

Description
A girl aged 16 years presented with primary amenorrhoea and absent secondary sexual characteristics. She had arhinia at birth and had undergone reconstruction twice in her childhood. Family history was unremarkable. On examination, she had a partially reconstructed nose with patent left nare, absent nasal root, hypertelorism, midfacial hypoplasia and anosmia (figure 1A and B). Her height was 156 cm (50th percentile for ethnicity) with sexual maturity rating of Tanner stage 1. Investigations showed follicle-stimulating hormone, 0.02 (normal, 2.5–10.2 mIU/mL), luteinising hormone, 0.01 (normal, 1.9–12.5 mIU/mL)and 17-β estradiol, 8 (normal, 27–143 pg/mL) suggestive of hypogonadotrophic hypogonadism (HH). Her thyroid function and prolactin were normal. MRI of the brain showed normal-sized pituitary, absent olfactory bulbs with aplastic (yellow arrow) and hypoplastic olfactory sulci (red arrow head) on the right and left side, respectively (figure 1C). With the above findings, a diagnosis of HH with arhinia was made. She was started on oestrogen replacement and showed good pubertal progression on follow-up. Anosmia associated with HH is commonly referred to as Kallmann syndrome, but arhinia is characteristically absent. The index case had arhinia with anosmia and HH differentiating it from Kallmann syndrome. Embryologically, the development of nose, olfactory pathway and gonadotropin-releasing hormone secreting nuclei are closely related. However, no specific mutations have been identified so far. To the best of our knowledge, our patient is the second girl reported with arhinia and HH in literature. Similar reports of arhinia with HH have been described in three boys1 2 and one girl3 previously.
Figure 1.
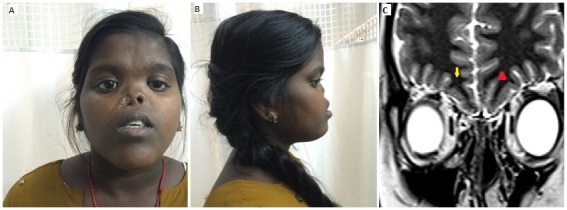
Clinical picture showing partially reconstructed nose with absent nasal root, patent left nare, midfacial hypoplasia and hypertelorism (A, B). MRI of the brain shows absence of olfactory bulbs bilaterally with aplastic (yellow arrow) and hypoplastic olfactory sulci (red arrow head) on the right and left side, respectively (C) in T2-weighted coronal view.
Learning points.
Anosmia or midline facial defects should always alert the clinician towards central aetiology of hypogonadism.
Kallmann syndrome is the most common cause of central hypogonadism.
It can be easily distinguished from other rare causes of central hypogonadism by a simple clinical sign of nasal developmental anomaly.
Footnotes
Contributors: ARA and SM were involved in diagnosis, management and manuscript preparation.
SB was involved in diagnosis and management of the patient.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Bosma JF, Henkin RI, Christiansen RL, et al. Hypoplasia of the nose and eyes, hyposmia, Hypogeusia, and hypogonadotrophic hypogonadism in two males. J Craniofac Genet Dev Biol 1981;1:153–84. [PubMed] [Google Scholar]
- 2. Tryggestad JB, Li S, Chernausek SD. Hypogonadotropic hypogonadism presenting with arhinia: a case report. J Med Case Rep 2013;7:52.doi:10.1186/1752-1947-7-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hunter JD, Davis MA, Law JR. Hypogonadotropic hypogonadism in a female patient with congenital arhinia. J Pediatr Endocrinol Metab 2017;30:1–4.doi:10.1515/jpem-2016-0082 [DOI] [PubMed] [Google Scholar]