an anchoring site for proteins that mediate and/or regulate metabolic, apoptotic, and other processes in normal and diseased cells. In addition, some of these proteins, such as Bcl-2, Bcl-xL, and HK, are overexpressed in many cancers (see VDAC Interaction with HK and Other Metabolism-Related Proteins, Interaction of VDAC1 with Bcl-2 Family Members). Importantly, we have been able to develop VDAC1-based peptides, which can interfere with these interactions, leading to impaired cell metabolism and apoptosis (18–20, 70, 71) (see VDAC1-Based Peptides As Potential Anti-Cancer Therapy).
Figure 1.

Schematic representation of VDAC1 as a hub protein and its associated proteins. VDAC1 interacting proteins (see Modulation of VDAC1-Mediated Apoptosis and Metabolism via Interacting Proteins), sub-grouped into those associated with metabolism, energy, apoptosis, anti-oxidant, cell receptors, or signal transduction, or localized to mitochondria, endoplasmic reticulum (ER), nucleus, and cell membrane.
VDAC1 as a Multi-Substrate Transporter
The transport of a variety of metabolites, nucleotides, and coenzymes across the IMM is mediated by about 53 secondary transport proteins called mitochondrial carriers. These transporters are substrate specific, such as the Pi carrier, the ADP/ATP carrier [adenine nucleotide translocase (ANT)] and the aspartate/glutamate carrier (72). On the other hand, VDAC1 is the sole channel mediating the fluxes of ions, nucleotides, and other metabolites up to ~5,000 Da, including hemes and cholesterol, across the OMM (1) (Figure 2). Thus, at the OMM, VDAC1 is perfectly positioned to function as gatekeeper for the entry and exit of substrates and products into and out of the mitochondria, and to interact with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities (1, 12, 14, 56, 61, 68, 73) (Figures 1 and 2).
Figure 2.

Schematic representation of VDAC1 as a multi-functional channel involved in cell survival and cell death. The various functions of VDAC1 include control of the metabolic cross-talk between the mitochondria and the rest of the cell, cellular energy production [by transporting ATP/ADP and reduced nicotinamide adenine dinucleotide (NADH) between the inter-membrane space and the cytosol and by binding hexokinase (HK)], and Ca2+ signaling (by transporting Ca2+, acyl-CoAs, and cholesterol); the Ca2+ influx and efflux transport systems in the OMMs and IMMs and Ca2+-mediated regulation of the tricarboxylic acid (TCA) cycle; activation of pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (α KGDH) by intra-mitochondrial Ca2+, leading to enhanced activity of the TCA cycle; and control of the electron transport chain and the ATP synthase (FoF1). VDAC1 in the OMM functions as a Ca2+ channel. In the IMM, the uptake of Ca2+ into the matrix is mediated by a Ca2+-selective transporter, the mitochondrial Ca2+ uniporter (MCU), regulated by a calcium-sensing accessory subunit (MICU1). Ca2+ efflux is mediated by NCLX, a Na+/Ca2+ exchanger. The accumulation of high levels of matrix Ca2+ triggers the opening of the permeability transition pore (PTP), a fast Ca2+ release channel. The role of VDAC1 in the transport of H2O2 is also presented. In addition, the figure shows the process of transfer of acyl-CoAs across the OMM by VDAC1 to the intermembrane space, where they are converted by carnitine palmitoyltransferase 1a (CPT1a) into acylcarnitine for further process during β-oxidation and cholesterol transport by a multi-protein complex, the transduceosome, containing steroidogenic acute regulatory protein (StAR)/translocator protein (TSPO)/VDAC1. Molecular fluxes are indicated by arrows.
VDAC1 As a Cellular Metabolite Transporter Controls Cell Energy and Metabolites
VDAC1 mediates the passage of metabolites, including pyruvate, malate, and succinate, into and out of mitochondria (1). In addition, VDAC1 also allows shuttling of ATP and ADP, and NAD+/reduced nicotinamide adenine dinucleotide, with mitochondria-generated ATP being transported to the cytosol in exchange for ADP, which is utilized in oxidative phosphorylation (OXPHOS) to generate ATP. As such, VDAC1 controls the ETC (1) (Figure 2) as well as the normal flow of metabolites (74). The importance of VDAC1 in cell energy and metabolism homeostasis is reflected in the findings that closure of VDAC1 (73), or downregulation of VDAC1 expression decreased metabolite exchange between mitochondria and the cytosol and inhibited cell growth (53, 75) [see Silencing VDAC1 Expression by Short Hairpin RNA (shRNA) or siRNA as a Tool to Reprogram Cancer Cell Metabolism].
The importance of VDAC1 in channeling ATP from the mitochondria to kinases has been presented in several studies. These showed that VDAC1 interacts with HK and creatine kinase (CrK) that produce high-energy metabolites, such as glucose-6-phosphate (G-6-P) and creatine phosphate in brain and muscle, respectively. The interaction of VDAC1 with HK mediates a coupling between OXPHOS and glycolysis (see Alterations in VDAC1 Expression Level in Cancer), while VDAC1 forms a complex with the ANT, and CrK at the contact sites between the IMM and OMM (76). Dimeric αβ-tubulin was proposed as a regulator of permeability of VDAC1 to ATP, with monomers of αβ-tubulin decreasing the passage of ATP through the channel (77). The function of VDAC1 in energy metabolism of cancer cells and the significance of the overexpression in many cancer cells (11) is discussed further below (see VDAC1 Expression Level and Cell Death Induction—a New Concept, Unraveling VDAC1-Based Therapies).
Cholesterol is another metabolite transported across the OMM (78) (Figure 2), with VDAC1 being a component of a multi-protein complex, the transduceosome, involved in the process. In addition to VDAC1, the transduceosome also includes the OMM high-affinity cholesterol-binding protein translocator protein (TSPO) and the steroidogenic acute regulatory protein (79) (Figure 2).
Cholesterol synthesis is highly elevated in various cancer cells, mainly in the OMM (80). In cancer cells, the increased mitochondria-bound HK is proposed to increase synthesis and uptake of cholesterol into the mitochondria. (81). Recently, it has been suggested that a glycine rich motif 21GYGFG25 sequence in the N-terminal part of VDAC1 is responsible for cholesterol binding (82). Cholesterol at high levels can reduce the activity of membrane-associated proteins and, thus, inhibit the metabolic functions of VDAC1 (83).
Thus, VDAC1 is involved in cholesterol synthesis and transport, and is regulated by cholesterol.
Finally, in rat liver mitochondria, VDAC1 is proposed as part of a complex mediating the transport of fatty acids through the OMM (84). In this case, the hypothesis is that VDAC1 allows docking of the long-chain acyl-CoA synthetase (ACSL) at the OMM and, thus, linking it to carnitine palmitoyltransferase 1a (CPT1a). According to this proposal, activation of VDAC1 by ACSL allows the transfers acyl-CoAs via VDAC1, thus acrossing the OMM to the IMS, where they are converted into acylcarnitine by CPT1a. Finally, it was recently proposed that VDAC serves as a lipid sensor (85).
VDAC1 As a Ca2+ and ROS Transporter
Mitochondria are also a major hub of cellular Ca2+ homeostasis that is fundamental for a wide range of cellular activities. Intra-mitochondrial Ca2+ controls energy metabolism modulation of critical enzymes, such as members of the tricarboxylic acid (TCA) cycle and enzymes responsible for fatty acid oxidation (FAO) (86) (Figure 2). Ca2+ overload in the mitochondria is involved in apoptotic cell death, triggering Cyto c release, and subsequent cell death (Figure 3).
Figure 3.

VDAC1 function in cell death. Different models proposed for the release of apoptogenic proteins, such as Cyto c (gold balls) and apoptosis-inducing factor (AIF), from the mitochondrial inter-membrane space to the cytosol, leading to apoptosis. These models include (a) VDAC1 closure and outer mitochondrial membrane (OMM) rupture serving as the Cyto c release pathway—prolonged VDAC1 closure leads to mitochondrial matrix swelling and OMM rupture, resulting in the appearance of a non-specific release pathway for apoptogenic proteins; (b) a permeability transition pore (PTP) provides the apoptogenic protein release pathway—a large conductance pore-forming complex, the PTP, composed of VDAC1 in the OMM, adenine nucleotide translocase in the inner mitochondrial membrane (IMM), and cyclophilin D (CyD) in the matrix, allows apoptogenic protein release; (c) Bax activation, followed by its oligomerization, results in OMM permeabilization—upon apoptosis induction, Bax becomes associated with mitochondria as a large oligomer/complex forming a Cyto c-conducting channel in the OMM; (d) a pore is formed by oligomerized forms of Bax and Bak—Bax/Bak oligomerization, supposedly activated by BH3-only proteins (e.g., Bid), results in OMM permeabilization and Cyto c release; (e) a Bax- and VDAC1-based hetro-oligomer mediates Cyto c release—the interaction of pro-apoptotic proteins (Bax/Bak) with VDAC1 forms a cytochrome c (Cyto c) release pathway; (f) mitochondrial apoptosis-inducing channel (MAC) as the release pathway—MAC offers a high-conductance channel and a putative Cyto c release channel; (g) oligomeric VDAC1 as a channel for the release of apoptotic proteins—a protein-conducting channel is formed within a VDAC1 homo-oligomer, allowing Cyto c release and apoptotic cell death; (h) mitochondrial Ca2+ overload induces apoptosis—following Ca2+ overload in the matrix, Ca2+ transport mediated by VDAC1 across the OMM and by the mitochondrial Ca2+ uniporter (MCU) in the IMM leads to dissipation of the membrane potential, mitochondria swelling, PTP opening, Cyto c release, and apoptotic cell death. PTP opening is also accompanied by an efflux of the accumulated Ca2+ into the cytosol.
Ca2+ transport across the IMM is mediated by several proteins, including the mitochondrial Ca2+ uniporter (87, 88) and the Ca2+ efflux mediator Na+/Ca2+ exchanger NCLX (89, 90) (Figure 2). A number of studies have reported that VDAC1 in the OMM can transport Ca2+ (91–94). VDAC1 possesses divalent cation-binding site(s) (91, 95) and it was proposed that VDAC1 activity is modulated by physiological [Ca2+]i (94) with the magnitude of transfer into the mitochondrial matrix regulated by Bcl-xL binding to VDAC (96, 97). VDAC1 also functions in the Ca2+ cross-talk between ER and mitochondria (98).
Inhibitors of VDAC1, such as 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), were shown to prevent the apoptosis stimulus following an increase in intracellular Ca2+ levels (99) or Ca2+-mediated oxidative stress and apoptosis, as induced by 5-aminolevulinic acid (100).
Mitochondria are a major source of ROS, that are linked to anti-tumor immunity, the tumor microenvironment, proliferation, and death of cancer cells (101). While ROS promote tumorigenicity through signaling, they can also selectively kill a number of cancer cell lines (102) as well as normal cells, attacking DNA, lipids, and proteins (103). VDAC1 has been proposed to mediate ROS release from the IMS to the cytosol (104) (Figure 2), with HK-I and HK-II bound to VDAC1 decreasing this release (105), and thereby reducing intracellular levels of ROS (106).
Cancer, Metabolism, Mitochondria, and VDAC1
It is now well accepted that in cancers, regardless of cellular or tissue origin, impaired cellular energy metabolism is the defining characteristic. Cancer cells exhibit significant metabolic alterations with respect to several critical substrates, including important changes in the metabolism of both glucose and glutamine that require plasticity of the metabolic machinery (107, 108). The view of cancer as a metabolic disease originated with the experiments of Otto Warburg in the 1920s, however, this view was gradually displaced by the concept of cancer as a genetic disease. Recently, although the Warburg effect and metabolic defects expressed in cancer cells are thought to arise primarily from genomic mutability selected during tumor progression (109, 110), the genetic origin of cancer has come into question. Accordingly, the genomic instability and essentially all hallmarks of cancer, including aerobic glycolysis were connected to mitochondria dysfunction and energy metabolism (111, 112). Indeed, supporting evidence suggests that cancer is primarily a mitochondrial metabolic disease (111).
Evidence for a metabolic rather than genetic origin includes the absence of a specific gene mutation or chromosomal abnormality that is common to all cancers (113), while nearly all cancers display aerobic glycolysis, regardless of their tissue or cellular origin. Cancer cells typically display high rates of glycolysis, even when fully oxygenated (aerobic glycolysis), and an altered redox balance (Warburg effect) (114–116). To increase glycolysis, cancer cells upregulate the transcription of genes involved in the glycolytic pathway (i.e., glucose transporters, glycolytic enzymes, etc.). Cancer cells actually use both glycolysis and OXPHOS, according to normoxic or hypoxic conditions and their capacity to regulate the expression of genes important for cell growth (117). By regulating the metabolic and energetic functions of mitochondria, VDAC1 can, therefore, control the fate of cancer cells. Mitochondrial-bound HK, considered the rate-limiting enzyme of glycolysis, is overexpressed in cancer (1, 23, 118) and, as discussed in Section “VDAC Interaction with HK and Other Metabolism-Related Proteins,” is associated with VDAC1, an interaction that offers several advantages to cancer cells (1, 11), as outlined below (see VDAC Interaction with HK and Other Metabolism-Related Proteins). The HK–VDAC1 complex formation is regulated by Akt (119) and glycogen synthase kinase 3 beta (GSK3β), while the HK–VDAC1 complex is disrupted by VDAC1 phosphorylation (24). In addition, it was shown that an increase in the amount of free cytoplasmic tubulin decreased VDAC conductance and mitochondrial membrane potential (ΔΨm) in all three VDAC isoforms (120).
The association of VDAC1 with the process of FAO is also important. Hetero-oligomeric complexes containing VDAC and CPT1a, a liver OMM protein catalyzing the first step in mitochondrial FAO and ACSL were detected and thought to transfer activated fatty acids through the OMM (84).
Specifically targeting metabolism in cancer cells presents a potential therapeutic strategy. However, although glucose metabolism is increased in cancer cells, they mostly use the same glycolytic enzymes as do normal cells so that the choice of glycolytic enzymes as a target for cancer treatment may increase the risk of affecting normal cells as well (121).
Mitochondria, Apoptosis, VDAC, and Cancer
Apoptosis can be initiated by two signaling cascades, the extrinsic and intrinsic pathways. The extrinsic pathway can be activated by binding of tumor necrosis factor (TNF), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and other ligands to their specific death receptor (122). Such interactions lead to cascade of events leading to activation of procaspase 8 which then activates cysteinyl/aspartate-specific protease (caspases)-3, -7, and -6, promoting apoptosis (123).
The intrinsic pathway can be activated by intracellular signals, such as oxidative stress, Ca2+ overload, DNA damage, and by various compounds, such as chemotherapeutic drugs (124). This leads to the release of IMS apoptogenic proteins [e.g., Cyto c, apoptosis-inducing factor (AIF), Smac/DIABLO] (124). The released Cyto c is a constituent of the apoptosome and activates procaspase-9, which in turn, activates the executioner caspases, caspase-3 and -7, leading to cell destruction (124). The AIF released is translocated to the nucleus, leading to chromatin condensation and DNA fragmentation (124). The intrinsic pathway is regulated by members of the B-cell lymphoma 2 (Bcl-2) family of proteins (125), and by the inhibitor of apoptosis protein (IAP) family of proteins (126).
The two pathways can be connected through the caspase-8-mediated cleavage of Bid into tBid (truncated Bid), which is translocated to the mitochondria, where it causes Cyto c release and subsequent cell death (123). In this way, tBid links extrinsic pathway to intrinsic, mitochondria-mediated apoptosis.
Cancer Cells Avoid Apoptosis
In many cancers, there is a deregulation of the balance between cell growth and death (115). Tumor cells avoid apoptosis by alterations in the expression levels of pro- and anti-apoptotic proteins, as well as because of reduced caspase function and impaired death receptor signaling (127). Overexpression of anti-apoptotic proteins, such as Bcl-2 and Bcl-xL, has been demonstrated in numerous cancers, including colon, thyroid, breast, and endometrial cancer (128). Moreover, Bcl-2 expression is correlated with the degree of aggressiveness and resistance to chemotherapy-induced apoptosis (129).
One of the most common apoptotic pathways involves transcription factor p53, which plays a role in promoting transcription of pro-apoptotic factors, such as Puma, Noxa, Bax, and apoptosis protease-activating factor 1 (APAF1). In response to a spectrum of apoptotic stimuli, such as oxidative stress, p53 translocates to the mitochondria (130), where it may regulate VDAC1-mediated apoptosis (131). As already discussed, cancer cells possess elevated levels of mitochondria-bound HK that not only enhances glycolysis but also protects against mitochondria-mediated apoptosis via direct interaction with VDAC1 (16, 17, 20, 21, 132, 133).
Although induction of apoptosis in cancer cells conceptually represents an effective therapeutic approach, conventional apoptosis-inducing chemotherapy is limited by a lack of specificity, by cancer cell resistance, and by side effects of cytotoxicity for normal cells.
VDAC1 Involvement in Apoptosis
VDAC1 is now well-accepted as an important player in apoptosis and is being explored as a new target for cancer therapy (1, 11, 13–15, 55, 134). Evidence supporting the activity of VDAC1 in apoptosis includes (a) Cyto c release, cell death, and Bax–VDAC1 interaction were all inhibited by anti-VDAC1 antibodies (135–137); (b) HK-I and HK-II interacting with VDAC1 inhibited staurosporine (STS)-induced Cyto c release and apoptosis in native but not mutated VDAC1-expressing cells (14, 17, 133); (c) the interaction of ruthenium red (RuR) with native but not mutated VDAC1 prevented Cyto c release and apoptosis (95, 133, 138); (d) siRNA-mediated downregulation of VDAC1 prevented cell death induced by cisplatin (139); (e) reducing the level of VDAC1 expression attenuated endostatin (ES)-induced apoptosis (140); (f) over-expression of VDAC1 induced apoptosis, regardless of cell type and the effect was antagonized by anti-apoptotic proteins (17, 133, 141); (g) VDAC1 mediated Cyto c release from proteoliposomes (25, 62, 135); (h) VDAC1-deficient mitochondria from mutant yeast did not exhibit Bax/Bak-induced Cyto c release (25, 142); (i) the anti-apoptotic effect of Bcl-2 and Bcl-xL was obtained in cells expressing native but not mutated VDAC1 (18, 19); (j) VDAC1 channel conductance inhibitors, such as DIDS, DPC (diphenylamine-2-carboxylate), and VBIT-4, inhibited apoptosis triggered by various inducers (99, 104, 133, 143); and finally, (k) cyathin-R, a cyathane-type diterpenoid from the medicinal fungus Cyathus africanus, could induce apoptosis in Bax/Bak-depleted cells but not when VDAC1 was depleted. Cyathin-R-induced apoptosis was inhibited by DPC (144).
Proposed Pathways and Mechanisms for Apoptogenic Factor Release from Mitochondria
All of the mitochondrial apoptotic proteins (Cyto c, AIF, Smac/DIABLO, and endonuclease G) that activate apoptosis in the cytosol are located in the IMS. Therefore, only increase in the OMM permeability is required for the release of apoptogenic proteins. It remains, however, unclear how these pro-apoptotic proteins cross the OMM for release into the cytosol. Based on a variety of approaches and strategies, several mechanisms describing the release of apoptotic proteins from the IMS were proposed [for reviews, see Ref. (1, 11–13, 61, 145)] (Figure 3). Some models propose that release of apoptotic proteins from the IMS is facilitated by a swelling of the mitochondrial matrix and subsequent rupture of OMM integrity (Figure 3a). Other models predict the formation of large channels that can allow the passage of Cyto c and other proteins and, thus, to be released from the IMS to the cytosol (Figure 3) such as:
-
(i)
Permeability transition pore (PTP): the mitochondrial PTP is a high-conductance non-specific pore activated by ROS, Ca2+ overload, and other agents, leading to mitochondrial swelling and the release of Cyto c into the cytosol. Initially, PTP was proposed to comprising VDAC1 in the OMM, ANT in the IMM, and cyclophilin D (CyD), a resident of the matrix (55, 146, 147) (Figure 3b,h). However, in knockout experiments performed in mice, even mitochondria examined from cells lacking some but not all ANT isoforms (148, 149), or VDAC1 (150) showed PTP formation. Recently, it was proposed that dimers of the ATP synthase complex can form the PTP (151).
-
(ii)
Bax/Bak complexes: Bax and Bak are pro-apoptotic proteins proposed to oligomerize to form a Cyto c release channel (152–154) (Figure 3c,d). Bcl-2 prevents Bax oligomerization and insertion into the OMM (152, 153). Bak can form a large pore due to its oligomerization (155) or due to formation of hetro-oligomers with Bax following their activation by tBid (156, 157).
-
(iii)
Bax/VDAC1 complexes: VDAC1 and Bax form hetero-oligomers forming the Cyto c release channel (26, 158) (Figure 3e).
-
(iv)
Mitochondrial apoptosis-inducing channel (MAC): MAC a supra-molecular complex forming channel at the OMM that can mediate Cyto c release (159) (Figure 3f). Recently, it was proposed that Bax and/or Bak form the MAC (160).
-
(v)
VDAC1 oligomerization: upon apoptosis induction, VDAC1 undergoes conformational changes and oligomerization, forming a channel within the homo-oligomer large enough to allow Cyto c release, and subsequent apoptosis (14, 16, 56, 60–62, 145, 161, 162) (Figure 3g) (see VDAC1 Homo-Oligomer Forming the Cyto c Release Pathway). It was also suggested that apoptosis stimuli lead to VDAC1 oligomerization by inducing upregulation of VDAC1 expression levels (see VDAC1 Expression Level and Cell Death Induction—a New Concept).
Finally, it should be noted that the release of Cyto c could be achieved by either of the above proposed mechanisms, depending on the nature of the apoptosis inducer and cell type (163).
VDAC1 Homo-Oligomer Forming the Cyto c Release Pathway
The determined VDAC1 pore diameter in its high conductance state is about 2.6–3.0 nm. This diameter is too small to allow the passage of Cyto c to be released to the cytosol. To overcome this issue, the formation of a large channel comprising VDAC1 monomers has been proposed to serve as the Cyto c release channel (56, 60–62, 145, 161, 162). VDAC1 is known to exist as higher-order oligomers (22, 60, 62, 145, 164–166) (see VDAC1 Structure, Channel Conductance, Properties, and Regulation) and both purified soluble and membrane-embedded VDAC1 have been shown to assemble into dimers, trimers, and tetramers in a dynamic process (62). Recently, the particular lipid composition of the OMM has been shown to significantly enhance VDAC1 oligomerization (167). Finally, the connection between VDAC1 oligomerization and Cyto c release was supported by the finding that VDAC1 oligomerization was highly enhanced by various inducers of apoptosis, including STS, curcumin, arsenic trioxide (As2O3), etoposide, cisplatin, selenite, TNF-α, H2O2, or UV light (60) reflecting a shift in VDAC1 status from the monomeric toward the oligomeric form. The oligomerization of VDAC1 was favored regardless of cell type or mechanism of action of the apoptosis inducer used, although all affected mitochondria. In addition, As2O3-induced homo-dimerization of VDAC1 was prevented by overexpression of the anti-apoptotic protein, Bcl-xL (137). The association between apoptosis induction and VDAC1 oligomerization gained support with the finding that both processes are inhibited by DIDS, SITS (4-acetamido-4′-isothiocyanato-stilbene-2,2′-disulfonic acid), H2DIDS (4,4′-diisothiocyanatodihydrostilbene-2,2′-disulfonic acid), DNDS (4,4′-dinitrostilbene-2,2′-disulfonic acid), and DPC, known anion transport inhibitors that all interact with VDAC1 (60, 99, 144, 145, 168).
The results presented above led us to propose a novel model in which VDAC1 exists in a dynamic equilibrium between the monomeric and oligomeric states, with apoptosis inducers shifting the equilibrium toward oligomers. VDAC1 oligomers form a large flexible pore between individual subunits of VDAC1, mediating the passage of released Cyto c across the OMM, leading to cell death (14, 56, 60–62, 144, 145, 161, 162) (Figure 4).
Figure 4.
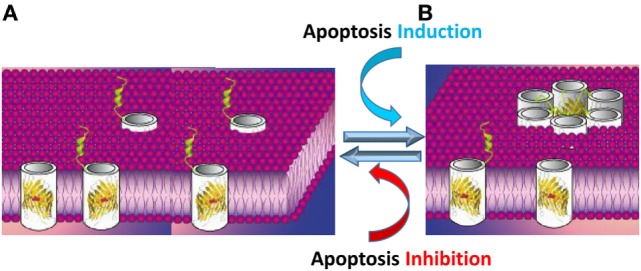
Apoptosis stimuli induce VDAC1 oligomerization: a proposed Cyto c release channel A. Before apoptosis induction, VDAC1 is in the monomeric state, with the amphipathic α-helix N-terminal region cytoplasmically exposed (169) or positioned within the pore (47–49). (B) Upon apoptotic signaling, VDAC1 oligomerizes to form a multimer and the amphipathic α-helix N-terminal region of each VDAC1 molecule flips into the hydrophobic pore formed by the β-barrels, forming a hydrophilic pore capable of conducting Cyto c.
The passage of Cyto c requires a channel formed by minimal number of six VDAC1 monomers. When arranged in a circle, the central pore should be 4.0 nm in diameter, allowing the transport of Cyto c (3.4 nm external diameter). The formation of VDAC1 hexamers and higher oligomers involves the formation of several contact sites between adjacent VDAC1 monomers, involving β-strands 1, 2, 18, and 19 from both VDAC1 monomers (47, 63).
The contact sites between VDAC1 molecules in dimers and higher oligomers were identified using structural- and computational-based approaches, in combination with site-directed mutagenesis, cysteine replacement and chemical cross-linking (170). A contact site involving β-strands 1, 2, and 19 that undergoes conformational changes following apoptosis induction and assembles into higher oligomeric states with contact sites involving β-strands 8 and 16 was identified in dimeric VDAC1 (170).
The proposed VDAC1 channel formed upon its oligomerization is composed of several β-barrels; thus, is expected to be hydrophobic, making it difficult for the positively charged Cyto c to cross such a pore. As discussed above (see VDAC1 As a Hub Protein), structural, biochemical, and functional studies revealed that the N-terminal domain is mobile and can be cytoplasmically exposed. Following apoptosis induction along with VDAC1 oligomerization, the amphipathic N-terminal segment is thought to be move out of the pore and interact with surface of the newly formed channel in the oligomeric structure (51, 56, 168) (Figure 4). The N-terminal domain is relocated in close proximity to Glu72 and is also surrounded by other hydrophilic residues, thereby converting the hydrophobic pore into a hydrophilic pore, allowing the passage of Cyto c and other apoptogenic proteins across the OMM (16, 51, 56, 60, 168). Importantly, the N-terminal domain is a target for anti-apoptotic proteins, interacting with HK, Bcl-2, and Bcl-xL (16, 18, 19, 51). Thus, anti-apoptotic proteins may function by interacting with the VDAC1 N-terminal domain and preventing the conversion of the pore to a hydrophilic environment and, hence, the translocation of apoptogenic proteins from the IMS to the cytosol to trigger apoptosis.
VDAC1 Expression Level and Cell Death Induction—A New Concept
Exogenous VDAC1 Overexpression Induces Cell Death
The expression level of VDAC1 plays a critical role in mitochondria-mediated apoptosis, as demonstrated using silencing or overexpression approaches (1, 16, 17, 133, 141, 161, 165, 171, 172). VDAC1 overexpression-induced apoptosis was inhibited by VDAC1-interacted compounds RuR (133, 138), Bcl-2, and DIDS (171), or by overexpression of HK-I (17, 21, 133). As further presented below (see Apoptosis Stimuli Induce VDAC1 Overexpression in Ca2+-Dependent Manner, Proposed Mechanism for Cell Death Induction by VDAC1 Overexpression—A New Concept), the cellular expression level of VDAC1, associated with its oligomerization is an important factor regulating mitochondria-mediated apoptosis.
Apoptosis Stimuli Induce VDAC1 Overexpression in Ca2+-Dependent Manner
Several studies have demonstrated that the induction of apoptosis is followed by an increase in the levels of VDAC1 expression (12), UV irradiation (173), ROS (174), and arsenic trioxide (As2O3) (175).
Other examples include arbutin (hydroquinone-O-beta-d-glucopyranoside), a tyrosinase inhibitor in A375 human malignant melanoma cells (176), and somatostatin, used in the treatment of advanced prostate cancer, which upregulated the expression of VDAC1 and VDAC2 in the LNCaP prostate cancer cell line (177). Upregulation of VDAC1 expression was noted in acute lymphoblastic leukemia cell lines following prednisolone treatment (178). Cisplatin-induced VDAC1 overexpression in a cisplatin-sensitive cervix squamous cell carcinoma cell line (A431) but downregulation VDAC1 in a cisplatin-resistant cell line (A431/Pt) (179).
The causal nature of the relationship between VDAC1 expression levels and sensitivity to various treatments was illustrated in several studies (12, 161) where the correlation with drug efficacy suggests that numerous anti-cancer drugs and treatments act via the regulation of VDAC1 expression.
Apoptosis induction was shown to disrupt [Ca2+]i homeostasis and energy production (180). Indeed, many anti-cancer drugs and other cytotoxic agents, such as thapsigargin, STS, As2O3, and selenite, induce apoptosis, and disrupt [Ca2+]i homeostasis (161, 162). The mechanism of upregulation of VDAC1 by the apoptosis induction is thought to involve an increase in [Ca2+]i (161, 162). The overexpressed VDAC1 then forms oligomers (as discussed in Section “VDAC1 Homo-Oligomer Forming the Cyto c Release Pathway”) and this triggers Cyto c release and, finally, cell death (161, 162). In support of this proposal, AKOS-022 and VBIT-4, compounds that interact directly with VDAC1 to prevent oligomerization, prevented the process of elevation of [Ca2+]i, Cyto c release, and apoptosis (143).
Thus, although mechanisms employed by apoptosis inducers may differ, they all induce VDAC1 overexpression in a Ca2+-dependent manner (161), suggesting that elevation [Ca2+] and upregulation of VDAC1 represent a common mechanism of apoptotic stimuli.
Proposed Mechanism for Cell Death Induction by VDAC1 Overexpression—A New Concept
Upregulation of the expression of VDAC1 may, therefore, represent a new common denominator for the mechanism of action of apoptosis. This proposal is based on the observation that several cancer drugs and treatments induce apoptosis as well as upregulating VDAC1 expression levels (176–179, 181).
Accordingly, a new concept of apoptosis induction can be formulated as: apoptosis inducers ⟶ enhanced VDAC1 expression levels ⟶ VDAC1 oligomerization ⟶ Cyto c release ⟶ apoptosis.
This novel mechanism provides a platform for developing a new class of drugs to treat cancer acting via modulating VDAC1 levels through action on the gene promoter.
Modulation of VDAC1-Mediated Apoptosis by Small Molecules Acting via Direct Interaction with VDAC1
Several compounds have been shown to interact with VDAC1 and thereby modulate the protein’s apoptotic function either positively or negatively. Some examples are given below.
Molecules Interacting with VDAC to Induce Apoptosis
Erastin is an anti-tumor agent selective for tumor cells bearing oncogenic RAS (30). The compound binds VDAC2 but not N-terminal truncated hVDAC2 (182).
Oblimersen (G3139), an 18-mer phosphorothioate anti-sense oligonucleotide targeting the initiation codon region of Bcl-2 mRNA, has been shown to bind to bilayer-reconstituted VDAC1 and reduces the channel conductance (183).
Avicins represent a family of triterpenoid saponins, which exhibit cytotoxic activity in tumor cells, as well as anti-inflammatory and anti-oxidant properties. Avicins interact with bilayer-reconstituted VDAC1 to reduce its channel conductance (184) and permeabilize the OMM to induce Cyto c release (185).
Cisplatin is a widely used anti-cancer drug that acts by inducing apoptosis through the formation of inter- and intra-strand nuclear DNA cross-links. Mitochondria have also been implicated as a cisplatin target. Cisplatin binds to VDAC1 (186) and modulates VDAC1 activity (179). It has been suggested that VDAC1 may serve as a cisplatin receptor in apoptotic pathways (187).
Endostatin has been shown to promote PTP opening via binding to VDAC1. Silencing VDAC1 expression by siRNA attenuated ES-induced apoptosis in endothelial cells (140).
Methyl jasmonate (MJ) is a natural cyclopentanone lipid belonging to the jasmonate family of plant oxylipin stress hormones (188). MJ interacts directly with VDAC1 to reduce the channel conductance and also detaches HK from VDAC1 (189).
Cyathin-R, a cyathane diterpenoid, was found to interact with VDAC1 to decrease channel conductance and induce VDAC1 oligomerization and apoptosis in Bax/Bak-deficient cells (144). Cyathin-R-induced VDAC1 oligomerization and apoptosis were inhibited by VDAC1-interacting molecules, such as DIDS, SITS, DNDS, and DPC (144). si-RNA targeting VDAC1 to reduce its expression prevented cyathin-R-induced apoptosis. Moreover, cyathin-R effectively attenuated tumor growth when Bax/Bak-deficient cells were implanted into a xenograft mouse model (144). Cyathin-R, thus, represents a potential lead candidate to be an effective anti-cancer drug, inducing cell death in cancerous cells with inactivated Bax/Bak.
Molecules Interacting with VDAC1 to Inhibit Apoptosis
DIDS, SITS, H2DIDS, DNDS, and DPC, which are known anion transport inhibitors, all interact with VDAC1, and inhibit apoptosis stimuli-induced apoptosis and VDAC1 oligomerization in many cancer cell lines (99, 144). DIDS blocked apoptosis triggered by overexpression of rice VDAC in mammalian cells (171) and prevented cisplatin-(139) and STS-induced (190) cell death.
AKOS-022 and VBIT-4 are newly developed apoptosis inhibitors whose direct interaction with VDAC1 reduces VDAC1 channel conductance and prevents VDAC1 oligomerization and apoptosis in a number of cell lines (143).
Ruthenium red (55, 91) and the ruthenium amine binuclear complex (Ru360) (191), which are proposed to specifically interact with Ca2+-binding proteins, decreased the channel conductance of VDAC1. RuR protects against cell death induced by various stimuli (91, 192), or by VDAC1 overexpression (133). RuR had no effect on cells expressing E72Q-mVDAC1, or on VDAC1 channel conductance, suggesting that RuR-mediated protection against apoptosis is exerted through its direct interaction with VDAC1 (138).
The action of these apoptosis inhibitors thus supports the concept of a tight coupling between VDAC1 oligomerization and apoptosis induction. Inhibiting apoptosis at an early stage, such as VDAC1 oligomerization, may be an effective approach to block or slow apoptosis in neurodegenerative disorders (193) and various cardiovascular diseases, where there is enhanced apoptosis (194, 195).
Modulation of VDAC1-Mediated Apoptosis and Metabolism via Interacting Proteins
The location of VDAC1 in the OMM positions it well to interact with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities (Figure 1). Here, we focus on the interactions of VDAC1 with proteins associated with cancer.
VDAC Interaction with HK and Other Metabolism-Related Proteins
The multiple regulatory roles of VDAC1 in cell metabolism are mediated not only through its function in the energy production and metabolic cross-talk between the mitochondria and the rest of the cell but also via interactions with other metabolism-related proteins. These proteins include ANT (196), CrK (76, 197), glycerol kinase (198), HK (17, 20, 21, 24, 165, 189), C-Raf kinase (199), glyceraldehyde 3-phosphate dehydrogenase (41, 200), TSPO (201), and tubulin (202). Here, we concentrate on the interaction of VDAC1 with HK, which effectively couples OXPHOS and glycolysis, an important factor in cancer cell energy homeostasis (the Warburg effect).
Cancer Cell Bioenergetics and Apoptosis Are Regulated by VDAC1–HK Interaction
Cancer cells are well characterized by their high rate of glycolysis, designed to satisfy the heavy demands of transformed cells for metabolic intermediates (203). The mitochondrial-bound isoforms HK-I and -II use newly synthesized ATP to catalyze the phosphorylation of glucose to G-6-P. These enzymes are overexpressed in many cancers, including colon, prostate, lymphoma, glioma, gastric adenomas, and breast cancers (118, 132, 204–206). Both HK-II and HK-I bind to mitochondria (16, 17, 81, 133) and possess the hydrophobic N-terminal domain necessary for binding to mitochondria. The isoforms have quite different mechanisms of expression regulation (207).
Various studies, including site-directed mutagenesis, have demonstrated that VDAC1 is the mitochondrial-binding site of HK (17, 18, 21, 133, 145, 208). HK binding to VDAC1 allows direct coupling of mitochondrially generated ATP to incoming glucose, permitting mitochondria to synchronize the glycolytic flux with that of the TCA cycle and ATP synthase (1, 23, 165). In this way, the VDAC1–HK complex can regulate not only the glycolytic pathway but also other seminal metabolic pathways, such as the pentose phosphate shunt. The VDAC1–HK interaction was shown to be regulated by phosphorylation, possibly as a result of upregulation of glycogen synthase kinase 3β (GSK3β) (24) by protein kinase C, or in response to the cholesterol content of the OMM (81).
Hexokinase-I and HK-II were shown to function as anti-apoptotic proteins via binding to VDAC1, with their detachment enabling apoptosis activation (14, 17, 20, 21, 24, 81, 165, 189, 209, 210). Thus, the interaction of HK with VDAC1 points to HK function not only in cell metabolism but also as regulator of apoptosis. This dual role of HK makes the VDAC1–HK complex an attractive target for anti-cancer therapy (see below).
Hexokinase interaction with VDAC1 protects cells against apoptosis as activated by Bax or Bak (81, 119, 132, 209) and HK-I acts at the mitochondria to block TNF-induced apoptosis while conversely HK-I depletion accelerates the process (211). Importantly, it has been shown that single mutations in VDAC1 or when N-terminal truncated VDAC1 were expressed, HK-I showed no anti-apoptotic effect or reduction of channel conductance of bilayer-reconstituted (16, 17, 133, 210). In addition, mitochondria-bound HK-II inhibited Cyto c release and apoptosis as induced by Bax (209). Finally, VDAC1-based peptides, interacting with purified HK, were shown to prevent HK protection against apoptosis (20) (see VDAC1-Based Peptides As Potential Anti-Cancer Therapy).
HK Interaction with VDAC1 Offers Advantages to Cancer Cells
The advantages to cancer cells of HK binding to VDAC1 have been reviewed previously (12).
-
(a)
Production and access to energy and metabolites: HK bound to VDAC1 has direct access to ATP newly produced in the mitochondria (212), facilitating the maintenance of a high glycolytic flux rate in tumors and, thus, increased energy and metabolite production (21). HK bound to VDAC1 is also less sensitive to inhibition by the product, G-6-P.
-
(b)
VDAC1-bound HK acts as an anti-apoptotic protein: As presented above, HK-I and HK-II bound to VDAC1 function as anti-apoptotic proteins preventing Cyto c release and subsequent apoptosis (14, 17, 20, 21, 24, 81, 165, 189, 209, 210). HK also protects against Bax- or Bak-mediated apoptosis (81, 119, 209).
-
(c)
Regulation of ROS production/efflux from the mitochondria by HK: ROS production is usually increased in cancer cells (213). HK, when associated with the mitochondria, reduced both mitochondrial ROS generation (214) and intracellular levels of ROS (105).
-
(d)
Increased synthesis and uptake of cholesterol: HK binding to the mitochondria mediates increased synthesis and uptake of cholesterol into the mitochondria of cancer cells (81).
Disrupting VDAC1–HK Interaction As a Strategy to Interfere with Cancer Cell Growth and Induce Cell Death
The advantages for cancer cells presented above make the HK–VDAC1 complex an attractive target for anti-cancer therapy.
A number of agents, including 2-deoxyglucose (2-DG), 3-bromopyruvate (3-BP), an alkylating reagent, and lonidamine have been used to inhibit HK activity and disrupt glycolysis (215). In addition, the anti-fungal agents clotrimazole and bifonazole were shown to disrupt the HK-VDAC1 complex (189, 216).
However, glycolysis inhibitors can affect not only cancer cells but also normal tissues that use glucose as their main energy source (brain, retinae, and testis). Despite these concerns, targeting VDAC1–HK interaction proposed as a promising target for anti-cancer therapy (215, 217). Detachment of HK from VDAC1 impairs energy and metabolic homeostasis, including the coupling between glycolysis and mitochondrial metabolism, as well as enabling activation of apoptosis. Disturbing the VDAC1–HK interaction could, furthermore, influence cholesterol synthesis and distribution in the OMM (83).
Several agents have been identified that can dissociate the VDAC1–HK complex including HK-I- (218) and HK-II-derived peptides, clotrimazole (165, 216), a cell-permeable HK-II-based peptide (119), and MJ (189). Recently, synthetic VDAC1-based peptides (20, 145, 210) were shown to interact directly with HK-I and HK-II and lead to their dissociation from VDAC1 (see VDAC1-Based Peptides As Potential Anti-Cancer Therapy).
Since VDAC1-bound HK is essential for tumor cells, the detachment of HK from VDAC1 represents a novel therapeutic strategy to impair cancer metabolism and augment apoptosis (11, 12, 73, 215).
Interaction of VDAC1 with Bcl-2 Family Members
The resistance of cancer cells to apoptosis involves a variety of strategies. One of these involves the overexpression of anti-apoptotic proteins of the Bcl-2 family, which contributes to disease progression and drug resistance (129, 219–221) The Bcl-2 family comprises pro-apoptotic (e.g., Bid, Bax, Bim, and Bak) and anti-apoptotic (e.g., Bcl-2 and Bcl-xL, Mcl-1) members that up- or downregulate apoptosis, respectively (222). Mcl-1 has been shown to interact directly with VDAC1 to increase mitochondrial Ca2+ uptake and ROS generation (223).
The mechanisms by which Bcl-2 family proteins regulate apoptosis involve interactions with mitochondria and control of OMM permeability. Regulation of apoptosis by interactions of VDAC1 with these proteins has been reported in a number of studies (16, 19, 20, 22, 139, 224, 225). VDAC1 has been shown to interact with Bax/Bak (142, 226), Bcl-2 (16, 19, 51), Bcl-xL (18, 19, 22, 25, 48, 96, 224, 225, 227), and with Bax and Bim (26, 158, 225), such that anti-VDAC antibodies inhibited Bax- and Bim-induced release of Cyto c (136). In addition, Bcl-2 and Bcl-xL block As2O3-induced VDAC1 dimerization (137). BH4 oligopeptides derived from Bcl-2 and Bcl-xL were able to inhibit VDAC1 activity in liposomes, even in the presence of a pro-apoptotic protein, such as Bax (224). Bcl-2 and Bcl-xL were shown to reduce the conductance of native but not mutated VDAC1, as well as to protect cells expressing native but not mutated VDAC1 against apoptosis (18, 19). In addition, activation of Bax by cisplatin was prevented in cells silenced for VDAC1 expression (25, 139). Finally, Bid was shown to interact with VDAC1 as reflected in the decrease in VDAC1 conductance (228).
Thus, VDAC1 binds members of the Bcl-2 family proteins thereby regulating their effects on apoptosis. Hence, interfering with such interactions could facilitate apoptosis induction and enhance the therapeutic effect of chemotherapeutic agents.
Interaction of VDAC1 with Other Proteins
Translocator protein is closely associated with VDAC1 (229) and this association allows ROS generated via TSPO to affect VDAC1 (201, 230). In addition, overexpression of TSPO inhibits VDAC1 expression, while VDAC1 expression level was increased upon silencing of TSPO in endothelial cells (231).
A number of cytoskeletal proteins have been reported to interact with and regulate VDAC1. These proteins include the following.
Gelsolin (Gsn) is a Ca2+-dependent protein that regulates actin assembly and disassembly. Human (h)Gsn has pro-apoptotic or anti-apoptotic activity, depending on the cell type (232). hGsn inhibited VDAC1 channel activity and interacted with VDAC1-containing liposomes in a Ca2+-dependent manner to inhibit Cyto c (232).
Tubulin, co-immunoprecipitated with VDAC1 (233) and the association was also demonstrated by tubulin-induced VDAC1 closure (202), an effect thought to sustain the Warburg effect (234). It is proposed that tubulin, VDAC1, and MtCK form a super-complex that is structurally and functionally coupled to the ATP synthasome (235).
Microtubule-associated protein 2 was shown by affinity chromatography to bind VDAC1 (236).
The mitochondrial anti-viral signaling protein, also known as IPS-1, VISA, or Cardif (237) and localized in the OMM, was demonstrated to modulate VDAC1 protein stability via the ubiquitin–proteasome pathway (238).
Superoxide dismutase 1 (SOD1) mutated protein, which is associated with ALS, and reduced VDAC1 channel conductance (239) and altered the interaction between VDAC1 and Bcl-2 (240).
Endothelial NO synthase (eNOS) was also found to bind VDAC1, with this amplifying eNOS activity intracellularly in a Ca2+-dependent manner (241).
Several additional proteins were shown or proposed to interact directly with VDAC1. These include PBP74 (heat-shock protein peptide-binding protein 74), also known as mtHSP70/GRP75/mortalin (242), and GRP78, a 78-kDa glucose-regulated protein that forms a complex with vaspin. The complex of GRP78, and VDAC on the plasma membrane, promotes proliferation, inhibits apoptosis, and protects against vascular injury in diabetes mellitus (243). Other proteins that can interact with VDAC are the ORDIC channel, actin (244), Nek1 (NIMA-related protein kinase 1) (173), aldolase (41), Tctex-1/DYNLT1 (dynein light chain) (242), CRYAB (α-crystallin B) (245), and α-synuclein (246).
The pro-apoptotic protein BNIP3 was shown to interact with VDAC1 to induce mitochondrial release of endonuclease G (247) and VDAC1 co-immunoprecipitated with the L-type Ca2+ channel (248).
A total of 44 VDAC1 interacting genes were identified as being commonly differentially expressed between normal and tumor tissues in human carcinomas (249).
In summary, VDAC1 serves as a central hub for responses to cellular signaling and the effects may be mediated by interaction with many proteins (Figure 1), indicating the central role played in cell metabolism and apoptosis.
Alterations in VDAC1 Expression Level in Cancer
Voltage-dependent anion channel 1 is highly expressed in many cancer types compared to the levels in normal cells. VDAC1 overexpressed in several cancer cell lines relative to fibroblast cell line (250) and, in ascites hepatoma AH130 cells, the three VDAC isoforms expression levels were significantly higher than in normal liver cells. Higher VDAC1 levels were connected to primary malignancies of the biliary tract (251), and were found in gastric cancer cells (252). Both VDAC1 mRNA and protein levels were upregulated in H358 cells (253). Induction of Cyto c release by G3139 in several melanoma and prostate cancer cell lines was found to be correlated with VDAC1 expression levels (254). In myeloma cells, CD45 expression was accompanied by elevated VDAC1 expression that sensitized the cells to a diverse set of apoptotic stimuli (255). These differences are not surprising in light of the of VDAC1 functions in cell metabolism and energy production, systems particularly important for cancer cells proliferation. Overexpressed VDAC1 presents anchoring sites for the cancer overexpressed HK and for Bcl-2 and Bcl-xL, interactions that are important for their anti-apoptotic activities (see Modulation of VDAC1-Mediated Apoptosis and Metabolism VIA Interacting Proteins).
Overexpression of VDAC1 was detected in tissue arrays for thyroid, lung, cervix, ovary, pancreas, melanoma, and glioblastoma cancers as well as in lung tissue samples taken from healthy and tumor-containing areas of the same patient (12). Similar results were obtained in other studies of breast, colon, liver, lung, pancreatic, and thyroid cancers (249) and lung tumors (256). VDAC1 was also found to be overexpressed in peripheral blood mononuclear cells (PBMCs) from chronic lymphocytic leukemia (CLL) patients, as compared to PBMCs from healthy donors (257). Cervical cancer patients with high VDAC1 displayed higher rates of recurrence and poorer overall survival than those with low VDAC1 (258). VDAC also appears to be a potential marker for the diagnosis of colorectal cancer (259) and gastric cancer (260). VDAC1 overexpression was demonstrated in lung cancer, where C-terminally truncated VDAC1 (VDAC1-ΔC) was present in tumor cells exposed to hypoxia in 50% of 46 patients with lung cancer (261). In addition, a significant positive correlation exists between the levels of VDAC1 and the histological grade of breast cancer (262) as well as poor prognosis of primary lung adenocarcinoma (263) and of non-small cell lung cancer (NSCLC) patients (264). In NSCLC VDAC1 was proposed as a potential predictor of poor outcome in the diseases early stage (264). In addition, tumor progression and sensitivity to chemotherapy was correlated with VDAC1 expression (263, 265). Thus, VDAC1 expression levels can serve as a biomarker for cancer development, treatment efficacy, and as a predictor of poor outcome.
Unraveling VDAC1-Based Therapies
As already discussed, VDAC1 offers a unique target for anti-cancer therapies because of its role as a key regulator of energy and metabolism and apoptosis. VDAC1-based therapeutic strategies include RNA interference (RNAi) designed to downregulate VDAC1 expression levels and cause growth arrest, as well as VDAC1-based peptides that impair energy homeostasis and minimize the self-defense mechanisms of cancer cells and small molecules that induce apoptosis. Together, such anti-cancer therapies are expected to be highly effective, even in drug-resistant tumors.
Silencing VDAC1 Expression by Short Hairpin RNA (shRNA) or siRNA As a Tool to Reprogram Cancer Cell Metabolism
The role of VDAC1 as a key regulator of the cellular metabolic and energy reprogramming processes essential to cancer survival (1, 11, 12, 61) makes targeting VDAC1 an attractive strategy for anti-cancer therapy. As one approach, overexpressed VDAC1 can be downregulated by using RNAi, including shRNA and siRNA.
VDAC1 Silencing Using shRNA and siRNA As a Strategy for Cancer Therapy
We have demonstrated that downregulation of VDAC1 expression by hVDAC1-shRNA disrupts energy production, arrests cell growth, and inhibits tumor development in an animal model, illustrating the essential role of VDAC1 in energy production and cell growth (53, 266). Furthermore, tumors developed into nude mice from HeLa cervical cancer cells stably expressing shRNA directed against hVDAC1, strongly inhibited the development of tumors (266). Indeed, silencing VDAC1 by shRNA blocked TRAIL-induced mitochondrial apoptosis, suggesting that expression of VDAC1 is required for caspase-8 activation (267). The use of hVDAC1-shRNA permitted the demonstration of VDAC1 involvement in arsenic trioxide-, ascorbic acid-, and disulfiram (AAA)-induced aponecrosis and the switch from apoptosis to the aponecrosis death pathways (268). The shRNA also reduced cell proliferation and migration of cervical cancer cells, and increased ROS production (258). Stable expression of hVDAC1-shRNA stimulated NLRP inflammasome activators and augmented caspase-1 and IL-1β secretion in THP cells (269). These multiple effects of VDAC1 silencing point to an additional role for VDAC1 as a central protein in regulating cell signaling.
Silencing VDAC1 expression by a single siRNA specific to the human VDAC1 (si-hVDAC1) sequence resulted in cell proliferation and cancer cell growth inhibition both in cell cultures and in vivo animal models (75, 270). In addition, siRNA at nanomolar concentrations silenced VDAC1 expression in many tested cell lines, inhibited cell growth (over 90%) and decreased ATP levels (53, 75, 266). In in vivo experiments using a xenograft lung cancer mouse model, si-hVDAC1 inhibited tumor growth and even caused tumor regression (75).
Recently (270), we demonstrated that depleting VDAC1 by si-hVDAC1 assaults critical functional nodes in the oncogenic network of GBM tumors, leading to a multi-pronged attack on cancer hallmarks, reversing cancer-reprogrammed metabolism, thereby inhibiting cell proliferation, tumor growth, EMT, and angiogenesis, and also targets GSCs, leading to their differentiation into neuronal-like cells.
Other studies from our group (271) using a GBM xenograft mouse model showed that upon VDAC1 depletion, several pro-apoptotic proteins were overexpressed yet apoptosis was not induced. This suggests that essential players of the apoptosis executioner pathways may serve dual functions, acting either to kill cells or to promote differentiation, depending on the energy level of the cell.
Another connection between VDAC1 and cell energy signaling was demonstrated in recent studies showing that VDAC1 is a direct target of the anti-fungal agent, itraconazole, and that VDAC1 is a key mediator of the inhibition of mTOR and endothelial cell proliferation by the AMPK signaling pathway (270, 272).
si-hVDAC also markedly decreased HIF1-α levels and tumor growth in U-87MG and U-118MG cancer cells (270). These findings suggest a relationship between HIF1-α and VDAC1 expression and tumor growth.
MicroRNA (miRNA) Acts via Modification of VDAC Expression Levels
MicroRNAs belong to a class of small, non-coding, regulatory RNAs which bind to the 3-UTR of target mRNAs to reduce target protein levels.
The level of miR-7 has been shown to be downregulated in various cancer cells, such as GBM (273), breast cancer (274), urothelial carcinoma (275), gastric tumors (276), pancreatic cancer (277), colorectal cancer (278), and in hepatocellular carcinoma tissues, as compared to adjacent non-tumor tissue (279). These findings suggest that miR-7 has a tumor suppressor function. Several studies have demonstrated that the miR-7 regulates the function of the mitochondrial PTP by downregulating VDAC1 expression. Overexpressing VDAC1 without the 3-UTR significantly abolished the protective effects of miR-7 against 1-methyl-4-phenylpyridinium ion (MPP)-induced cytotoxicity and mitochondrial dysfunction (280).
Proteomic profiling of cells showed that overexpression of miR-29a also resulted in downregulation of VDAC1 and the VDAC2 protein (281).
Thus, considering the high expression level of VDAC1 in tumors and the specificity of si-hVDAC1 in inhibiting cancer cell and tumor growth, silencing VDAC1 expression can be considered as a novel strategic therapeutic approach to treat cancer.
VDAC1-Based Peptides As Potential Anti-Cancer Therapy
VDAC1-Based Peptides—Development and Cell Death Induction
Cancer cells share several features that distinguish them from normal cells, including avoiding apoptosis, thereby drug resistance (282). Indeed, defects in the regulation or even evasion of apoptosis are hallmarks of cancer (1, 11, 61, 115). To avoid apoptosis, cancer cells developed several strategies, such as overexpression of anti-apoptotic proteins, such as the Bcl-2 family of proteins and HK, to prevent the release of Cyto c from mitochondria (1, 11, 124).
To mediate their anti-apoptotic activities, HK, Bcl-2, and Bcl-xL interact with VDAC1 (16–20, 133). Therefore, we engineered VDAC1-based peptides designed to interfere with these interactions. We identified those VDAC1 domains and amino acid residues important for the interaction of VDAC1 with HK, Bcl-2, and Bcl-xL and designed VDAC1-based peptides specifically targeting these interactions (15–20, 22, 133, 224, 225). These peptides are designed to serve as “decoy” peptides that compete with VDAC1 for the HK-, Bcl-2-, and Bcl-xL–VDAC1 interactions and consequently abolish their anti-apoptotic activities. As the VDAC1-based peptide target intracellular proteins, several cell-penetrating peptides were developed.
VDAC1 derived sequences corresponding to the N-terminal domain (N-Ter) and a VDAC1 sequence partially exposed to the cytosol (LP4) were fused to Antp (penetrating), a 16 residue-long sequence from the Drosophila antennapedia-homeodomain, to yield the Antp-LP4 and N-Ter-Antp peptides (Figure 5). These peptides promoted cell death in a variety of genetically characterized cell lines derived from different human cancers (70, 283).
Figure 5.
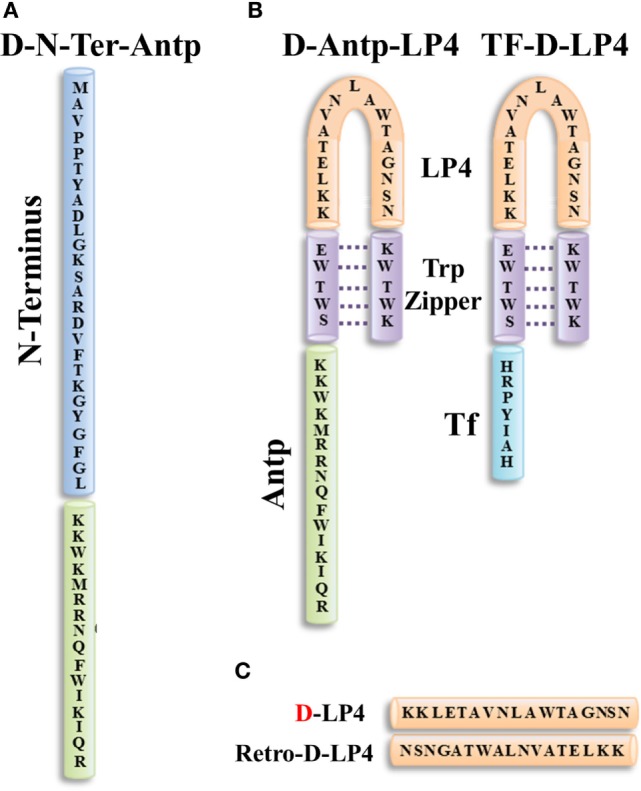
Structure of VDAC1-based peptides. Schematic illustration of D-N-terminus-Antp (D-N-Ter-Antp) (A) and D-Antp-LP4 and Tf-D-LP4 (B) peptides. The VDAC1-derived sequences N-terminus and LP4 are in blue and orange, respectively. The cell-penetrating peptide Antp in green and Tf is in light blue. The loop shape of LP4 stabilized by a tryptophan zipper (Trp) is in purple. The N-terminus of LP4 and Antp is composed of d-amino acids. (C) The sequences of D-LP4 and retro-D-LP4.
Antp-LP4—this is a loop-shaped cell-penetrating peptide comprising the SWTWE sequence at the N-terminal end and the KWTWK sequence at the C-terminal end of a VDAC1-derived sequence (LP4, residues 199–216). This generates a tryptophan zipper and a stable β-hairpin conformation (284), mimicking the LP4 loop in the native VDAC1 protein, which is fused to Antp. Antp-LP4 peptide prevented the anti-apoptotic effects of HK, Bcl-2, or Bcl-xL (16–20, 133) and induced cell death in several cancer cell lines, while being less effective in non-cancerous cells (70).
N-Ter-Antp—in this peptide, the N-terminal sequence was selected based on the findings that N-terminal domain-truncated VDAC1 had lost the ability to bind HK, Bcl-2, or Bcl-xL (16–20, 133). N-Ter-Antp peptide interacted with HK, Bcl-2, and Bcl-xL and inhibited their anti-apoptotic effects (16–20, 133).
Tf-D-LP4 peptide—this peptide comprises a VDAC1-derived sequence (LP4), with the tryptophan zipper fused to a cell penetrating peptide, a human transferrin receptor (hTfR) (CD71)-recognition sequence, HAIYPRH (Tf) (Figure 5). hTfR is overexpressed (up to 100-fold) in liver, pancreatic, prostate, GBM, and other cancers (285), relative to their normal counterparts and is also highly expressed in the BBB (286).
Based on results obtained using over 40 versions of cell-penetrating peptides, several modifications were introduced into the VDAC1-based peptides to in order to address peptide efficacy, stability, cell penetration, and specificity toward cancer cells as described below.
The effects of the selected designed VDAC1-based peptides were tested on 42 different cancer cell lines and found to induce cell death regardless of cancer type or mutation status, with specificity toward cancerous cells (16, 17, 19, 20, 70, 133). VDAC1-based peptides (N-Ter and LP4 derived sequences) were shown to limit Ca2+ uptake into the mitochondrial matrix and inhibit ROS generation in lung cancer cells (223). In addition, the use of VDAC1-based peptides (residues 10–30) suggests that VDAC1 is part of a system controlling cell proliferation in neuroblastoma cells via its interaction with glucose-regulated protein 78 (GRP78) (287).
The Mode of Action of VDAC1-Based Peptides
Our results suggest that the VDAC1-based peptides lead to: (i) impaired cell metabolism and energy homeostasis; (ii) negation of the anti-apoptotic activities of HK, Bcl-2, and Bcl-xL, and (iii) induction of massive apoptosis (16, 17, 70) (Figure 6).
Figure 6.
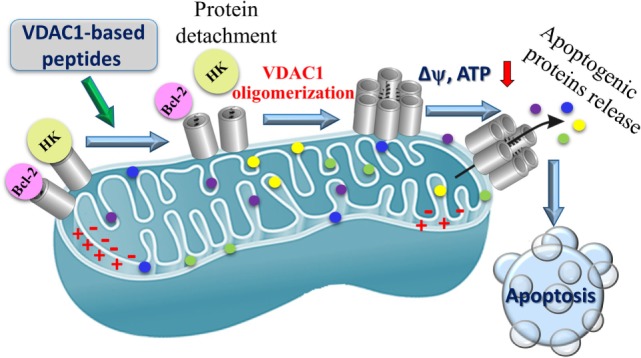
Proposed mode of action of VDAC1-based peptides leading to mitochondria-mediated cell death. VDAC1 is overexpressed in the mitochondria of cancer cells and associated with hexokinase (HK) and Bcl-2, Δψ is maintained, the cell remains in homeostasis with respect to energy production and is protected from apoptosis. VDAC1-based peptides interact with anti-apoptotic proteins HK and Bcl-2 causing the proteins to disassociate from VDAC1, and leading to Δψ dissipation, decreased ATP production, mitochondrial dysfunction, VDAC1 oligomerization, and Cyto c release. These events ultimately lead to cell death.
Voltage-dependent anion channel 1-based peptides were found to impair cell metabolism and energy homeostasis. Altered energy metabolism, including the use of glucose via glycolysis as an energy source, is a common feature of most malignant tumors (115, 288). Mitochondrial-bound HK is highly expressed in glycolytic cancer cells (289), supporting aerobic glycolysis (23) but also conferring stability to mitochondria (290) and resistance to apoptosis (14, 17, 20, 21, 24, 81, 165, 189, 209, 210). As already discussed, VDAC1 is overexpressed in many cancer types (see Alterations in VDAC1 Expression Level in Cancer) (12) and, thus, presents anchoring sites for overexpressed HK, allowing direct access to mitochondrial ATP and an increased glycolytic rate (1). The VDAC1-based peptides interact with and detach HK from its binding site in VDAC1, leading to decreased glycolysis, and decreases in ΔΨ and cellular ATP levels (16, 20). Thus, detachment of mitochondria-bound HK leads to a disruption of the cellular energetics status of cancer cells.
VDAC1-based peptides were found to prevent the anti-apoptotic activity of anti-apoptotic proteins to induce apoptosis. As already described above, cancer cells utilize a variety of strategies to limit or avoid apoptosis, including overexpression of anti-apoptotic proteins, such as members of the Bcl-2 family of proteins and HK. VDAC1 binds HK, Bcl-2, and Bcl-xL (16, 18–20, 51, 96, 142, 224, 227). The VDAC1-based cell-penetrating peptides N-Ter and LP4 were shown to interact directly with purified anti-apoptotic proteins and, after penetrating into cells, antagonized their anti-apoptotic activities (18–20). These findings suggest that VDAC1-based peptides interfere with the binding of anti-apoptotic proteins to VDAC1 thereby permitting apoptosis induction.
VDAC1-based peptides induced VDAC1 oligomerization, release of Cyto c and other molecular hallmarks of apoptosis, promoting membrane blebbing, phosphatidylserine surface exposure, and nuclear condensation and fragmentation (20, 70) (Figure 6). This multiple mode of peptide activities may explain their high potency and specificity toward tumor cells.
The Effects of VDAC1-Based Peptides on Cancer—Ex Vivo and In Vivo Studies
Peptide activity was tested in an ex vivo study using samples from CLL patients (70). CLL is characterized by a clonal accumulation of mature neoplastic B cells that are resistant to apoptosis (291). VDAC1-based peptides (Antp-LP4 and N-terminal-Antp) selectively killed PBMCs obtained from CLL patients but not from healthy donors (70). The ability of the peptides to induce cell death was well correlated with the amount of the cancer cells. The mode of action of the peptides on CLL involves inhibition of energy production and apoptosis induction.
In sub-cutaneous and intracranial xenograft mouse models of GBM, the VDAC1-based peptides, N-Ter and Tf-D-LP4, were found to disrupt cell metabolism and energy homeostasis to inhibit tumor growth, invasion, stemness, and induce apoptosis (71). Peptide-treated tumors downregulated metabolism-related enzymes and transporters, and elevated the levels of apoptotic proteins, such as p53, Cyto c, and caspases. In agreement with the results obtained with cells in culture, the peptides acts by impairing energy and metabolism, interfering with the actions of anti-apoptotic proteins, and inducing cell death (71). VDAC1-based peptides, thus, offer an affordable and innovative new conceptual therapeutic paradigm that can potentially overcome chemoresistant, invasive, GBM cancer stem cells, and reduce relapse rates.
To summarize, VDAC1-based peptides act relatively fast and at low concentrations to induce cell death in a variety of cancer cell lines, irrespective of the origin of the cancer or carried mutations. This is very important in view of tumor heterogeneity, metastatic transformation, and modifications acquired during tumor development.
VDAC Involvement in Diseases Other Than Cancer
Neurodegenerative Diseases, Mitochondria, Apoptosis, and VDAC
Impaired mitochondrial function has been reported for most neurodegenerative diseases, such as Parkinson’s disease, Huntington’s disease, ALS, and AD (292). Recent studies have shown that such disorders share characteristics of mitochondria-mediated apoptotic death (293).
Mitochondrial dysfunction is an early event in AD pathogenesis, as reflected by reduced metabolism, disruption of Ca2+-homeostasis, increased free radical production, and lipid peroxidation (294–296). Aβ also affects mitochondrial respiration (297) and activates Cyto c release, resulting in apoptosis (298). Importantly, Aβ does not cause toxicity in cells depleted of mitochondria (299). High-levels of VDAC1 were demonstrated in the dystrophic neurites of Aβ deposits in AD post-mortem brains and amyloid precursor protein transgenic mice (300). VDAC1 was shown to participate in Aβ-induced toxicity (45, 301, 302) where Aβ–VDAC1 interactions are toxic to AD-affected neurons (303) and VDAC1 interactions with Aβ and phosphorylated Tau lead to mitochondrial dysfunction (302). Recently (45), we demonstrated that Aβ interacts directly with VDAC1, specifically with the N-terminal region. Moreover, VDAC1 is required for Aβ entry into the cell, as well as Aβ-mediated apoptosis, since Aβ cell penetration and toxicity were prevented in cells depleted of VDAC1 using siRNA. Finally, an increase in nitrated VDAC1 in AD was reported, reflecting oxidative damage to VDAC (304), and possibly affecting cell energy and metabolite homeostasis (305). The involvement of plasmalemmal VDAC in AD was also proposed (301, 306).
These findings point to VDAC1 as a potential target for novel therapeutic strategies for AD.
Cardiovascular Diseases, Mitochondria, Apoptosis, and VDAC
The loss of cardiac myocytes plays a critical role in the pathogenesis of cardiovascular disorders. Activation of the mitochondrial pathway of apoptotic cell death has been implicated in ischemia/reperfusion injury involving the release of Cyto c from mitochondria, followed by activation of caspase-9 in the myocardium (307). VDAC1 levels were increased in cardiomyoblast H9c2 cells that were differentiated in the presence of all-trans retinoic acid (308). The increased susceptibility of differentiated cells to mitochondrial-mediated cell death may be related to the increase in VDAC1 levels.
Type 2 Diabetes (T2D) and VDAC1
In T2D, beta cell decompensation develops when insulin secretion fails to balance insulin resistance. Hyperglycemia was found to increase VDAC1 expression in pancreatic β-cells (309) and in the kidney (310). VDAC1 levels were also increased in mouse coronary vascular endothelial cells isolated from diabetic mice. This was associated with increased mitochondrial Ca2+ concentration, mitochondrial O2− production, and PTP opening activity (311). Finally, since glucose-stimulated insulin secretion depends on the generation of ATP and other metabolites in the mitochondria (312) and since VDAC1 regulates energy and metabolism, it follows that VDAC1 is required for insulin secretion.
Perspectives
In this review, we highlighted the cancer–mitochondria–metabolism–apoptosis-VDAC1 axis. As Otto Warburg noted almost a century ago, cancer cells frequently share bioenergetics abnormalities, regardless of their cellular or tissue origin. Furthermore, the view of cancer as primarily a metabolic disease can impact any approach to cancer management and prevention. As a mitochondrial gatekeeper and overexpressed in cancer VDAC1 is a very attractive emerging anti-cancer drug target.
Another hallmark of cancer cells is their ability to avoid apoptosis by activating anti-apoptotic mechanisms associated with drug resistance, including overexpression of anti-apoptotic proteins that interacts with VDAC1. As such, interfering with this association would allow for activation of the mitochondrial pathway of apoptosis and allow for apoptosis induction by anti-cancer drugs.
Most importantly, VDAC1, in the OMM, serves as a hub protein, with its interactome, including over 150 proteins involved in metabolism, apoptosis, signal transduction, and anti-oxidation, as well as DNA- and RNA-associated proteins, that together mediate and/or regulate metabolic, apoptotic, and other processes in normal and diseased cells. Thus, VDAC1 can be considered as a key protein not only in metabolism and apoptosis regulation but also as a link between the energy, redox, and signaling pathways in mitochondria and other cell compartments. Thus, targeting these VDAC1–protein interactions may interfere with a variety of processes, such as cellular energy homeostasis, apoptosis, and other activities and signaling pathways in cancer.
Finally, VDAC1 silencing in tumors leads to reprogramed metabolism and results in alterations in the transcription factors that regulate signaling pathways associated with cancer hallmarks affecting angiogenesis, EMT, invasiveness, stemness, and induced differentiation. This suggests that VDAC1 plays a key role in cancer cell fate by controlling the cross-talk between metabolism and oncogenic signaling networks, most likely affecting the interplay between metabolism and epigenetics. VDAC1 overexpression is, moreover, a signature of most cancers and can serve as a prognostic biomarker to predict clinical outcome in diversified human cancers. Thus, a comprehensive understanding of the exact molecular basis of the complex signaling networks activated by VDAC1 over- or downregulation in the tumor that forms a pro- or anti-tumorigenic milieu is required.
Author Contributions
All authors contributed to this review by writing certain sections of the review and preparing the illustrations. All the authors read and approved the final manuscript and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This research was supported by grants from the Israel Science Foundation (307/13), Ezra and Yafa Yeruham and by Sima and Philip Needleman research funds.
Abbreviations
AIF, apoptosis-inducing factor; ANT, adenine nucleotide translocase; APAF-1, apoptosis protease-activating factor 1; Bcl-2, B-cell lymphoma 2; caspase, cysteinyl/aspartate-specific protease; CLL, chronic lymphocytic leukemia; CyP D, cyclophilin D; Cyto c, cytochrome c; DIDS, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid; ER, endoplasmic reticulum; FNQs, furanonaphthoquinones; G-6-P, glucose-6-phosphate; HK, hexokinase; IMM, inner mitochondrial membrane; IP3Rs, inositol-1,4,5-trisphosphate receptors; LDH, lactate dehydrogenase; MAC, mitochondrial apoptosis-inducing channel; NADH, reduced nicotinamide adenine dinucleotide; NSCLC, non-small cell lung cancer; OMM, outer mitochondrial membrane; PDH, pyruvate dehydrogenase; PLB, planar lipid bilayer; PT, permeability transition; PTP, permeability transition pore; RNAi, RNA interference, shRNA, short-hairpin RNA; ROS, reactive oxygen species; RuR, ruthenium red; SR, sarcoplasmic reticulum; STS, staurosporine; TNF-α, tumor necrosis factor alpha; TSPO, translocator protein; VDAC, voltage-dependent anion channel.
References
- 1.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med (2010) 31:227–85. 10.1016/j.mam.2010.03.002 [DOI] [PubMed] [Google Scholar]
- 2.Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochim Biophys Acta (2012) 1818:1466–76. 10.1016/j.bbamem.2011.10.005 [DOI] [PubMed] [Google Scholar]
- 3.De Pinto V, Guarino F, Guarnera A, Messina A, Reina S, Tomasello FM, et al. Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim Biophys Acta (2010) 1797:1268–75. 10.1016/j.bbabio.2010.01.031 [DOI] [PubMed] [Google Scholar]
- 4.De Pinto V, Reina S, Gupta A, Messina A, Mahalakshmi R. Role of cysteines in mammalian VDAC isoforms’ function. Biochim Biophys Acta (2016) 1857:1219–27. 10.1016/j.bbabio.2016.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukada K, Zhang F, Vien A, Cashman NR, Zhu H. Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis. Mol Cell Proteomics (2004) 3:1211–23. 10.1074/mcp.M400094-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang W, Du B, Chi Z, Ma L, Wang S, Zhang X, et al. Preliminary explorations of the role of mitochondrial proteins in refractory epilepsy: some findings from comparative proteomics. J Neurosci Res (2007) 85:3160–70. 10.1002/jnr.21384 [DOI] [PubMed] [Google Scholar]
- 7.Reddy PH. Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim Biophys Acta (2013) 1832:67–75. 10.1016/j.bbadis.2012.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anflous-Pharayra K, Lee N, Armstrong DL, Craigen WJ. VDAC3 has differing mitochondrial functions in two types of striated muscles. Biochim Biophys Acta (2011) 1807:150–6. 10.1016/j.bbabio.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raghavan A, Sheiko T, Graham BH, Craigen WJ. Voltage-dependant anion channels: novel insights into isoform function through genetic models. Biochim Biophys Acta (2012) 1818:1477–85. 10.1016/j.bbamem.2011.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurya SR, Mahalakshmi R. VDAC-2: mitochondrial outer membrane regulator masquerading as a channel? FEBS J (2016) 283:1831–6. 10.1111/febs.13637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shoshan-Barmatz V, Golan M. Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr Med Chem (2012) 19:714–35. 10.2174/092986712798992110 [DOI] [PubMed] [Google Scholar]
- 12.Shoshan-Barmatz V, Ben-Hail D, Admoni L, Krelin Y, Tripathi SS. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim Biophys Acta (2015) 1848:2547–75. 10.1016/j.bbamem.2014.10.040 [DOI] [PubMed] [Google Scholar]
- 13.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator – thinking outside the box. Biochim Biophys Acta (2006) 1762:181–90. 10.1016/j.bbadis.2005.10.006 [DOI] [PubMed] [Google Scholar]
- 14.Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr Pharm Des (2006) 12:2249–70. 10.2174/138161206777585111 [DOI] [PubMed] [Google Scholar]
- 15.Tsujimoto Y, Shimizu S. The voltage-dependent anion channel: an essential player in apoptosis. Biochimie (2002) 84:187–93. 10.1016/S0300-9084(02)01370-6 [DOI] [PubMed] [Google Scholar]
- 16.Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci (2009) 122:1906–16. 10.1242/jcs.040188 [DOI] [PubMed] [Google Scholar]
- 17.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem (2008) 283:13482–90. 10.1074/jbc.M708216200 [DOI] [PubMed] [Google Scholar]
- 18.Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J Biol Chem (2012) 287:23152–61. 10.1074/jbc.M112.345918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem (2010) 285:6053–62. 10.1074/jbc.M109.082990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J Biol Chem (2009) 284:3946–55. 10.1074/jbc.M803614200 [DOI] [PubMed] [Google Scholar]
- 21.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J (2004) 377:347–55. 10.1042/bj20031465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malia TJ, Wagner G. NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry (2007) 46:514–25. 10.1021/bi061577h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene (2006) 25:4777–86. 10.1038/sj.onc.1209603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res (2005) 65:10545–54. 10.1158/0008-5472.CAN-05-1925 [DOI] [PubMed] [Google Scholar]
- 25.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature (1999) 399:483–7. 10.1038/20959 [DOI] [PubMed] [Google Scholar]
- 26.Shimizu S, Tsujimoto Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci U S A (2000) 97:577–82. 10.1073/pnas.97.7.3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science (2003) 301:513–7. 10.1126/science.1083995 [DOI] [PubMed] [Google Scholar]
- 28.Roy SS, Ehrlich AM, Craigen WJ, Hajnoczky G. VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep (2009) 10:1341–7. 10.1038/embor.2009.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandra D, Choy G, Daniel PT, Tang DG. Bax-dependent regulation of Bak by voltage-dependent anion channel 2. J Biol Chem (2005) 280:19051–61. 10.1074/jbc.M501391200 [DOI] [PubMed] [Google Scholar]
- 30.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447:864–8. 10.1038/nature05859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabirov RZ, Merzlyak PG. Plasmalemmal VDAC controversies and maxi-anion channel puzzle. Biochim Biophys Acta (2012) 1818:1570–80. 10.1016/j.bbamem.2011.09.024 [DOI] [PubMed] [Google Scholar]
- 32.Bathori G, Parolini I, Tombola F, Szabo I, Messina A, Oliva M, et al. Porin is present in the plasma membrane where it is concentrated in caveolae and caveolae-related domains. J Biol Chem (1999) 274:29607–12. 10.1074/jbc.274.42.29607 [DOI] [PubMed] [Google Scholar]
- 33.Shoshan-Barmatz V, Hadad N, Feng W, Shafir I, Orr I, Varsanyi M, et al. VDAC/porin is present in sarcoplasmic reticulum from skeletal muscle. FEBS Lett (1996) 386:205–10. 10.1016/0014-5793(96)00442-5 [DOI] [PubMed] [Google Scholar]
- 34.Shoshan-Barmatz V, Zalk R, Gincel D, Vardi N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim Biophys Acta (2004) 1657:105–14. 10.1016/j.bbabio.2004.02.009 [DOI] [PubMed] [Google Scholar]
- 35.Bathori G, Parolini I, Szabo I, Tombola F, Messina A, Oliva M, et al. Extramitochondrial porin: facts and hypotheses. J Bioenerg Biomembr (2000) 32:79–89. 10.1023/A:1005516513313 [DOI] [PubMed] [Google Scholar]
- 36.Shafir I, Feng W, Shoshan-Barmataz V. Voltage-dependent anion channel proteins in synaptosomes of the torpedo electric organ: immunolocalization, purification, and characterization. J Bioenerg Biomembr (1998) 30:499–510. 10.1023/A:1020598315287 [DOI] [PubMed] [Google Scholar]
- 37.Hinsch KD, De Pinto V, Aires VA, Schneider X, Messina A, Hinsch E. Voltage-dependent anion-selective channels VDAC2 and VDAC3 are abundant proteins in bovine outer dense fibers, a cytoskeletal component of the sperm flagellum. J Biol Chem (2004) 279:15281–8. 10.1074/jbc.M313433200 [DOI] [PubMed] [Google Scholar]
- 38.Buettner R, Papoutsoglou G, Scemes E, Spray DC, Dermietzel R. Evidence for secretory pathway localization of a voltage-dependent anion channel isoform. Proc Natl Acad Sci U S A (2000) 97:3201–6. 10.1073/pnas.97.7.3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thinnes FP, Gotz H, Kayser H, Benz R, Schmidt WE, Kratzin HD, et al. [Identification of human porins. I. Purification of a porin from human B-lymphocytes (porin 31HL) and the topochemical proof of its expression on the plasmalemma of the progenitor cell]. Biol Chem Hoppe Seyler (1989) 370:1253–64. [PubMed] [Google Scholar]
- 40.De Pinto V, Messina A, Lane DJ, Lawen A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett (2010) 584:1793–9. 10.1016/j.febslet.2010.02.049 [DOI] [PubMed] [Google Scholar]
- 41.Shoshan-Barmatz V, Israelson A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: characterization, modulation and possible function. J Membr Biol (2005) 204:57–66. 10.1007/s00232-005-0749-4 [DOI] [PubMed] [Google Scholar]
- 42.Reymann S, Florke H, Heiden M, Jakob C, Stadtmuller U, Steinacker P, et al. Further evidence for multitopological localization of mammalian porin (VDAC) in the plasmalemma forming part of a chloride channel complex affected in cystic fibrosis and encephalomyopathy. Biochem Mol Med (1995) 54:75–87. 10.1006/bmme.1995.1011 [DOI] [PubMed] [Google Scholar]
- 43.Dermietzel R, Hwang TK, Buettner R, Hofer A, Dotzler E, Kremer M, et al. Cloning and in situ localization of a brain-derived porin that constitutes a large-conductance anion channel in astrocytic plasma membranes. Proc Natl Acad Sci U S A (1994) 91:499–503. 10.1073/pnas.91.2.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okada SF, O’Neal WK, Huang P, Nicholas RA, Ostrowski LE, Craigen WJ, et al. Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J Gen Physiol (2004) 124:513–26. 10.1085/jgp.200409154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smilansky A, Dangoor L, Nakdimon I, Ben-Hail D, Mizrachi D, Shoshan-Barmatz V. The voltage-dependent anion channel 1 mediates amyloid beta toxicity and represents a potential target for Alzheimer disease therapy. J Biol Chem (2015) 290:30670–83. 10.1074/jbc.M115.691493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thinnes FP. Apoptogenic interactions of plasmalemmal type-1 VDAC and Abeta peptides via GxxxG motifs induce Alzheimer’s disease – a basic model of apoptosis? Wien Med Wochenschr (2011) 161:274–6. 10.1007/s10354-011-0887-5 [DOI] [PubMed] [Google Scholar]
- 47.Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, et al. Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci U S A (2008) 105:15370–5. 10.1073/pnas.0808115105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science (2008) 321:1206–10. 10.1126/science.1161302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, et al. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci U S A (2008) 105:17742–7. 10.1073/pnas.0809634105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Colombini M. The published 3D structure of the VDAC channel: native or not? Trends Biochem Sci (2009) 34:382–9. 10.1016/j.tibs.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 51.Geula S, Ben-Hail D, Shoshan-Barmatz V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem J (2012) 444:475–85. 10.1042/BJ20112079 [DOI] [PubMed] [Google Scholar]
- 52.Mannella CA. Minireview: on the structure and gating mechanism of the mitochondrial channel, VDAC. J Bioenerg Biomembr (1997) 29:525–31. 10.1023/A:1022489832594 [DOI] [PubMed] [Google Scholar]
- 53.Abu-Hamad S, Sivan S, Shoshan-Barmatz V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci U S A (2006) 103:5787–92. 10.1073/pnas.0600103103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Junankar PR, Dulhunty AF, Curtis SM, Pace SM, Thinnes FP. Porin-type 1 proteins in sarcoplasmic reticulum and plasmalemma of striated muscle fibres. J Muscle Res Cell Motil (1995) 16:595–610. 10.1007/BF00130241 [DOI] [PubMed] [Google Scholar]
- 55.Shoshan-Barmatz V, Gincel D. The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem Biophys (2003) 39:279–92. 10.1385/CBB:39:3:279 [DOI] [PubMed] [Google Scholar]
- 56.Shoshan-Barmatz V, Mizrachi D, Keinan N. Oligomerization of the mitochondrial protein VDAC1: from structure to function and cancer therapy. Prog Mol Biol Transl Sci (2013) 117:303–34. 10.1016/B978-0-12-386931-9.00011-8 [DOI] [PubMed] [Google Scholar]
- 57.De Pinto V, Reina S, Guarino F, Messina A. Structure of the voltage dependent anion channel: state of the art. J Bioenerg Biomembr (2008) 40:139–47. 10.1007/s10863-008-9140-3 [DOI] [PubMed] [Google Scholar]
- 58.Zachariae U, Schneider R, Briones R, Gattin Z, Demers JP, Giller K, et al. Beta-barrel mobility underlies closure of the voltage-dependent anion channel. Structure (2012) 20:1540–9. 10.1016/j.str.2012.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shuvo SR, Ferens FG, Court DA. The N-terminus of VDAC: structure, mutational analysis, and a potential role in regulating barrel shape. Biochim Biophys Acta (2016) 1858:1350–61. 10.1016/j.bbamem.2016.03.017 [DOI] [PubMed] [Google Scholar]
- 60.Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol (2010) 30:5698–709. 10.1128/MCB.00165-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shoshan-Barmatz V, Mizrachi D. VDAC1: from structure to cancer therapy. Front Oncol (2012) 2:164. 10.3389/fonc.2012.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zalk R, Israelson A, Garty E, Azoulay-Zohar H, Shoshan-Barmatz V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem J (2005) 386:73–83. 10.1042/BJ20041356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ujwal R, Cascio D, Chaptal V, Ping P, Abramson J. Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels (Austin) (2009) 3:167–70. 10.4161/chan.3.3.9196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schredelseker J, Paz A, Lopez CJ, Altenbach C, Leung CS, Drexler MK, et al. High resolution structure and double electron-electron resonance of the zebrafish voltage-dependent anion channel 2 reveal an oligomeric population. J Biol Chem (2014) 289:12566–77. 10.1074/jbc.M113.497438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ben-Hail D, Shoshan-Barmatz V. Purification of VDAC1 from rat liver mitochondria. Cold Spring Harb Protoc (2014) 2014:94–9. 10.1101/pdb.prot073130 [DOI] [PubMed] [Google Scholar]
- 66.Gincel D, Silberberg SD, Shoshan-Barmatz V. Modulation of the voltage-dependent anion channel (VDAC) by glutamate1. J Bioenerg Biomembr (2000) 32:571–83. 10.1023/A:1005670527340 [DOI] [PubMed] [Google Scholar]
- 67.Rostovtseva T, Colombini M. VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophys J (1997) 72:1954–62. 10.1016/S0006-3495(97)78841-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colombini M. VDAC structure, selectivity, and dynamics. Biochim Biophys Acta (2012) 1818:1457–65. 10.1016/j.bbamem.2011.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manna B, Bhattacharya T, Kahali B, Ghosh TC. Evolutionary constraints on hub and non-hub proteins in human protein interaction network: insight from protein connectivity and intrinsic disorder. Gene (2009) 434:50–5. 10.1016/j.gene.2008.12.013 [DOI] [PubMed] [Google Scholar]
- 70.Prezma T, Shteinfer A, Admoni L, Raviv Z, Sela I, Levi I, et al. VDAC1-based peptides: novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis (2013) 4:e809. 10.1038/cddis.2013.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shteinfer A, Arif A, Krelin Y, SharanTripathi S, Paul A, Shoshan-Barmatz V. Mitochondrial VDAC1-based peptides: attacking glioblastoma oncogenic properties. Oncotarget (2017) 8:31329–46. 10.18632/oncotarget.15455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palmieri F, Pierri CL. Mitochondrial metabolite transport. Essays Biochem (2010) 47:37–52. 10.1042/bse0470037 [DOI] [PubMed] [Google Scholar]
- 73.Aram L, Geula S, Arbel N, Shoshan-Barmatz V. VDAC1 cysteine residues: topology and function in channel activity and apoptosis. Biochem J (2010) 427:445–54. 10.1042/BJ20091690 [DOI] [PubMed] [Google Scholar]
- 74.Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc Natl Acad Sci U S A (2000) 97:4666–71. 10.1073/pnas.090082297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arif T, Vasilkovsky L, Refaely Y, Konson A, Shoshan-Barmatz V. Silencing VDAC1 expression by siRNA inhibits cancer cell proliferation and tumor growth in vivo. Mol Ther Nucleic Acids (2014) 3:e159. 10.1038/mtna.2014.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dolder M, Wendt S, Wallimann T. Mitochondrial creatine kinase in contact sites: interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol Signals Recept (2001) 10:93–111. 10.1159/000046878 [DOI] [PubMed] [Google Scholar]
- 77.Gurnev PA, Rostovtseva TK, Bezrukov SM. Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett (2011) 585:2363–6. 10.1016/j.febslet.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rone MB, Fan J, Papadopoulos V. Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim Biophys Acta (2009) 1791:646–58. 10.1016/j.bbalip.2009.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aghazadeh Y, Rone MB, Blonder J, Ye X, Veenstra TD, Hales DB, et al. Hormone-induced 14-3-3gamma adaptor protein regulates steroidogenic acute regulatory protein activity and steroid biosynthesis in MA-10 Leydig cells. J Biol Chem (2012) 287:15380–94. 10.1074/jbc.M112.339580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, Michikawa M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J Biol Chem (2005) 280:11731–9. 10.1074/jbc.M412898200 [DOI] [PubMed] [Google Scholar]
- 81.Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr (2008) 40:171–82. 10.1007/s10863-008-9148-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thinnes FP, Burckhardt G. On a fully closed state of native human type-1 VDAC enriched in Nonidet P40. Mol Genet Metab (2012) 107:632–3. 10.1016/j.ymgme.2012.08.015 [DOI] [PubMed] [Google Scholar]
- 83.Campbell AM, Chan SH. Mitochondrial membrane cholesterol, the voltage dependent anion channel (VDAC), and the Warburg effect. J Bioenerg Biomembr (2008) 40:193–7. 10.1007/s10863-008-9138-x [DOI] [PubMed] [Google Scholar]
- 84.Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem (2011) 286:25655–62. 10.1074/jbc.M111.228692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qiu J, Tan Y-W, Hagenston AM, Martel M-A, Kneisel N, Skehel PA, et al. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun (2013) 4:2034. 10.1038/ncomms3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta (2009) 1787:1309–16. 10.1016/j.bbabio.2009.01.005 [DOI] [PubMed] [Google Scholar]
- 87.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A (2010) 107:436–41. 10.1073/pnas.0908099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol (2013) 59:205–13. 10.1016/j.yjmcc.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J (2001) 358:147–55. 10.1042/bj3580147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol (2002) 159:613–24. 10.1083/jcb.200205091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta (2007) 1768:2510–5. 10.1016/j.bbamem.2007.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem (2006) 281:17347–58. 10.1074/jbc.M600906200 [DOI] [PubMed] [Google Scholar]
- 95.Israelson A, Abu-Hamad S, Zaid H, Nahon E, Shoshan-Barmatz V. Localization of the voltage-dependent anion channel-1 Ca2+-binding sites. Cell Calcium (2007) 41:235–44. 10.1016/j.ceca.2006.06.005 [DOI] [PubMed] [Google Scholar]
- 96.Huang H, Hu X, Eno CO, Zhao G, Li C, White C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol Chem (2013) 288:19870–81. 10.1074/jbc.M112.448290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Monaco G, Decrock E, Arbel N, van Vliet AR, La Rovere RM, De Smedt H, et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem (2015) 290:9150–61. 10.1074/jbc.M114.622514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ (2012) 19:267–73. 10.1038/cdd.2011.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ben-Hail D, Shoshan-Barmatz V. VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim Biophys Acta (2016) 1863:1612–23. 10.1016/j.bbamcr.2016.04.002 [DOI] [PubMed] [Google Scholar]
- 100.Chen H, Gao W, Yang Y, Guo S, Wang H, Wang W, et al. Inhibition of VDAC1 prevents Ca(2)(+)-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages. Apoptosis (2014) 19:1712–26. 10.1007/s10495-014-1045-5 [DOI] [PubMed] [Google Scholar]
- 101.Manda G, Nechifor MT, Neagu TM. Reactive oxygen species, cancer and anti-cancer therapies. Curr Chem Biol (2009) 3:342–66. 10.2174/187231309787158271 [DOI] [Google Scholar]
- 102.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell (2006) 10:241–52. 10.1016/j.ccr.2006.08.009 [DOI] [PubMed] [Google Scholar]
- 103.Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal (2012) 16:1323–67. 10.1089/ars.2011.4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem (2003) 278:5557–63. 10.1074/jbc.M210269200 [DOI] [PubMed] [Google Scholar]
- 105.Sun L, Shukair S, Naik TJ, Moazed F, Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol Cell Biol (2008) 28:1007–17. 10.1128/MCB.00224-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ahmad A, Ahmad S, Schneider BK, Allen CB, Chang LY, White CW. Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am J Physiol Lung Cell Mol Physiol (2002) 283:L573–84. 10.1152/ajplung.00410.2001 [DOI] [PubMed] [Google Scholar]
- 107.Wenger JB, Chun SY, Dang DT, Luesch H, Dang LH. Combination therapy targeting cancer metabolism. Med Hypotheses (2011) 76:169–72. 10.1016/j.mehy.2010.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. 10.1038/nrc2981 [DOI] [PubMed] [Google Scholar]
- 109.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell (2008) 134:703–7. 10.1016/j.cell.2008.08.021 [DOI] [PubMed] [Google Scholar]
- 110.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev (2009) 23:537–48. 10.1101/gad.1756509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond) (2010) 7:7. 10.1186/1743-7075-7-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seyfried TN. Cancer as a mitochondrial metabolic disease. Front Cell Dev Biol (2015) 3:43. 10.3389/fcell.2015.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Loeb LA. A mutator phenotype in cancer. Cancer Res (2001) 61:3230–9. [PubMed] [Google Scholar]
- 114.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. 10.1038/nrc1478 [DOI] [PubMed] [Google Scholar]
- 115.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 116.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. 10.1038/nrc3038 [DOI] [PubMed] [Google Scholar]
- 117.Majeed R, Hamid A, Qurishi Y, Qazi AK, Hussain A, Ahmed M, et al. Therapeutic targeting of cancer cell metabolism: role of metabolic enzymes, oncogenes and tumor suppressor genes. J Cancer Sci Ther (2012) 4:281–91. 10.4172/1948-5956.1000156 [DOI] [Google Scholar]
- 118.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta (2002) 1555:14–20. 10.1016/S0005-2728(02)00248-7 [DOI] [PubMed] [Google Scholar]
- 119.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol (2004) 24:730–40. 10.1128/MCB.24.2.730-740.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maldonado EN, Lemasters JJ. Warburg revisited: regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J Pharmacol Exp Ther (2012) 342:637–41. 10.1124/jpet.112.192153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer (2016) 16:635–49. 10.1038/nrc.2016.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rahman A, Sultan T, Islam R. Apoptosis and cancer: insights molecular mechanisms and treatments. Int J Biomol Biomed (2012) 2:1–16. [Google Scholar]
- 123.Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta (2011) 1813:558–63. 10.1016/j.bbamcr.2011.01.026 [DOI] [PubMed] [Google Scholar]
- 124.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev (2007) 87:99–163. 10.1152/physrev.00013.2006 [DOI] [PubMed] [Google Scholar]
- 125.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer (2002) 2:647–56. 10.1038/nrc883 [DOI] [PubMed] [Google Scholar]
- 126.Altieri DC. Survivin and IAP proteins in cell-death mechanisms. Biochem J (2010) 430:199–205. 10.1042/BJ20100814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res (2000) 256:50–7. 10.1006/excr.2000.4839 [DOI] [PubMed] [Google Scholar]
- 128.Reed JC. Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin Hematol (1997) 34:9–19. [PubMed] [Google Scholar]
- 129.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev (2011) 21:12–20. 10.1016/j.gde.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Galluzzi L, Morselli E, Kepp O, Vitale I, Pinti M, Kroemer G. Mitochondrial liaisons of p53. Antioxid Redox Signal (2011) 15:1691–714. 10.1089/ars.2010.3504 [DOI] [PubMed] [Google Scholar]
- 131.Wolff S, Erster S, Palacios G, Moll UM. p53’s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res (2008) 18:733–44. 10.1038/cr.2008.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bryson JM, Coy PE, Gottlob K, Hay N, Robey RB. Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J Biol Chem (2002) 277:11392–400. 10.1074/jbc.M110927200 [DOI] [PubMed] [Google Scholar]
- 133.Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ (2005) 12:751–60. 10.1038/sj.cdd.4401599 [DOI] [PubMed] [Google Scholar]
- 134.Granville DJ, Gottlieb RA. The mitochondrial voltage-dependent anion channel (VDAC) as a therapeutic target for initiating cell death. Curr Med Chem (2003) 10:1527–33. 10.2174/0929867033457214 [DOI] [PubMed] [Google Scholar]
- 135.Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol (2001) 155:1003–15. 10.1083/jcb.200105057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shimizu S, Matsuoka Y, Shinohara Y, Yoneda Y, Tsujimoto Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J Cell Biol (2001) 152:237–50. 10.1083/jcb.152.2.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, et al. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene (2004) 23:1239–47. 10.1038/sj.onc.1207205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Israelson A, Zaid H, Abu-Hamad S, Nahon E, Shoshan-Barmatz V. Mapping the ruthenium red-binding site of the voltage-dependent anion channel-1. Cell Calcium (2008) 43:196–204. 10.1016/j.ceca.2007.05.006 [DOI] [PubMed] [Google Scholar]
- 139.Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene (2008) 27:4221–32. 10.1038/onc.2008.63 [DOI] [PubMed] [Google Scholar]
- 140.Yuan S, Fu Y, Wang X, Shi H, Huang Y, Song X, et al. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J (2008) 22:2809–20. 10.1096/fj.08-107417 [DOI] [PubMed] [Google Scholar]
- 141.Ghosh T, Pandey N, Maitra A, Brahmachari SK, Pillai B. A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS One (2007) 2:e1170. 10.1371/journal.pone.0001170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shimizu S, Shinohara Y, Tsujimoto Y. Bax and Bcl-xL independently regulate apoptotic changes of yeast mitochondria that require VDAC but not adenine nucleotide translocator. Oncogene (2000) 19:4309–18. 10.1038/sj.onc.1203788 [DOI] [PubMed] [Google Scholar]
- 143.Ben-Hail D, Begas-Shvartz R, Shalev M, Shteinfer-Kuzmine A, Gruzman A, Reina S, et al. Novel compaunds targeting the mitochondrial protein VDAC1 inhibit apoptosis and protect against mitochondrial dysfunction. J Biol Chem (2016) 291:24986–5003. 10.1074/jbc.M116.744284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang L, Han J, Ben-Hail D, He L, Li B, Chen Z, et al. Diterpene induces VDAC1-dependent apoptosis in Bax/Bak-deficient cells. J Biol Chem (2015) 290:23563–78. 10.1074/jbc.M115.648774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shoshan-Barmatz V, Arbel N, Arzoine L. VDAC, the voltage-dependent anion channel: function, regulation & mitochondrial signaling in cell life and death. Cell Sci (2008) 4:74–118. [Google Scholar]
- 146.Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis (2007) 12:835–40. 10.1007/s10495-006-0525-7 [DOI] [PubMed] [Google Scholar]
- 147.Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta (2011) 1813:616–22. 10.1016/j.bbamcr.2010.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature (2004) 427:461–5. 10.1038/nature02229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature (2005) 434:658–62. 10.1038/nature03434 [DOI] [PubMed] [Google Scholar]
- 150.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol (2007) 9:550–5. 10.1038/ncb1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bernardi P, Di Lisa F, Fogolari F, Lippe G. From ATP to PTP and back: a dual function for the mitochondrial ATP synthase. Circ Res (2015) 116:1850–62. 10.1161/CIRCRESAHA.115.306557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bleicken S, Classen M, Padmavathi PV, Ishikawa T, Zeth K, Steinhoff HJ, et al. Molecular details of Bax activation, oligomerization, and membrane insertion. J Biol Chem (2010) 285:6636–47. 10.1074/jbc.M109.081539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang Z, Zhu W, Lapolla SM, Miao Y, Shao Y, Falcone M, et al. Bax forms an oligomer via separate, yet interdependent, surfaces. J Biol Chem (2010) 285:17614–27. 10.1074/jbc.M110.113456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem (2001) 276:11615–23. 10.1074/jbc.M010810200 [DOI] [PubMed] [Google Scholar]
- 155.Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci (2009) 122:2801–8. 10.1242/jcs.038166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Antignani A, Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr Opin Cell Biol (2006) 18:685–9. 10.1016/j.ceb.2006.10.004 [DOI] [PubMed] [Google Scholar]
- 157.Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell (2008) 135:1074–84. 10.1016/j.cell.2008.11.010 [DOI] [PubMed] [Google Scholar]
- 158.Banerjee J, Ghosh S. Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem Biophys Res Commun (2004) 323:310–4. 10.1016/j.bbrc.2004.08.094 [DOI] [PubMed] [Google Scholar]
- 159.Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis (2007) 12:857–68. 10.1007/s10495-007-0722-z [DOI] [PubMed] [Google Scholar]
- 160.Peixoto PM, Lue JK, Ryu SY, Wroble BN, Sible JC, Kinnally KW. Mitochondrial apoptosis-induced channel (MAC) function triggers a Bax/Bak-dependent bystander effect. Am J Pathol (2011) 178:48–54. 10.1016/j.ajpath.2010.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Weisthal S, Keinan N, Ben-Hail D, Arif T, Shoshan-Barmatz V. Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim Biophys Acta (2014) 1843:2270–81. 10.1016/j.bbamcr.2014.03.021 [DOI] [PubMed] [Google Scholar]
- 162.Keinan N, Pahima H, Ben-Hail D, Shoshan-Barmatz V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim Biophys Acta (2013) 1833:1745–54. 10.1016/j.bbamcr.2013.03.017 [DOI] [PubMed] [Google Scholar]
- 163.Gogvadze V, Orrenius S, Zhivotovsky B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim Biophys Acta (2006) 1757:639–47. 10.1016/j.bbabio.2006.03.016 [DOI] [PubMed] [Google Scholar]
- 164.Zeth K, Meins T, Vonrhein C. Approaching the structure of human VDAC1, a key molecule in mitochondrial cross-talk. J Bioenerg Biomembr (2008) 40:127–32. 10.1007/s10863-008-9144-z [DOI] [PubMed] [Google Scholar]
- 165.Shoshan-Barmatz V, Keinan N, Zaid H. Uncovering the role of VDAC in the regulation of cell life and death. J Bioenerg Biomembr (2008) 40:183–91. 10.1007/s10863-008-9147-9 [DOI] [PubMed] [Google Scholar]
- 166.Hoogenboom BW, Suda K, Engel A, Fotiadis D. The supramolecular assemblies of voltage-dependent anion channels in the native membrane. J Mol Biol (2007) 370:246–55. 10.1016/j.jmb.2007.04.073 [DOI] [PubMed] [Google Scholar]
- 167.Betaneli V, Petrov EP, Schwille P. The role of lipids in VDAC oligomerization. Biophys J (2012) 102:523–31. 10.1016/j.bpj.2011.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shoshan-Barmatz V, Keinan N, Abu-Hamad S, Tyomkin D, Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim Biophys Acta (2010) 1797:1281–91. 10.1016/j.bbabio.2010.03.003 [DOI] [PubMed] [Google Scholar]
- 169.De Pinto V, Messina A, Accardi R, Aiello R, Guarino F, Tomasello MF, et al. New functions of an old protein: the eukaryotic porin or voltage dependent anion selective channel (VDAC). Ital J Biochem (2003) 52:17–24. [PubMed] [Google Scholar]
- 170.Geula S, Naveed H, Liang J, Shoshan-Barmatz V. Structure-based analysis of VDAC1 protein: defining oligomer contact sites. J Biol Chem (2012) 287:2179–90. 10.1074/jbc.M111.268920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Godbole A, Varghese J, Sarin A, Mathew MK. VDAC is a conserved element of death pathways in plant and animal systems. Biochim Biophys Acta (2003) 1642:87–96. 10.1016/S0167-4889(03)00102-2 [DOI] [PubMed] [Google Scholar]
- 172.Lu AJ, Dong CW, Du CS, Zhang QY. Characterization and expression analysis of Paralichthys olivaceus voltage-dependent anion channel (VDAC) gene in response to virus infection. Fish Shellfish Immunol (2007) 23:601–13. 10.1016/j.fsi.2007.01.007 [DOI] [PubMed] [Google Scholar]
- 173.Chen Y, Craigen WJ, Riley DJ. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle (2009) 8:257–67. 10.4161/cc.8.2.7551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jung JY, Han CR, Jeong YJ, Kim HJ, Lim HS, Lee KH, et al. Epigallocatechin gallate inhibits nitric oxide-induced apoptosis in rat PC12 cells. Neurosci Lett (2007) 411:222–7. 10.1016/j.neulet.2006.09.089 [DOI] [PubMed] [Google Scholar]
- 175.Sharaf el dein O, Gallerne C, Brenner C, Lemaire C. Increased expression of VDAC1 sensitizes carcinoma cells to apoptosis induced by DNA cross-linking agents. Biochem Pharmacol (2012) 83:1172–82. 10.1016/j.bcp.2012.01.017 [DOI] [PubMed] [Google Scholar]
- 176.Nawarak J, Huang-Liu R, Kao SH, Liao HH, Sinchaikul S, Chen ST, et al. Proteomics analysis of A375 human malignant melanoma cells in response to arbutin treatment. Biochim Biophys Acta (2009) 1794:159–67. 10.1016/j.bbapap.2008.09.023 [DOI] [PubMed] [Google Scholar]
- 177.Liu Z, Bengtsson S, Krogh M, Marquez M, Nilsson S, James P, et al. Somatostatin effects on the proteome of the LNCaP cell-line. Int J Oncol (2007) 30:1173–9. 10.3892/ijo.30.5.1173 [DOI] [PubMed] [Google Scholar]
- 178.Jiang N, Kham SK, Koh GS, Suang Lim JY, Ariffin H, Chew FT, et al. Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). J Proteomics (2011) 74:843–57. 10.1016/j.jprot.2011.02.034 [DOI] [PubMed] [Google Scholar]
- 179.Castagna A, Antonioli P, Astner H, Hamdan M, Righetti SC, Perego P, et al. A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics (2004) 4:3246–67. 10.1002/pmic.200400835 [DOI] [PubMed] [Google Scholar]
- 180.Anis Y. Involvement of Ca2+ in the Apoptotic Process – Friends or Foes. Jerusalem: Pathways; (2006). p. 2–7. [Google Scholar]
- 181.Cheng SL, Liu RH, Sheu JN, Chen ST, Sinchaikul S, Tsay GJ. Toxicogenomics of A375 human malignant melanoma cells treated with arbutin. J Biomed Sci (2007) 14:87–105. 10.1007/s11373-006-9130-6 [DOI] [PubMed] [Google Scholar]
- 182.Sahasrabudhe SR, Lai S, Pierce M, Clemens C, Venkat R, Rebentisch M, et al. Selective in vitro and in vivo anti-tumor activity of PRLX 93936 in biological models of melanoma and ovarian cancer. American Society of Clinical Oncology. J Clin Oncol (2008) 15:14586. 10.1200/jco.2008.26.15_suppl.14586 [DOI] [Google Scholar]
- 183.Tan W, Loke YH, Stein CA, Miller P, Colombini M. Phosphorothioate oligonucleotides block the VDAC channel. Biophys J (2007) 93:1184–91. 10.1529/biophysj.107.105379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Haridas V, Li X, Mizumachi T, Higuchi M, Lemeshko VV, Colombini M, et al. Avicins, a novel plant-derived metabolite lowers energy metabolism in tumor cells by targeting the outer mitochondrial membrane. Mitochondrion (2007) 7:234–40. 10.1016/j.mito.2006.12.005 [DOI] [PubMed] [Google Scholar]
- 185.Lemeshko VV, Haridas V, Quijano Perez JC, Gutterman JU. Avicins, natural anticancer saponins, permeabilize mitochondrial membranes. Arch Biochem Biophys (2006) 454:114–22. 10.1016/j.abb.2006.08.008 [DOI] [PubMed] [Google Scholar]
- 186.Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res (2006) 12:5817–25. 10.1158/1078-0432.CCR-06-1037 [DOI] [PubMed] [Google Scholar]
- 187.Thinnes FP. Human type-1 VDAC, a cisplatin target involved in either apoptotic pathway. Mol Genet Metab (2009) 97:163. 10.1016/j.ymgme.2009.01.014 [DOI] [PubMed] [Google Scholar]
- 188.Raviv Z, Cohen S, Reischer-Pelech D. The anti-cancer activities of jasmonates. Cancer Chemother Pharmacol (2013) 71:275–85. 10.1007/s00280-012-2039-z [DOI] [PubMed] [Google Scholar]
- 189.Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, et al. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene (2008) 27:4636–43. 10.1038/onc.2008.108 [DOI] [PubMed] [Google Scholar]
- 190.Liu AH, Cao YN, Liu HT, Zhang WW, Liu Y, Shi TW, et al. DIDS attenuates staurosporine-induced cardiomyocyte apoptosis by PI3K/Akt signaling pathway: activation of eNOS/NO and inhibition of Bax translocation. Cell Physiol Biochem (2008) 22:177–86. 10.1159/000149795 [DOI] [PubMed] [Google Scholar]
- 191.Gincel D, Vardi N, Shoshan-Barmatz V. Retinal voltage-dependent anion channel: characterization and cellular localization. Invest Ophthalmol Vis Sci (2002) 43:2097–104. [PubMed] [Google Scholar]
- 192.Bae JH, Park JW, Kwon TK. Ruthenium red, inhibitor of mitochondrial Ca2+ uniporter, inhibits curcumin-induced apoptosis via the prevention of intracellular Ca2+ depletion and cytochrome c release. Biochem Biophys Res Commun (2003) 303:1073–9. 10.1016/S0006-291X(03)00479-0 [DOI] [PubMed] [Google Scholar]
- 193.Obulesu M, Lakshmi MJ. Apoptosis in Alzheimer’s disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem Res (2014) 39:2301–12. 10.1007/s11064-014-1454-4 [DOI] [PubMed] [Google Scholar]
- 194.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res (2003) 92:715–24. 10.1161/01.RES.0000067471.95890.5C [DOI] [PubMed] [Google Scholar]
- 195.Marunouchi T, Tanonaka K. Cell death in the cardiac myocyte. Biol Pharm Bull (2015) 38:1094–7. 10.1248/bpb.b15-00288 [DOI] [PubMed] [Google Scholar]
- 196.Vyssokikh MY, Brdiczka D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim Pol (2003) 50:389–404. [PubMed] [Google Scholar]
- 197.Schlattner U, Dolder M, Wallimann T, Tokarska-Schlattner M. Mitochondrial creatine kinase and mitochondrial outer membrane porin show a direct interaction that is modulated by calcium. J Biol Chem (2001) 276:48027–30. 10.1074/jbc.M106524200 [DOI] [PubMed] [Google Scholar]
- 198.Adams V, Griffin L, Towbin J, Gelb B, Worley K, McCabe ER. Porin interaction with hexokinase and glycerol kinase: metabolic microcompartmentation at the outer mitochondrial membrane. Biochem Med Metab Biol (1991) 45:271–91. 10.1016/0885-4505(91)90032-G [DOI] [PubMed] [Google Scholar]
- 199.Le Mellay V, Troppmair J, Benz R, Rapp UR. Negative regulation of mitochondrial VDAC channels by C-Raf kinase. BMC Cell Biol (2002) 3:14. 10.1186/1471-2121-3-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, et al. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene (2007) 26:2606–20. 10.1038/sj.onc.1210074 [DOI] [PubMed] [Google Scholar]
- 201.Veenman L, Papadopoulos V, Gavish M. Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des (2007) 13:2385–405. 10.2174/138161207781368710 [DOI] [PubMed] [Google Scholar]
- 202.Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, et al. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci U S A (2008) 105:18746–51. 10.1073/pnas.0806303105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pedersen PL. Warburg, me and hexokinase 2: multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr (2007) 39:211–22. 10.1007/s10863-007-9094-x [DOI] [PubMed] [Google Scholar]
- 204.Brown RS, Goodman TM, Zasadny KR, Greenson JK, Wahl RL. Expression of hexokinase II and Glut-1 in untreated human breast cancer. Nucl Med Biol (2002) 29:443–53. 10.1016/S0969-8051(02)00288-3 [DOI] [PubMed] [Google Scholar]
- 205.Katabi MM, Chan HL, Karp SE, Batist G. Hexokinase type II: a novel tumor-specific promoter for gene-targeted therapy differentially expressed and regulated in human cancer cells. Hum Gene Ther (1999) 10:155–64. 10.1089/10430349950018959 [DOI] [PubMed] [Google Scholar]
- 206.Mathupala SP, Ko YH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta (2010) 1797:1225–30. 10.1016/j.bbabio.2010.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol (2003) 206:2049–57. 10.1242/jeb.00241 [DOI] [PubMed] [Google Scholar]
- 208.Neumann D, Buckers J, Kastrup L, Hell SW, Jakobs S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys (2010) 3:4. 10.1186/1757-5036-3-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem (2002) 277:7610–8. 10.1074/jbc.M109950200 [DOI] [PubMed] [Google Scholar]
- 210.Shoshan-Barmatz V, Zakar M, Rosenthal K, Abu-Hamad S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim Biophys Acta (2009) 1787:421–30. 10.1016/j.bbabio.2008.11.009 [DOI] [PubMed] [Google Scholar]
- 211.Schindler A, Foley E. Hexokinase 1 blocks apoptotic signals at the mitochondria. Cell Signal (2013) 25:2685–92. 10.1016/j.cellsig.2013.08.035 [DOI] [PubMed] [Google Scholar]
- 212.Pedersen PL. Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr (2008) 40:123–6. 10.1007/s10863-008-9165-7 [DOI] [PubMed] [Google Scholar]
- 213.Zhang T, Tang SS, Jin X, Liu FY, Zhang CM, Zhao WX, et al. c-Myc influences olaquindox-induced apoptosis in human hepatoma G2 cells. Mol Cell Biochem (2011) 354:253–61. 10.1007/s11010-011-0825-2 [DOI] [PubMed] [Google Scholar]
- 214.da-Silva WS, Gomez-Puyou A, de Gomez-Puyou MT, Moreno-Sanchez R, De Felice FG, de Meis L, et al. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J Biol Chem (2004) 279:39846–55. 10.1074/jbc.M403835200 [DOI] [PubMed] [Google Scholar]
- 215.Krasnov GS, Dmitriev AA, Lakunina VA, Kirpiy AA, Kudryavtseva AV. Targeting VDAC-bound hexokinase II: a promising approach for concomitant anti-cancer therapy. Expert Opin Ther Targets (2013) 17:1221–33. 10.1517/14728222.2013.833607 [DOI] [PubMed] [Google Scholar]
- 216.Penso J, Beitner R. Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur J Pharmacol (1998) 342:113–7. 10.1016/S0014-2999(97)01507-0 [DOI] [PubMed] [Google Scholar]
- 217.Ganapathy-Kanniappan S. Chapter 15: Targeting glycolytic adaptations of cancer cells: from molecular mechanisms to therapeutic opportunities. In: Wondrak GT, editor. Stress Response Pathways in Cancer. Dordrecht: Springer; (2015). p. 331–44. [Google Scholar]
- 218.Gelb BD, Adams V, Jones SN, Griffin LD, MacGregor GR, McCabe ER. Targeting of hexokinase 1 to liver and hepatoma mitochondria. Proc Natl Acad Sci U S A (1992) 89:202–6. 10.1073/pnas.89.1.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell (2006) 9:351–65. 10.1016/j.ccr.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 220.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene (2007) 26:1324–37. 10.1038/sj.onc.1210220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood (1993) 81:151–7. [PubMed] [Google Scholar]
- 222.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol (2008) 9:47–59. 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 223.Huang H, Shah K, Bradbury NA, Li C, White C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis (2014) 5:e1482. 10.1038/cddis.2014.419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A (2000) 97:3100–5. 10.1073/pnas.97.7.3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sugiyama T, Shimizu S, Matsuoka Y, Yoneda Y, Tsujimoto Y. Activation of mitochondrial voltage-dependent anion channel by a pro-apoptotic BH3-only protein Bim. Oncogene (2002) 21:4944–56. 10.1038/sj.onc.1205621 [DOI] [PubMed] [Google Scholar]
- 226.Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol (2007) 9:487–9. 10.1038/ncb0507-487 [DOI] [PubMed] [Google Scholar]
- 227.Shi Y, Chen J, Weng C, Chen R, Zheng Y, Chen Q, et al. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem Biophys Res Commun (2003) 305:989–96. 10.1016/S0006-291X(03)00871-4 [DOI] [PubMed] [Google Scholar]
- 228.Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM. Bid, but not Bax, regulates VDAC channels. J Biol Chem (2004) 279:13575–83. 10.1074/jbc.M310593200 [DOI] [PubMed] [Google Scholar]
- 229.McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci U S A (1992) 89:3170–4. 10.1073/pnas.89.8.3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Golani I, Weizman A, Leschiner S, Spanier I, Eckstein N, Limor R, et al. Hormonal regulation of peripheral benzodiazepine receptor binding properties is mediated by subunit interaction. Biochemistry (2001) 40:10213–22. 10.1021/bi010431+ [DOI] [PubMed] [Google Scholar]
- 231.Joo HK, Lee YR, Lim SY, Lee EJ, Choi S, Cho EJ, et al. Peripheral benzodiazepine receptor regulates vascular endothelial activations via suppression of the voltage-dependent anion channel-1. FEBS Lett (2012) 586:1349–55. 10.1016/j.febslet.2012.03.049 [DOI] [PubMed] [Google Scholar]
- 232.Kusano H, Shimizu S, Koya RC, Fujita H, Kamada S, Kuzumaki N, et al. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene (2000) 19:4807–14. 10.1038/sj.onc.1203868 [DOI] [PubMed] [Google Scholar]
- 233.Carre M, Andre N, Carles G, Borghi H, Brichese L, Briand C, et al. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem (2002) 277:33664–9. 10.1074/jbc.M203834200 [DOI] [PubMed] [Google Scholar]
- 234.Rostovtseva TK, Bezrukov SM. VDAC inhibition by tubulin and its physiological implications. Biochim Biophys Acta (2012) 1818:1526–35. 10.1016/j.bbamem.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Saks V, Guzun R, Timohhina N, Tepp K, Varikmaa M, Monge C, et al. Structure-function relationships in feedback regulation of energy fluxes in vivo in health and disease: mitochondrial interactosome. Biochim Biophys Acta (2010) 1797:678–97. 10.1016/j.bbabio.2010.01.011 [DOI] [PubMed] [Google Scholar]
- 236.Linden M, Karlsson G. Identification of porin as a binding site for MAP2. Biochem Biophys Res Commun (1996) 218:833–6. 10.1006/bbrc.1996.0148 [DOI] [PubMed] [Google Scholar]
- 237.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell (2005) 19:727–40. 10.1016/j.molcel.2005.08.014 [DOI] [PubMed] [Google Scholar]
- 238.Guan K, Zheng Z, Song T, He X, Xu C, Zhang Y, et al. MAVS regulates apoptotic cell death by decreasing K48-linked ubiquitination of voltage-dependent anion channel 1. Mol Cell Biol (2013) 33:3137–49. 10.1128/MCB.00030-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron (2010) 67:575–87. 10.1016/j.neuron.2010.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Tan W, Naniche N, Bogush A, Pedrini S, Trotti D, Pasinelli P. Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS. J Neurosci (2013) 33:11588–98. 10.1523/JNEUROSCI.5385-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sun J, Liao JK. Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc Natl Acad Sci U S A (2002) 99:13108–13. 10.1073/pnas.202260999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Schwarzer C, Barnikol-Watanabe S, Thinnes FP, Hilschmann N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int J Biochem Cell Biol (2002) 34:1059–70. 10.1016/S1357-2725(02)00026-2 [DOI] [PubMed] [Google Scholar]
- 243.Nakatsuka A, Wada J, Iseda I, Teshigawara S, Higashio K, Murakami K, et al. Visceral adipose tissue-derived serine proteinase inhibitor inhibits apoptosis of endothelial cells as a ligand for the cell-surface GRP78/voltage-dependent anion channel complex. Circ Res (2013) 112:771–80. 10.1161/CIRCRESAHA.111.300049 [DOI] [PubMed] [Google Scholar]
- 244.Xu X, Forbes JG, Colombini M. Actin modulates the gating of Neurospora crassa VDAC. J Membr Biol (2001) 180:73–81. 10.1007/s002320010060 [DOI] [PubMed] [Google Scholar]
- 245.Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S. Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis (2013) 4:e582. 10.1038/cddis.2013.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lu L, Zhang C, Cai Q, Lu Q, Duan C, Zhu Y, et al. Voltage-dependent anion channel involved in the alpha-synuclein-induced dopaminergic neuron toxicity in rats. Acta Biochim Biophys Sin (Shanghai) (2013) 45:170–8. 10.1093/abbs/gms114 [DOI] [PubMed] [Google Scholar]
- 247.Zhang X, Bian X, Kong J. The proapoptotic protein BNIP3 interacts with VDAC to induce mitochondrial release of endonuclease G. PLoS One (2014) 9:e113642. 10.1371/journal.pone.0113642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Viola HM, Adams AM, Davies SM, Fletcher S, Filipovska A, Hool LC. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc Natl Acad Sci U S A (2014) 111:E2905–14. 10.1073/pnas.1402544111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ko JH, Gu W, Lim I, Zhou T, Bang H. Expression profiling of mitochondrial voltage-dependent anion channel-1 associated genes predicts recurrence-free survival in human carcinomas. PLoS One (2014) 9:e110094. 10.1371/journal.pone.0110094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Simamura E, Shimada H, Ishigaki Y, Hatta T, Higashi N, Hirai K. Bioreductive activation of quinone antitumor drugs by mitochondrial voltage-dependent anion channel 1. Anat Sci Int (2008) 83:261–6. 10.1111/j.1447-073X.2008.00241.x [DOI] [PubMed] [Google Scholar]
- 251.Yoo HJ, Yun BR, Kwon JH, Ahn HS, Seol MA, Lee MJ, et al. Genetic and expression alterations in association with the sarcomatous change of cholangiocarcinoma cells. Exp Mol Med (2009) 41:102–15. 10.3858/emm.2009.41.2.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Bai Z, Ye Y, Liang B, Xu F, Zhang H, Zhang Y, et al. Proteomics-based identification of a group of apoptosis-related proteins and biomarkers in gastric cancer. Int J Oncol (2011) 38:375–83. 10.3892/ijo.2010.873 [DOI] [PubMed] [Google Scholar]
- 253.Ge Y, Yan D, Deng H, Chen W, An G. Novel molecular regulators of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in NSCLC cells. Clin Lab (2015) 61:1855–63. 10.7754/Clin.Lab.2015.150328 [DOI] [PubMed] [Google Scholar]
- 254.Lai JC, Tan W, Benimetskaya L, Miller P, Colombini M, Stein CA. A pharmacologic target of G3139 in melanoma cells may be the mitochondrial VDAC. Proc Natl Acad Sci U S A (2006) 103:7494–9. 10.1073/pnas.0602217103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Liu S, Ishikawa H, Tsuyama N, Li FJ, Abroun S, Otsuyama KI, et al. Increased susceptibility to apoptosis in CD45(+) myeloma cells accompanied by the increased expression of VDAC1. Oncogene (2006) 25:419–29. 10.1038/sj.onc.1208982 [DOI] [PubMed] [Google Scholar]
- 256.Ko JH, Gu W, Lim I, Bang H, Ko EA, Zhou T. Ion channel gene expression in lung adenocarcinoma: potential role in prognosis and diagnosis. PLoS One (2014) 9:e86569. 10.1371/journal.pone.0086569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Admoni-Elisha L, Nakdimon I, Shteinfer A, Prezma T, Arif T, Arbel N, et al. Novel biomarker proteins in chronic lymphocytic leukemia: impact on diagnosis, prognosis and treatment. PLoS One (2016) 11:e0148500. 10.1371/journal.pone.0148500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wu CH, Lin YW, Wu TF, Ko JL, Wang PH. Clinical implication of voltage-dependent anion channel 1 in uterine cervical cancer and its action on cervical cancer cells. Oncotarget (2016) 7:4210–25. 10.18632/oncotarget.6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Alfonso P, Canamero M, Fernandez-Carbonie F, Nunez A, Casal JI. Proteome analysis of membrane fractions in colorectal carcinomas by using 2D-DIGE saturation labeling. J Proteome Res (2008) 7:4247–55. 10.1021/pr800152u [DOI] [PubMed] [Google Scholar]
- 260.Gao W, Xua J, Wang F, Zhang L, Peng R, Zhu Y, et al. Mitochondrial proteomics approach reveals voltage-dependent anion channel 1 (VDAC1) as a potential biomarker of gastric cancer. Cell Physiol Biochem (2015) 37:2339–54. 10.1159/000438588 [DOI] [PubMed] [Google Scholar]
- 261.Brahimi-Horn MC, Ben-Hail D, Ilie M, Gounon P, Rouleau M, Hofman V, et al. Expression of a truncated active form of VDAC1 in lung cancer associates with hypoxic cell survival and correlates with progression to chemotherapy resistance. Cancer Res (2012) 72:2140–50. 10.1158/0008-5472.CAN-11-3940 [DOI] [PubMed] [Google Scholar]
- 262.Ko JH, Ko EA, Gu W, Lim I, Bang H, Zhou T. Expression profiling of ion channel genes predicts clinical outcome in breast cancer. Mol Cancer (2013) 12:106. 10.1186/1476-4598-12-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pernemalm M, De Petris L, Branca RM, Forshed J, Kanter L, Soria JC, et al. Quantitative proteomics profiling of primary lung adenocarcinoma tumors reveals functional perturbations in tumor metabolism. J Proteome Res (2013) 12:3934–43. 10.1021/pr4002096 [DOI] [PubMed] [Google Scholar]
- 264.Grills C, Jithesh PV, Blayney J, Zhang SD, Fennell DA. Gene expression meta-analysis identifies VDAC1 as a predictor of poor outcome in early stage non-small cell lung cancer. PLoS One (2011) 6:e14635. 10.1371/journal.pone.0014635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Simamura E, Shimada H, Hatta T, Hirai K. Mitochondrial voltage-dependent anion channels (VDACs) as novel pharmacological targets for anti-cancer agents. J Bioenerg Biomembr (2008) 40:213–7. 10.1007/s10863-008-9158-6 [DOI] [PubMed] [Google Scholar]
- 266.Koren I, Raviv Z, Shoshan-Barmatz V. Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol Ther (2010) 9:1046–52. 10.4161/cbt.9.12.11879 [DOI] [PubMed] [Google Scholar]
- 267.Chacko AD, Liberante F, Paul I, Longley DB, Fennell DA. Voltage dependent anion channel-1 regulates death receptor mediated apoptosis by enabling cleavage of caspase-8. BMC Cancer (2010) 10:380. 10.1186/1471-2407-10-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Dinnen RD, Mao Y, Qiu W, Cassai N, Slavkovich VN, Nichols G, et al. Redirecting apoptosis to aponecrosis induces selective cytotoxicity to pancreatic cancer cells through increased ROS, decline in ATP levels, and VDAC. Mol Cancer Ther (2013) 12:2792–803. 10.1158/1535-7163.MCT-13-0234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- 270.Arif T, Krelin Y, Nakdimon I, Benharroch D, Paul A, Dadon-Klein D, et al. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol (2017) 19:951–64. 10.1093/neuonc/now297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Arif T, Krelin Y, Shoshan-Barmatz V. Reducing VDAC1 expression induces a non-apoptotic role for pro-apoptotic proteins in cancer cell differentiation. Biochim Biophys Acta (2016) 1857:1228–42. 10.1016/j.bbabio.2016.04.005 [DOI] [PubMed] [Google Scholar]
- 272.Head SA, Shi W, Zhao L, Gorshkov K, Pasunooti K, Chen Y, et al. Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells. Proc Natl Acad Sci U S A (2015) 112:E7276–85. 10.1073/pnas.1512867112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Skalsky RL, Cullen BR. Reduced expression of brain-enriched microRNAs in glioblastomas permits targeted regulation of a cell death gene. PLoS One (2011) 6:e24248. 10.1371/journal.pone.0024248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Foekens JA, Sieuwerts AM, Smid M, Look MP, de Weerd V, Boersma AW, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci U S A (2008) 105:13021–6. 10.1073/pnas.0803304105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Veerla S, Lindgren D, Kvist A, Frigyesi A, Staaf J, Persson H, et al. miRNA expression in urothelial carcinomas: important roles of miR-10a, miR-222, miR-125b, miR-7 and miR-452 for tumor stage and metastasis, and frequent homozygous losses of miR-31. Int J Cancer (2009) 124:2236–42. 10.1002/ijc.24183 [DOI] [PubMed] [Google Scholar]
- 276.Kong D, Piao YS, Yamashita S, Oshima H, Oguma K, Fushida S, et al. Inflammation-induced repression of tumor suppressor miR-7 in gastric tumor cells. Oncogene (2012) 31:3949–60. 10.1038/onc.2011.558 [DOI] [PubMed] [Google Scholar]
- 277.Ikeda Y, Tanji E, Makino N, Kawata S, Furukawa T. MicroRNAs associated with mitogen-activated protein kinase in human pancreatic cancer. Mol Cancer Res (2012) 10:259–69. 10.1158/1541-7786.MCR-11-0035 [DOI] [PubMed] [Google Scholar]
- 278.Zhang N, Li X, Wu CW, Dong Y, Cai M, Mok MT, et al. MicroRNA-7 is a novel inhibitor of YY1 contributing to colorectal tumorigenesis. Oncogene (2013) 32:5078–88. 10.1038/onc.2012.526 [DOI] [PubMed] [Google Scholar]
- 279.Ma C, Qi Y, Shao L, Liu M, Li X, Tang H. Downregulation of miR-7 upregulates Cullin 5 (CUL5) to facilitate G1/S transition in human hepatocellular carcinoma cells. IUBMB Life (2013) 65:1026–34. 10.1002/iub.1231 [DOI] [PubMed] [Google Scholar]
- 280.Chaudhuri AD, Choi DC, Kabaria S, Tran A, Junn E. MicroRNA-7 regulates the function of mitochondrial permeability transition pore by targeting VDAC1 expression. J Biol Chem (2016) 291:6483–93. 10.1074/jbc.M115.691352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Bargaje R, Gupta S, Sarkeshik A, Park R, Xu T, Sarkar M, et al. Identification of novel targets for miR-29a using miRNA proteomics. PLoS One (2012) 7:e43243. 10.1371/journal.pone.0043243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell (2002) 108:153–64. 10.1016/S0092-8674(02)00625-6 [DOI] [PubMed] [Google Scholar]
- 283.Shoshan-Barmatz V, Krelin Y, Chen Q. VDAC1 as a player in mitochondria-mediated apoptosis and target for modulating apoptosis. Curr Med Chem (2017). 10.2174/0929867324666170616105200 [DOI] [PubMed] [Google Scholar]
- 284.Cochran AG, Tong RT, Starovasnik MA, Park EJ, McDowell RS, Theaker JE, et al. A minimal peptide scaffold for beta-turn display: optimizing a strand position in disulfide-cyclized beta-hairpins. J Am Chem Soc (2001) 123:625–32. 10.1021/ja003369x [DOI] [PubMed] [Google Scholar]
- 285.Yoon DJ, Kwan BH, Chao FC, Nicolaides TP, Phillips JJ, Lam GY, et al. Intratumoral therapy of glioblastoma multiforme using genetically engineered transferrin for drug delivery. Cancer Res (2010) 70:4520–7. 10.1158/0008-5472.CAN-09-4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Johnsen KB, Moos T. Revisiting nanoparticle technology for blood-brain barrier transport: unfolding at the endothelial gate improves the fate of transferrin receptor-targeted liposomes. J Control Release (2016) 222:32–46. 10.1016/j.jconrel.2015.11.032 [DOI] [PubMed] [Google Scholar]
- 287.Gonzalez-Gronow M, Gomez CF, de Ridder GG, Ray R, Pizzo SV. Binding of tissue-type plasminogen activator to the glucose-regulated protein 78 (GRP78) modulates plasminogen activation and promotes human neuroblastoma cell proliferation in vitro. J Biol Chem (2014) 289:25166–76. 10.1074/jbc.M114.589341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Kaelin WG, Jr, Thompson CB. Q&A: cancer: clues from cell metabolism. Nature (2010) 465:562–4. 10.1038/465562a [DOI] [PubMed] [Google Scholar]
- 289.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol (2009) 19:17–24. 10.1016/j.semcancer.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wenner CE. Cell signaling and cancer-possible targets for therapy. J Cell Physiol (2010) 223:299–308. 10.1002/jcp.22021 [DOI] [PubMed] [Google Scholar]
- 291.Montserrat E, Moreno C. Chronic lymphocytic leukaemia: a short overview. Ann Oncol (2008) 19(Suppl 7):vii320–5. 10.1093/annonc/mdn460 [DOI] [PubMed] [Google Scholar]
- 292.Lezi E, Swerdlow RH. Mitochondria in neurodegeneration. Adv Exp Med Biol (2012) 942:269–86. 10.1007/978-94-007-2869-1_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron (2008) 60:748–66. 10.1016/j.neuron.2008.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med (2004) 5:147–62. 10.1385/NMM:5:2:147 [DOI] [PubMed] [Google Scholar]
- 295.Benek O, Aitken L, Hroch L, Kuca K, Gunn-Moore F, Musilek K. A direct interaction between mitochondrial proteins and amyloid-beta peptide and its significance for the progression and treatment of Alzheimer’s disease. Curr Med Chem (2015) 22:1056–85. 10.2174/0929867322666150114163051 [DOI] [PubMed] [Google Scholar]
- 296.Devi L, Ohno M. Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis (2012) 45:417–24. 10.1016/j.nbd.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci (2001) 21:3017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Aleardi AM, Benard G, Augereau O, Malgat M, Talbot JC, Mazat JP, et al. Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J Bioenerg Biomembr (2005) 37:207–25. 10.1007/s10863-005-6631-3 [DOI] [PubMed] [Google Scholar]
- 299.Morais Cardoso S, Swerdlow RH, Oliveira CR. Induction of cytochrome c-mediated apoptosis by amyloid beta 25-35 requires functional mitochondria. Brain Res (2002) 931:117–25. 10.1016/S0006-8993(02)02256-4 [DOI] [PubMed] [Google Scholar]
- 300.Perez-Gracia E, Torrejon-Escribano B, Ferrer I. Dystrophic neurites of senile plaques in Alzheimer’s disease are deficient in cytochrome c oxidase. Acta Neuropathol (2008) 116:261–8. 10.1007/s00401-008-0370-6 [DOI] [PubMed] [Google Scholar]
- 301.Marin R, Ramirez CM, Gonzalez M, Gonzalez-Munoz E, Zorzano A, Camps M, et al. Voltage-dependent anion channel (VDAC) participates in amyloid beta-induced toxicity and interacts with plasma membrane estrogen receptor alpha in septal and hippocampal neurons. Mol Membr Biol (2007) 24:148–60. 10.1080/09687860601055559 [DOI] [PubMed] [Google Scholar]
- 302.Reddy PH. Amyloid beta-induced glycogen synthase kinase 3 beta phosphorylated VDAC1 in Alzheimer’s disease: implications for synaptic dysfunction and neuronal damage. Biochim Biophys Acta (2013) 1832:1913–21. 10.1016/j.bbadis.2013.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Manczak M, Reddy PH. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum Mol Genet (2012) 21:5131–46. 10.1093/hmg/dds360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, et al. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis (2006) 22:76–87. 10.1016/j.nbd.2005.10.004 [DOI] [PubMed] [Google Scholar]
- 305.Ferrer I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J Bioenerg Biomembr (2009) 41:425–31. 10.1007/s10863-009-9243-5 [DOI] [PubMed] [Google Scholar]
- 306.Ramirez CM, Gonzalez M, Diaz M, Alonso R, Ferrer I, Santpere G, et al. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer’s disease. Mol Cell Neurosci (2009) 42:172–83. 10.1016/j.mcn.2009.07.001 [DOI] [PubMed] [Google Scholar]
- 307.Jiang X, Wang X. Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to Apaf-1. J Biol Chem (2000) 275:31199–203. 10.1074/jbc.C000405200 [DOI] [PubMed] [Google Scholar]
- 308.Branco AF, Pereira SL, Moreira AC, Holy J, Sardao VA, Oliveira PJ. Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9c2 cell line. Cardiovasc Toxicol (2011) 11:191–203. 10.1007/s12012-011-9111-5 [DOI] [PubMed] [Google Scholar]
- 309.Ahmed M, Muhammed SJ, Kessler B, Salehi A. Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic beta-cells exposed to high glucose. Islets (2010) 2:283–92. 10.4161/isl.2.5.12639 [DOI] [PubMed] [Google Scholar]
- 310.Gong D, Chen X, Middleditch M, Huang L, Vazhoor Amarsingh G, Reddy S, et al. Quantitative proteomic profiling identifies new renal targets of copper(II)-selective chelation in the reversal of diabetic nephropathy in rats. Proteomics (2009) 9:4309–20. 10.1002/pmic.200900285 [DOI] [PubMed] [Google Scholar]
- 311.Sasaki K, Donthamsetty R, Heldak M, Cho YE, Scott BT, Makino A. VDAC: old protein with new roles in diabetes. Am J Physiol Cell Physiol (2012) 303:C1055–60. 10.1152/ajpcell.00087.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science (2005) 307:384–7. 10.1126/science.1104343 [DOI] [PubMed] [Google Scholar]