

Abstract
UBE3A is a HECT domain E3 ubiquitin ligase whose dysfunction is linked to autism, Angelman syndrome, and cancer. Recently, we characterized a de novo autism-linked UBE3A mutant (UBE3AT485A) that disrupts phosphorylation control of UBE3A activity. Through quantitative proteomics and reporter assays, we found that the UBE3AT485A protein ubiquitinates multiple proteasome subunits, reduces proteasome subunit abundance and activity, stabilizes nuclear β-catenin, and stimulates canonical Wnt signaling more effectively than wild-type UBE3A. We also found that UBE3AT485A activates Wnt signaling to a greater extent in cells with low levels of ongoing Wnt signaling, suggesting that cells with low basal Wnt activity are particularly vulnerable to UBE3AT485A mutation. Ligase-dead UBE3A did not stimulate Wnt pathway activation. Overexpression of several proteasome subunits reversed the effect of UBE3AT485A on Wnt signaling. We also observed that subunits that interact with UBE3A and affect Wnt signaling are located along one side of the 19S regulatory particle, indicating a previously unrecognized spatial organization to the proteasome. Altogether, our findings indicate that UBE3A regulates Wnt signaling in a cell context-dependent manner and that an autism-linked mutation exacerbates these signaling effects. Our study has broad implications for human disorders associated with UBE3A gain or loss of function and suggests that dysfunctional UBE3A might affect additional proteins and pathways that are sensitive to proteasome activity.
Keywords: autism, β-catenin, proteasome, proteomics, Wnt signaling, E6-AP, UBE3A
Introduction
Loss of UBE3A causes a debilitating neurodevelopmental disorder called Angelman syndrome (1–3), whereas excess UBE3A, through gene duplication or gain-of-function mutation, increases the risk for autism (4–7). Moreover, ectopic activation of UBE3A (also known as E6AP), via the human papillomavirus E6 oncoprotein, contributes to cervical cancers (8). UBE3A is an E3 ubiquitin ligase that targets several substrate proteins, including itself, for proteasomal degradation (9). Although much has been learned from studying postnatal functions for UBE3A in mature neurons (10–13), precisely how ubiquitination of these proteins, or other yet to be identified substrates, affects brain development is largely unclear. Here, we sought to gain new insights into developmental functions for UBE3A by exploring a novel connection among UBE3A, the proteasome, and the Wnt signaling pathway.
Wnt signaling is a principal determinant of cell fate and proliferation during development. Abnormal Wnt signaling is implicated in autism pathogenesis (14–17), and Wnt signaling is dysregulated in other diseases, including cancer (18, 19). Induction of the canonical Wnt pathway centers on the dynamics of β-catenin degradation by the proteasome. In the absence of Wnt ligand, a complex of cytosolic proteins, referred to as the destruction complex, sequentially phosphorylates and ubiquitinates β-catenin, resulting in its proteasomal degradation (Fig. 1) (20, 21). Conversely, the presence of Wnt induces the formation of a Frizzled receptor and low-density lipoprotein receptor-related protein co-receptor complex (22). This stabilizes β-catenin and allows its translocation to the nucleus where β-catenin functions as a transcriptional co-activator with members of the T cell factor/lymphoid enhancer factor family of transcription factors (Fig. 1).
Figure 1.
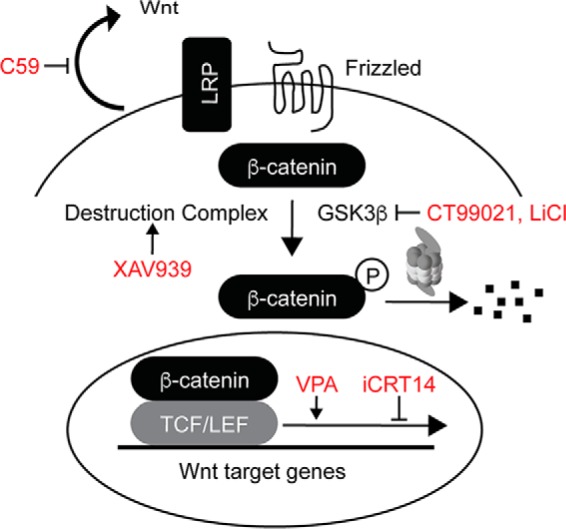
The canonical Wnt signaling pathway. Drugs used in epistasis experiments are indicated (red). C59 is a porcupine inhibitor that suppresses Wnt secretion, XAV939 is a tankyrase inhibitor that stabilizes the destruction complex, LiCl and CT99021 are inhibitors of GSK3β, valproic acid (VPA) is an activator of β-catenin-dependent transcription, and iCRT14 inhibits β-catenin-dependent transcription. TCF/LEF, T cell factor/lymphoid enhancer factor; LRP, low-density lipoprotein receptor-related protein.
It is well known that UBE3A reversibly associates with the proteasome and impacts its processivity (23–27). Moreover, proteasome subunits and proteasome-associated proteins physically interact with and are ubiquitinated by UBE3A (28–34). The relevance of these UBE3A substrates to nervous system development is largely unclear with the notable exception of UCHL5 whose deletion leads to disorganized brain development (35) and PSMD4 (S5A/RPN10), which promotes dendrite development (36). It is also well known that proteasome inhibitors, such as MG-132, block proteasomal processing of β-catenin and potently activate Wnt signaling (37, 38). As part of a genome-wide small interfering RNA screen, we found that Wnt pathway activation is highly sensitive to knockdown of specific proteasomal subunits (Ref. 39 and see new analyses of these data below). Moreover, others found that wild-type (WT) UBE3A can stimulate Wnt pathway activation and protect β-catenin from proteasomal degradation with these activities dependent on the ubiquitin ligase activity of UBE3A (40–42).
Recently, we characterized a de novo autism-linked UBE3AT485A mutation that disrupts phosphorylation control of UBE3A and enhances UBE3A ubiquitin ligase activity (6). This UBE3AT485A mutation, along with an engineered UBE3AT485E mutation that inhibits UBE3A by mimicking phosphorylation, gave us new molecular tools to probe the link between UBE3A activity and Wnt signaling. Through extensive proteomic and functional experiments, we show that UBE3A and Wnt signaling converge at the proteasome with UBE3A impacting overall protein homeostasis, including β-catenin turnover, by ubiquitinating multiple proteasome subunits. Intriguingly, subunits that interact with UBE3A and affect Wnt signaling are located along one side of the 19S regulatory particle, suggesting functional organization of the proteasome. The UBE3AT485A mutant activated Wnt signaling more effectively than WT UBE3A, and ligase-dead UBE3A failed to activate Wnt signaling, raising the novel possibility that abnormal Wnt signaling contributes to neurodevelopmental disorders involving UBE3A loss or gain of function.
Results
UBE3AT485A enhances Wnt signaling in a cell-context dependent manner
WT UBE3A, but not ligase-dead (LD) 6 UBE3A, was found previously to stimulate Wnt reporter gene expression in HEK293T cells and to do so independently of Wnt ligand (40, 42). Given that a significant number of autism-linked de novo mutations are found in genes associated with the Wnt pathway (14, 17), we sought to determine whether the autism-linked UBE3AT485A mutation, which disables phosphorylation control and hyperactivates ubiquitin ligase activity (6), had an equal or greater effect on Wnt pathway activation. To test this possibility, we transfected HEK293T cells with the β-catenin-activated luciferase reporter (BAR) (43) along with the following UBE3A expression constructs: WT UBE3A, UBE3A-LD, UBE3AT485A, and UBE3AT485E (phosphomimetic mutant; reduces UBE3A activity to near UBE3A-LD levels). We previously characterized the protein level and ubiquitin ligase activity of each construct in HEK293T cells (6). Cells were then acutely (12–16 h) treated with control (L-cell) or Wnt3a-conditioned medium (CM) prior to quantifying luciferase activity. We found that WT UBE3A and the UBE3AT485A mutant strongly stimulated Wnt pathway activation in the absence (Fig. 2A) or in the presence of Wnt ligand (Fig. 2B). The autism-linked UBE3AT485A construct activated Wnt signaling to a greater extent than WT UBE3A, particularly as the amount of plasmid transfected was increased (Fig. 2C). The phosphomimetic UBE3AT485E mutant, which impairs UBE3A ubiquitin ligase activity, was much less effective at stimulating Wnt pathway activation. Consistent with a previous study (40), UBE3A-LD did not activate the pathway (Fig. 2, A and B), indicating that UBE3A ubiquitin ligase activity is required to stimulate Wnt pathway activation. Lastly, we found that UBE3AT485A stimulated Wnt pathway activation more than 11-fold (relative to T485E) in cells with low baseline Wnt pathway activation (L-cell control medium) and was relatively less effective in cells treated acutely (12–16 h; 2.6-fold relative to T485E) or chronically (one passage prior to transfection through to lysis; 1.8-fold relative to T485E) with Wnt ligand (Fig. 2D). These data suggest that UBE3A stimulates Wnt signaling in a cell-context dependent manner and is relatively more effective in cells with low levels of ongoing Wnt signaling, consistent with findings by Sherman and co-workers (42).
Figure 2.

The autism-linked UBE3AT485A mutant exacerbates Wnt signaling. A and B, HEK293T cells were transfected with the BAR reporter, CMV-Renilla, and empty vector or the indicated UBE3A plasmid. Cells were serum-deprived and treated with control (L-cell) CM (A) or Wnt3a CM (B). Mean values are shown for firefly:Renilla ratios (n = 12). Error bars represent S.D. Statistical analysis was performed using one-way analysis of variance with Bonferroni post hoc correction. ***, p < 0.0005. C, HEK293T cells were transfected with increasing amounts of plasmid encoding WT UBE3A or UBE3AT485A. Mean values are shown for firefly:Renilla ratios (n = 3). Error bars represent S.D. Statistical analysis was performed using two-way analysis of variance with Bonferroni post hoc correction. ***, p < 0.0005. D, transfected HEK293T cells were grown for 12–16 h in L-cell CM or Wnt3a CM (acute) or in Wnt3a CM for one passage prior to transfection through lysis (chronic). Values are shown as box and whisker plots for firefly:Renilla ratios (n = 6). Whiskers represent the range of maximum and minimum values obtained in our experiments. Statistical analysis was performed using a two-sample t test (two-tailed). *, p < 0.05; ***, p < 0.0005.
UBE3A and the Wnt pathway converge at the proteasome
Epistasis experiments suggested that UBE3A stimulated the Wnt pathway independently of GSK3β or adenomatous polyposis coli protein (41). We performed additional epistasis experiments, probing different parts of the pathway (Fig. 1), with the O-acyltransferase inhibitor C59 (inhibits Wnt palmitoylation and secretion), tankyrase inhibitor XAV939 (stabilizes the destruction complex), GSK3β inhibitors CT99021 and LiCl, the β-catenin transcriptional activator valproic acid, and the β-catenin transcriptional inhibitor iCRT14 (supplemental Fig. S1). These studies suggested that UBE3A acts at or downstream of the destruction complex and influences the transcriptional activity of β-catenin, consistent with work by Kuslansky et al. (42).
We next sought to identify a molecular link between UBE3A and the Wnt/β-catenin signaling pathway. Given that UBE3A is an E3 ubiquitin ligase that targets proteins for destruction, we hypothesized that any proteins that 1) interact with UBE3A, 2) are ubiquitinated by UBE3A, and 3) activate the Wnt pathway when knocked down could represent bona fide UBE3A substrates and serve as core components of this signaling pathway. Published data sets addressed two of these criteria. Martínez-Noël et al. (24) identified a comprehensive list of proteins that selectively interact with UBE3A in HEK293T cells (relative to NEDD4, another HECT domain E3 ubiquitin ligase). Of these proteins, 63 interact with all three UBE3A isoforms (WT and LD; Fig. 3A and supplemental Table S1). We recently identified a comprehensive list of positive and negative regulators of Wnt signaling in HEK293T cells from a near-saturation genome-wide siRNA screen (39). Of these regulators, 1,958 proteins significantly increased Wnt pathway activation when knocked down (Fig. 3A and supplemental Table S2). Database for Annotation, Visualization and Integrated Discovery (DAVID) analysis predicted that “proteasome” was the top gene ontology term for both protein lists (p = 3 × 10−54 and 8 × 10−8, respectively). Remarkably, only 24 proteins were shared between these two lists, and each of these proteins was a component of the 19S regulatory particle, the 20S core complex, or a proteasome accessory protein (Fig. 3B). Intriguingly, when mapped to the structure of the proteasome (44), the shared 19S regulatory subunits were located on one side of the complex (Fig. 3C, red). Subunits on the opposite side largely did not alter Wnt pathway signaling when knocked down (Fig. 3C, blue). These data suggest that proteasome subunits are functionally organized with one side of the 19S regulatory particle associated with Wnt/β-catenin signaling.
Figure 3.

UBE3A and Wnt signaling converge at the proteasome. A, high-confidence unique UBE3A-interacting proteins identified by proteomics (63; white) were cross-referenced with negative regulators of Wnt signaling identified from a genome-wide siRNA screen (1,958; red). The Venn diagram shows 24 common proteins (pink) that complex with UBE3A and negatively regulate Wnt signaling. B, list of the 24 common proteins. Each protein was a component of the 19S regulatory particle of the proteasome, the 20S core complex, or an accessory protein associated with the proteasome (Other). Proteins in bold were identified as putative UBE3A substrates from ubiquitin remnant immunoaffinity profiling (see supplemental Table S4). C, spatial organization of Wnt regulators in the 19S proteasome complex. The cryo-EM structure shows the 19S and 20S proteasome complex (Protein Data Bank code 5GJQ). 20S core complex subunits are shown in gray. 19S subunits that negatively regulated Wnt signaling when knocked down in HEK293T cells are shown in red, whereas subunits that did not affect Wnt signaling are shown in blue.
UBE3A physically associates with the proteasome (24–26), and this association can be reversibly altered by certain stimuli (23). To evaluate whether Thr-485 phosphorylation affects the association of UBE3A with the proteasome, we isolated UBE3A protein complexes from HEK293T cells overexpressing LD versions of UBE3AT485A, which cannot be phosphorylated, and UBE3AT485E, a mutant that mimics phosphorylation. Using mass spectrometry and spectral counts to quantify interactions, we found that the UBE3AT485A and UBE3AT485E mutants interact with the same proteasome subunits with a slight bias for UBE3AT485A–proteasome subunit interactions (supplemental Table S3 and supplemental Fig. S2A). To further evaluate whether phosphorylation at Thr-485 affects UBE3A binding to the proteasome, we biochemically purified proteasomes from HEK293T cells expressing LD versions of UBE3AT485A and UBE3AT485E mutants. UBE3A abundance was measured in proteasome-bound and unbound fractions by Western blot analysis. We found no difference between UBE3AT485A and UBE3AT485E mutants, suggesting that phosphorylation does not strongly influence UBE3A–proteosome interaction (supplemental Fig. S2, B and C).
Multiple proteasome subunits are ubiquitinated by UBE3A
We next sought to comprehensively identify proteins that are ubiquitinated by UBE3A in HEK293T cells. We combined quantitative stable isotope labeling of amino acids in cell culture (SILAC) with ubiquitin remnant immunoaffinity profiling (45, 46). This approach allowed us to isolate ubiquitinated peptides containing the characteristic diglycine motif exposed after tryptic digestion and to perform ratiometric analysis in cells expressing active UBE3AT485A relative to inactive UBE3AT485E (supplemental Table S4). We found that UBE3A ubiquitinated multiple components of the proteasome, including eight core proteasome subunits that physically interact with UBE3A and that negatively regulate Wnt signaling (Fig. 3B, shown in bold).
Of these eight subunits, we examined endogenous protein abundance of five (PSMA2, PSMB1, PSMD2, PSMD4, and PSMD11) by Western blot analysis in cells transfected with UBE3AT485A or UBE3AT485E expression constructs. Protein levels of most of these proteasome subunits were reduced in UBE3AT485A-expressing cells relative to T485E controls in the absence or presence of Wnt ligand (Fig. 4, A–C). We also examined endogenous levels of these five subunits in immortalized human lymphocytes derived from the UBE3AT485A autism proband and the unaffected (WT UBE3A) parents (47). We previously found that UBE3A ubiquitin ligase activity is elevated in cells from this proband using PSMD4 (also known as S5A or RPN10), RAD23A (HHR23A), and UBE3A as endogenous substrates (6). Several proteasome subunits were reduced in cells from the proband relative to the unaffected parents (Fig. 4, D and E). Note that these lymphocytes showed no evidence of Wnt pathway activation based on quantitative PCR experiments with Wnt target genes AXIN2, BMP4, NKD1, and SOX17 (supplemental Fig. S3, A–D), likely because these non-adherent cells have undetectable levels of β-catenin protein (supplemental Fig. S3E). Thus, we could not evaluate the extent to which endogenous Wnt signaling was affected in this patient-derived cell line.
Figure 4.

UBE3AT485A depletes multiple endogenous proteasome subunits. A, protein lysates from HEK293T cells expressing UBE3AT485A or UBE3AT485E mutants were analyzed by Western blotting for proteasome subunit protein levels. Analysis was performed on cells grown in L-cell CM or Wnt3a CM. B and C, quantification of Western blots for cells grown in L-cell CM (B) or Wnt3a CM (C). Mean percent abundance values in UBE3AT485A samples relative to UBE3AT485E samples are shown (n = 6). Error bars indicate S.D. Statistical analysis was performed using a one-sample t test (two-tailed). *, p < 0.05; **, p < 0.005. PSMD11 was not analyzed due to its low abundance in HEK293T cells. D and E, representative Western blot (D) and quantification (E) of proteasome subunits in immortalized lymphocyte cell lines from the father, mother, and autism proband (Simon's Simplex Collection; Family ID 13873). Mean percent values are shown relative to the father's level (n = 3). Error bars indicate S.D. *, p < 0.05, one-sample t test.
PSMD2 is a substrate of UBE3A
Our experiments suggested that PSMD2 (also known as RPN1) was a novel UBE3A substrate. Unlike other 19S components, PSMD2 is located at the edge of the regulatory complex (Fig. 5A, green) and anchors several accessory proteins to the proteasome (48). These accessory proteins include multiple UBE3A substrates, RAD23A and RAD23B (28, 49), ADRM1, PSMC3, UCHL5 (25), PSMD4 (6), and MCM7 (50), as well as proteins that associate with UBE3A, including the E2 enzyme UBE2L3 (51).
Figure 5.

PSMD2 is a substrate of UBE3A. A, cryo-EM structure of the proteasome showing the location of PSMD2 (green) in the 19S regulatory particle (Protein Data Bank code 5GJQ). B and C, PSMD2 dose-dependently rescues UBE3A-stimulated Wnt signaling. HEK293T cells were co-transfected with plasmids encoding UBE3AT485A and increasing quantities of PSMD2 (0, 20, 40, and 60 ng). Cells were then grown in L-cell CM (B) or Wnt3a CM (C). Mean percent values for firefly:Renilla ratios are shown relative to cells transfected with UBE3A + empty vector (n = 6). Error bars indicate S.D. ***, p < 0.0005, one-sample t test (two-tailed). D, HEK293T cells transfected with the indicated UBE3A, Myc-DDK-PSMD2, and HA-ubiquitin constructs were treated with the proteasome inhibitor MG-132 (30 μm; 4 h). PSMD2 was immunoprecipitated (IP) using an anti-FLAG antibody, and the Western blot was probed with an anti-HA antibody (left panel) or PSMD2 antibody (right panels) to detect ubiquitinated PSMD2. E, an in vitro ubiquitin assay was performed using recombinant E1, E2 (UBE2D3), UBE3A, and PSMD2 expressed and purified from HEK293T cells. Reactions were stopped at the indicated time, and the formation of ubiquitinated PSMD2 was monitored using a PSMD2 antibody. WT UBE3A was omitted from the reaction as a negative control (first lane).
In titration experiments with increasing amounts of PSMD2 plasmid, we found that PSMD2 reduced UBE3AT485A-mediated Wnt pathway activation in the absence or presence of Wnt ligand (Fig. 5, B and C). These findings suggest that overexpression of PSMD2 can functionally rescue or compensate for the loss of endogenous PSMD2 in UBE3AT485A-expressing cells. Moreover, high-molecular-weight ubiquitin-conjugated forms of PSMD2 were detected in HEK293T cells expressing UBE3AT485A, PSMD2, and HA-tagged ubiquitin after treatment with the proteasome inhibitor MG-132 (30 μm; 4 h) (Fig. 5D). In contrast, polyubiquitinated PSMD2 was not detected in cells expressing the inactive UBE3AT485E mutant (Fig. 5D). We also performed in vitro ubiquitination reactions with recombinant ubiquitin, E1, E2, and UBE3A enzymes along with PSMD2 purified from HEK293T cells. A single distinct ubiquitin-conjugated PSMD2 species was detected in reactions containing UBE3A (Fig. 5E) that differed from what was observed in cells (Fig. 5D), suggesting that additional cellular factors promote polyubiquitin chain formation on PSMD2. Collectively, these data indicate that PSMD2 is a substrate of UBE3A and that overexpression of PSMD2 dampens Wnt pathway activation in UBE3AT485A-expressing cells.
UBE3A regulates Wnt signaling through multiple proteasome subunits
We next evaluated the extent to which additional proteasomal subunits rescue Wnt pathway activation in UBE3AT485A-expressing cells. Plasmids encoding subunits that interact with UBE3A were transfected into HEK293T cells together with UBE3AT485A at 1:1 and 1:2 UBE3A:subunit ratios or alone to confirm overexpression (supplemental Fig. S4). We were particularly interested in PSMD4 as PSMD4 is a well characterized UBE3A substrate (6, 25, 26, 29). However, PSMD4 failed to reduce Wnt pathway activation in UBE3AT485A-expressing cells at the 1:1 or 1:2 transfection ratio (Fig. 6; ranked results shown in supplemental Table S5). Instead, PSMD4 augmented Wnt signaling by nearly 200% at the 1:2 transfection ratio. Of the remaining subunits tested, PSMB1, PSMC2, PSMD2, and PSMD7 consistently reduced Wnt pathway activation in UBE3AT485A-expressing cells, whereas PSMC6 augmented the Wnt response (at 1:1 and 1:2 ratios; Fig. 6 and supplemental Table S5). PSMD2 was one of the most effective subunits to rescue UBE3A-dependent Wnt activation in HEK293T cells (supplemental Table S6). PSMD1, PSMD3, PSMD6, and PSMD11 had no effect or inconsistent effects across the four conditions. Our experiments collectively suggest that UBE3A stimulates Wnt pathway activation by interacting with, ubiquitinating, and reducing the levels of multiple (PSMB1, PSMC2, PSMD2, and PSMD7) proteasome subunits.
Figure 6.

Overexpression of proteasome subunits reversed the effect of UBE3AT485A on Wnt signaling. A and B, BAR assays were performed in HEK293T cells expressing the UBE3AT485A mutant and the indicated proteasome subunits. Cells were transfected using a 1:1 UBE3A:subunit ratio and grown in L-cell CM (A) or Wnt3a CM (B). Values are shown as box and whisker plots for percent firefly:Renilla ratios relative to cells transfected with UBE3AT458A + empty vector. C and D, cells were transfected using a 1:2 UBE3A:subunit ratio and grown in L-cell CM (C) or Wnt3a CM (D). Values are shown as box and whisker plots for percent firefly:Renilla ratios relative to cells transfected with UBE3AT458A + empty vector. A–D, n = 9 (three independent experiments; three biological replicates/experiment). *, p < 0.05; **, p < 0.005; ***, p < 0.0005, one-sample t test (two-tailed). Whiskers represent the range of maximum and minimum values obtained in our experiments.
UBE3A inhibits proteasome function and stabilizes β-catenin
Several groups found that WT UBE3A inhibits the proteasome in cells (25, 34). To determine whether UBE3AT485A similarly affected proteasome activity, we transfected HEK293T cells with UBE3AT485A or UBE3AT485E mutants along with a destabilized G76V-ubiquitin (UbG76V)-GFP fusion reporter protein and mCherry as the normalization control. This UbG76V-GFP construct was used previously to quantify proteasome-dependent activity in cells (52). We found that the levels of this UbG76V-GFP reporter were higher in cells transfected with UBE3AT485A relative to the inactive T485E mutant, suggesting that elevated UBE3AT485A activity inhibits proteasome function in cells (Fig. 7, A and B). This difference in UbG76V-GFP reporter levels was abolished in cells treated with MG-132 (10 μm; 12 h; Fig. 7, A and B), further suggesting a proteasome-dependent mechanism.
Figure 7.

UBE3A inhibits the proteasome and increases nuclear β-catenin accumulation. A and B, representative Western blot (A) and quantification (B) of protein lysates from HEK293T cells expressing UBE3AT485A or UBE3AT485E, the destabilized UbG76V-GFP reporter protein, and mCherry as a transfection and loading control. The proteasome was inhibited with MG-132 (10 μm; 12 h). Mean percent values relative to UBE3AT458E-expressing cells are shown (n = 5). Error bars indicate S.D. *, p < 0.05, two-sample t test (two-tailed). C and D, representative Western blot (C) and quantification (D) of nuclear lysates from HEK293T cells expressing UBE3AT485A or UBE3AT485E mutants. Cells were grown in L-cell CM or Wnt3a CM and analyzed for β-catenin abundance. Mean percent values relative to UBE3AT458E-expressing cells are shown (n = 6). Error bars indicate S.D. *, p < 0.05, two-sample t test (two-tailed). E, β-catenin degradation is required for UBE3A-dependent Wnt pathway activation. BAR assays were performed with HEK293T cells expressing UBE3AT485A or UBE3AT485E and co-transfected with plasmids encoding GFP, WT β-catenin, or a stabilized β-catenin mutant. Values are shown as box and whisker plots for percent firefly:Renilla ratios relative to cells transfected with GFP alone (n = 9). Whiskers represent the range of maximum and minimum values obtained in our experiments. ***, p < 0.0005, two-sample t test (two-tailed). n.s., not significant. F, mechanism. Enhanced UBE3A activity at the proteasome reduces protein turnover, leading to β-catenin stabilization and enhanced transcription of Wnt target genes. TCF/LEF, T cell factor/lymphoid enhancer factor; LRP, low-density lipoprotein receptor-related protein.
β-Catenin is ubiquitinated and then degraded by the proteasome, which limits entry to the nucleus and transcriptional activity (Fig. 1). Given that UBE3AT485A reduces proteasome activity, we hypothesized that nuclear β-catenin levels might be elevated in UBE3AT485A-expressing cells. Indeed, we found that nuclear β-catenin levels were slightly elevated in cells expressing the UBE3AT485A mutant relative to cells expressing UBE3AT485E in the absence or presence of Wnt ligand (Fig. 7, C and D). Additionally, UBE3AT485A less effectively stimulated Wnt signaling (relative to UBE3AT485E) in cells overexpressing WT β-catenin and did not augment Wnt signaling (relative to UBE3AT485E) in cells overexpressing a constitutively active β-catenin construct (Fig. 7E). These data further demonstrate that UBE3AT485A most effectively stimulates Wnt signaling in cells with subsaturating levels of active β-catenin, that is when baseline Wnt pathway activation is low. Altogether, our results suggest a mechanism whereby UBE3A ubiquitinates multiple subunits of the proteasome, leading to impairment of proteasome-mediated degradation of β-catenin and increased β-catenin/Wnt signaling (Fig. 7F).
Discussion
Despite the well known association between excess UBE3A and autism risk (4, 5, 53), including autism-linked mutations that elevate UBE3A alone (6, 7), and extensive research on UBE3A (10), it is currently unclear how excess UBE3A affects mechanisms linked to autism and neurodevelopment. UBE3A is thought to be a promiscuous enzyme (54), and this seeming promiscuity has confounded attempts to identify the physiological substrates of UBE3A. Recently, we found that an autism-linked point mutation in UBE3A (UBE3AT485A) disables a phosphorylation control switch and elevates UBE3A ubiquitin ligase activity (6). Here, we found that UBE3AT485A exacerbates Wnt signaling by inhibiting the proteasome and stabilizing nuclear β-catenin. Numerous autism-linked genes are implicated in Wnt signaling (14, 15, 17); valproic acid, an environmental risk for autism, activates Wnt signaling (supplemental Fig. S1E) (55, 56); and Wnt signaling regulates proliferation and differentiation of neuronal progenitors (57, 58). Our study and the work of others (40–42) firmly place UBE3A within the Wnt signaling pathway and provide fundamental new insights into how excess UBE3A could impair brain development and increase risk for autism.
Our biochemical and proteomics findings suggest that UBE3A stimulates Wnt signaling by ubiquitinating multiple proteasome subunits and reducing proteasome activity, which ultimately stabilize β-catenin. UBE3AT485A-mediated stimulation of Wnt signaling was reduced in cells overexpressing different proteasome subunits, suggesting that proteasome subunits are functionally relevant UBE3A substrates. Multiple laboratories have found that UBE3A can ubiquitinate proteasome subunits and inhibit proteasome function (25, 34), although one group found that UBE3A stimulated proteasome proteolytic activity (27). Discrepancies could relate to differences in cell context or experimental approach. Jacobson et al. (25) found that UBE3A inhibited the proteasome by in situ ubiquitination provided the levels of polyubiquitinated proteins in the cell were low. Cellular stresses that increase polyubiquitinated protein levels (oxidative stress and serum starvation) blocked UBE3A from ubiquitinating the proteasome, leading to increased proteasome activity (25). In a simplistic sense, the proteasome can “sense” whether global polyubiquitinated protein levels are low or high and, via UBE3A, adjust proteasomal activity accordingly. Thus, differences in cell culture conditions (such as differences in serum composition, oxygen tension, and plating density) have the potential to influence polyubiquitinated protein levels and alter proteasome activity via UBE3A.
We found that knockdown of different proteasome subunits or overexpression of UBE3AT485A led to activation of Wnt signaling in HEK293T cells, a cell line with low baseline Wnt pathway activation. In contrast, UBE3AT485A was relatively less effective at stimulating Wnt signaling in HEK293T cells exposed (acutely or chronically) to Wnt ligand or in HEK293T cells expressing elevated levels of β-catenin. It is well known that cell context can affect Wnt signaling (59, 60). Our study suggests that the proteasome, which lies at the heart of the Wnt signaling pathway (Fig. 1), and UBE3A, which regulates proteasome function and subunit composition, can influence active β-catenin levels in a cell context-dependent manner.
This cell context-dependent effect of UBE3A has implications for brain development. Wnt signaling weakens over the course of development in cortical neuron progenitors, which express UBE3A biallelically (61), and varies by cell and tissue type (62, 63). It is thus conceivable that cells with latent but low levels of Wnt pathway activation may be more vulnerable to UBE3A excess. Future experiments will be needed to test this possibility.
To our knowledge, we are the first to identify a proteasomal subdomain that associates with the Wnt signaling pathway. Proteasome subunits that enhance Wnt signaling upon siRNA-mediated knockdown localize to one side of the 19S regulatory complex. Although this observation is based on data from a primary siRNA screen and will need to be independently verified, we speculate that such an arrangement could spatially restrict docking or shuttling of Wnt pathway components to the proteasome. The destruction complex scaffolding protein AXIN1 can directly bind to β-catenin and PSMD1, whereas β-catenin stabilization is dependent on its sequestration away from the proteasome (64, 65). Post-translational modifications of proteasome subunits, such as ubiquitination, could block the association of β-catenin with the proteasome, which would stabilize β-catenin and promote Wnt signaling. Thus, cells might physically segregate Wnt signaling from other ubiquitin-dependent signaling pathways to permit independent control of cellular processes that depend on proteasomal proteolysis.
Numerous pathways require protein degradation, but it has long remained unknown how the proteasome simultaneously processes signaling components to maintain homeostasis. Our study raises the possibility that distinct pathways are compartmentalized at the proteasome, and this provides the necessary molecular platforms to execute multiple parallel functions.
Lastly, our study has broader implications and suggests that UBE3A could affect additional proteins, signaling pathways, and cellular functions that are particularly sensitive to ubiquitination and proteasomal degradation. Our findings hint that the apparent promiscuity of UBE3A may relate to its inhibitory effect on the proteasome as proteasome inhibition would elevate the levels of many ubiquitinated proteins, including those that are not direct UBE3A substrates.
Experimental procedures
Molecular biology
All UBE3A constructs used in this study were derived from human UBE3A isoform II (NCBI accession number NP_000453.2). Myc epitope tags were placed on the N terminus of UBE3A by polymerase chain reaction and cloned into pCIG2 using SacI and XmaI or into pEGFP-N1 using BamHI and NotI. All constructs were verified by sequencing. HA-tagged ubiquitin, pGL3-BAR, TK-Renilla, pHAGE β-catenin WT, and pHAGE β-catenin mutant constructs were described previously (39, 66, 67). UbG76V-GFP was obtained from Addgene (23969). pCMV6 Myc-DDK-tagged constructs for PSMB1 (RC201798), PSMC2 (RC200945), PSMC5 (RC201251), PSMC6 (RC202809), PSMD1 (RC210486), PSMD2 (RC203204), PSMD3 (RC202307), PSMD6 (RC202292), PSMD7 (RC203133), PSMD11 (RC201201), and untagged PSMD4 (SC111678) were all purchased from Origene.
Antibodies and reagents
Primary antibodies used were mouse anti-Myc (1:1,000; EMD Millipore, 05-724), mouse anti-HA (1:1,000; EMD Millipore, 05-904), rabbit anti-GAPDH (1:1,000; Genetex, GTX100118), mouse anti-FLAG M2 (1:1,000; Sigma, F3165), rabbit anti-PSMA2 (1:1,000; Thermo Fisher, PA5-17294), rabbit anti-PSMB1 (1:1,000; Thermo Fisher, PA5-49648), rabbit anti-PSMD2 (1:1,000; Thermo Fisher, PA5-27663), goat anti-PSMD4 (1:1000; Boston Biochem, AF5540), rabbit anti-PSMD11 (1:1,000; Thermo Fisher, PA5-27447); mouse anti-GFP (1:5,000; Clontech, 632281), rabbit anti-mCherry (1:2,500; Abcam, ab167453), rabbit anti-β-catenin (1:1,000, Cell Signaling Technology, 9562), and mouse anti-HDAC1 (1:1,000; Cell Signaling Technology, 5356). LiCl, XAV939, iCRT14, and valproic acid were purchased from Sigma; CT99021 was purchased from Axon Med Chem; and C59 was purchased from Cellagen Technology. MG-132 was purchased from Calbiochem. Wnt3a medium was made according to the ATCC protocol (68).
Tissue culture
HEK293T cells (ATCC) were maintained in a 5% CO2 humidified incubator in DMEM (Thermo Fisher) containing 4.5 g/liter glucose, 584 mg/liter glutamine, and 110 mg/liter sodium pyruvate supplemented with 10% (v/v) fetal bovine serum (Hyclone) and 1× antibiotic-antimycotic (penicillin, streptomycin, amphotericin B; Life Technologies). For biochemical analyses, transfections were performed in 6-well dishes using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. HEK293T cells were lysed in NuPAGE lithium dodecyl sulfate sample buffer (Life Technologies) supplemented with 1% β-mercaptoethanol and 1× Complete Mini protease inhibitor mixture (Roche Applied Science). Lysates were boiled, and proteins were resolved by 4–20% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences) and probed with the appropriate primary antibodies overnight. Protein bands were visualized using the Odyssey CLx infrared imaging system (LI-COR Biosciences) with the following secondary antibodies: donkey anti-rabbit 800CW (926-32213), donkey anti-rabbit 680RD (925-68073), and donkey anti-mouse 680RD (926-68072; all from LI-COR Biosciences and used at 1:10,000). Nuclear fractionation was performed using the NE-PER kit (Thermo Fisher) according to the manufacturer's instructions.
Immortalized human lymphocytes (Simons Simplex Collection) were maintained in a 5% CO2 humidified incubator in RPMI 1640 medium supplemented with 15% (v/v) fetal bovine serum (Hyclone), 1× GlutaMAX (Gibco), and 1× antibiotic-antimycotic. The following cell lines were used for our study: 13873.Fa, 13873.Mo, and 13873.P1. For protein analysis, 10 million lymphocytes were collected by centrifugation and lysed for 20 min on ice with periodic rocking in a lysis buffer consisting of 20 mm HEPES, pH 7.4, 1% Nonidet P-40, and 50 mm KCl supplemented with protease and phosphatase inhibitors. Samples were cleared at 1,000 × g for 5 min, and the supernatant was collected and subjected to a protein assay (Bio-Rad) to determine concentration. Samples were prepared by diluting in sample buffer supplemented with 1% β-mercaptoethanol, boiled, and resolved by SDS-PAGE.
Luciferase assays
BAR assays were performed in 96-well plates for all experiments except those shown in supplemental Fig. S1, B–F (see below). HEK293T cells were plated at a density of 10,000/well. Cells were transiently transfected with 10 ng of pRL-TK-Renilla, 30 ng of BAR-pGL3, and 60 or 80 ng of the indicated constructs using the TransIT®-2020 transfection reagent (Mirus) or TransIT-LT1 transfection reagent (Mirus). After 2 h, medium containing transfection reagents was replaced with fresh medium. The following day, cells were serum-deprived (0.2% FBS) for 6 h and stimulated with Wnt3a-conditioned medium or control L-cell medium for 12–16 h. Reporter gene expression was assessed using the Dual-Luciferase reporter assay system (Promega) and measured on an Enspire plate reader from PerkinElmer Life Sciences. For supplemental Fig. S1, B–F, HEK293T cells were plated at a density of 5,000/well in 384-well plates, and drugs were added 4 h postplating. 24 h after plating, cells were transfected with 5 ng of pRL-TK-Renilla, 15 ng of BAR-pGL3, and 30 ng of the indicated UBE3A expression constructs using FuGENE HD (Promega). Reporter gene expression was assessed 24 h post-transfection using the Dual-Glo luciferase assay system (Promega). For all experiments, the luciferase activity was normalized against Renilla activity.
Ubiquitin remnant immunoaffinity profiling
HEK293T cells were grown in high-glucose DMEM (Caisson Laboratories) supplemented with 10% dialyzed FBS (Gibco), 3.7 g/liter sodium bicarbonate, 1× GlutaMAX, and 1× antibiotic-antimycotic. For heavy medium, 50 μg/ml l-lysine (Lys8) and 40 μg/ml l-arginine (Arg10) were added (Cambridge Isotope Laboratories). 50 μg/ml standard l-lysine and 40 μg/ml standard l-arginine (Thermo Fisher) were added for light medium. For complete labeling, cells were passaged for at least seven generations in the above medium. Cells were lysed in lysis buffer (8 m urea in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 2 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm PMSF, 50 μm PR-619, and 1 mm iodoacetamide) and sonicated to clarify the lysate. The extract was centrifuged at 20,000 rpm at 4 °C for 15 min, and protein was estimated using the method of Bradford (69). Both heavy and light protein samples were mixed in 1:1 ratio (mg:mg) and diluted in 50 mm Tris-HCl, pH 7.5, to reduce the urea concentration to 2 m. The total protein was digested overnight with sequencing grade trypsin (Promega). The trypsin:protein ratio was maintained at 1:100. Total peptides were purified on a Sep-Pak column (Waters) and eluted with a 40-ml step gradient of 10–50% acetonitrile and 0.1% trifluoroacetic acid gradient (10, 15, 20, 25, 30, 35, 40 and 50%; 5 ml each). The eluent fractions were lyophilized overnight until dry powdery flakes were obtained.
A diglycine antibody kit was purchased from Cell Signaling Technology (PTMScan® Ubiquitin Remnant Motif (K-ϵ-GG) kit 5562). Dried total cell peptide samples were solubilized in 1× immunoaffinity purification buffer, and peptide immunoprecipitations were performed at 4 °C for 4–6 h. The bead slurry was washed three times with PBS and two times with water. K-ϵ-GG peptides were eluted with two washes of 50 μl of 0.15% trifluoroacetic acid (TFA). Eluted peptides were further purified on Pierce C18 spin columns (Cat 89870) using the manufacturer's protocol. Peptides were eluted using 70% acetonitrile and 0.1% TFA solution in 50-μl volumes twice and dried in a SpeedVac at room temperature.
Mass spectrometry
Mass spectrometry analysis was performed at the Michael Hooker Proteomics Center at the University of North Carolina. Dried peptide samples were reconstituted in 1% acetonitrile and 0.1% formic acid. Each sample was analyzed by LC/MS/MS using an Easy nLC 1000 coupled to a QExactive HF mass spectrometer (Thermo Scientific). Samples were injected onto an Easy Spray PepMap C18 column (75-μm inner diameter × 25 cm; 2-μm particle size) (Thermo Scientific) and separated over a 2-h method. The gradient for separation consisted of 5–35% mobile phase B at a 250 nl/min flow rate where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in acetonitrile. The QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent fragmentation. Resolution for the precursor scan (m/z 400–1600) was set to 120,000 with a target value of 3 × 106 ions. MS/MS scan resolution was set to 30,000 with a target value of 1 × 105 ions. The normalized collision energy was set to 27% for higher-energy collisional dissociation, and the isolation window was set to 1.6 m/z. Peptide match was set to preferred, and precursors with unknown charge or charge states of 1 and ≥7 were excluded.
Raw data files were processed using Proteome Discoverer version 2.1 (Thermo Scientific). Peak lists were searched against a reviewed human UniProt database (containing 20,203 entries), appended with a contaminant database, using Sequest. The following parameters were used to identify tryptic peptides for protein identification: 10-ppm precursor ion mass tolerance; 0.02-Da product ion mass tolerance; up to two missed trypsin cleavage sites; carbamidomethylation of Cys was set as a fixed modification; and oxidation of Met, Gly-Gly of Lys, phosphorylation of Ser/Thr/Tyr, [13C6]Arg, [13C6]Lys, and acetylation of the N terminus were set as variable modifications. Percolator was used to estimate the number of false-positive identifications, and a q-value (false discovery rate) was assigned, a q-value of <0.05 (5% false discovery rate), which was used to filter all results. The ptmRS node was used to assign the post-translational modification site localization probability. Peptide quantitation was enabled to determine SILAC ratios for ubiquitin-modified peptides.
Ubiquitination assays
In vitro ubiquitination assays were performed using immunopurified PSMD2 in combination with a commercially available recombinant UBE3A ubiquitin ligase kit (Boston Biochem, K-230) according to the manufacturer's instructions. In brief, HEK293T cells grown in 25-cm dishes were transfected with a plasmid encoding Myc-DDK-PSMD2 and allowed to grow for 48 h. Cells were lysed on ice for 20 min with gentle agitation using an immunoprecipitation buffer containing 20 mm HEPES, pH 7.4, 1% Nonidet P-40, 50 mm KCl, and 100 mm NaCl. Lysates were clear by centrifugation, and PSMD2 was immunoprecipitated using an anti-Myc affinity gel (Sigma, A7470) at 4 °C for 2 h. The resulting beads were washed two times each (eight washes total) in immunoprecipitation buffer containing 100, 250, and 500 mm NaCl as well as two final washes in PBS. PSMD2 was eluted from the beads in PBS containing 10 μg/ml Myc peptide (Sigma).
For cell-based assays, HEK293T cells were transfected with constructs encoding UBE3A, Myc-DDK-PSMD2, and HA-ubiquitin. Cells were allowed to grow for 48 h and treated with 30 μm MG-132 for 4 h. The cells were then lysed in radioimmune precipitation assay buffer containing 1% SDS and 30 μm MG-132. Cell lysates were boiled for 20 min and clarified by centrifugation at 15,000 × g for 10 min. The resulting supernatant was diluted 1:10 (v/v) in an immunoprecipitation buffer (20 mm HEPES, pH 7.4, 50 mm KCl, and 1% Triton X-100), and PSMD2 was immunoprecipitated using the EZ View anti-FLAG affinity gel (Sigma, F2426) at 4 °C for 1 h. The final complex was washed three times with immunoprecipitation buffer containing 125 mm NaCl, resuspended in sample buffer, and processed for SDS-PAGE and immunoblot analysis.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. Statistical treatments for each experiment are indicated in the figure legends. p < 0.05 was considered significant.
Author contributions
J. J. Y. conceived, designed, and performed experiments and wrote the manuscript. S. R. P. performed luciferase experiments. M. P. W. and J. M. W. performed epistasis experiments. R. C. performed ubiquitin remnant immunoaffinity profiling. G. F. performed quantitative PCR. M. J. E., M. B. M., and M. J. Z. conceived and designed experiments and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Laura Herring for performing mass spectrometry analysis. Immortalized lymphocytes from the Simons Simplex Collection were acquired from the National Institute of Mental Health Center for Genetic Studies via the Rutgers University Cell and DNA Repository.
Note added in proof
In the version of the article that was published as a Paper in Press on May 30, 2017, the PSMA2 and PSMB1 immunoblots shown in Fig. 4D were derived from the same physical membrane. As these two proteins are the same size, the two images appeared to be similar. To avoid any confusion, the PSMB1 immunoblot was replaced using an image from a replicate immunoblot.
This work was supported in part by grants from the Angelman Syndrome Foundation (to M. J. Z.), National Institute of Mental Health Grant R01MH093372 (to M. J. Z.), and National Institutes of Health Pioneer Award DP1ES024088 (to M. J. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S6 and Figs. S1–S4.
- LD
- ligase-dead
- BAR
- β-catenin-activated luciferase reporter
- CM
- conditioned medium
- SILAC
- stable isotope labeling of amino acids in cell culture
- Ub
- ubiquitin.
References
- 1. Jiang Y. H., Armstrong D., Albrecht U., Atkins C. M., Noebels J. L., Eichele G., Sweatt J. D., and Beaudet A. L. (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811 [DOI] [PubMed] [Google Scholar]
- 2. Kishino T., Lalande M., and Wagstaff J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73 [DOI] [PubMed] [Google Scholar]
- 3. Mabb A. M., Judson M. C., Zylka M. J., and Philpot B. D. (2011) Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glessner J. T., Wang K., Cai G., Korvatska O., Kim C. E., Wood S., Zhang H., Estes A., Brune C. W., Bradfield J. P., Imielinski M., Frackelton E. C., Reichert J., Crawford E. L., Munson J., et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hogart A., Wu D., LaSalle J. M., and Schanen N. C. (2010) The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 38, 181–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yi J. J., Berrios J., Newbern J. M., Snider W. D., Philpot B. D., Hahn K. M., and Zylka M. J. (2015) An autism-linked mutation disables phosphorylation control of UBE3A. Cell 162, 795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Noor A., Dupuis L., Mittal K., Lionel A. C., Marshall C. R., Scherer S. W., Stockley T., Vincent J. B., Mendoza-Londono R., and Stavropoulos D. J. (2015) 15q11.2 duplication encompassing only the UBE3A gene is associated with developmental delay and neuropsychiatric phenotypes. Hum. Mutat. 36, 689–693 [DOI] [PubMed] [Google Scholar]
- 8. Scheffner M., Huibregtse J. M., Vierstra R. D., and Howley P. M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505 [DOI] [PubMed] [Google Scholar]
- 9. Sell G. L., and Margolis S. S. (2015) From UBE3A to Angelman syndrome: a substrate perspective. Front. Neurosci. 9, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. LaSalle J. M., Reiter L. T., and Chamberlain S. J. (2015) Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics 7, 1213–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Margolis S. S., Salogiannis J., Lipton D. M., Mandel-Brehm C., Wills Z. P., Mardinly A. R., Hu L., Greer P. L., Bikoff J. B., Ho H. Y., Soskis M. J., Sahin M., and Greenberg M. E. (2010) EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 143, 442–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wallace M. L., Burette A. C., Weinberg R. J., and Philpot B. D. (2012) Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74, 793–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yashiro K., Riday T. T., Condon K. H., Roberts A. C., Bernardo D. R., Prakash R., Weinberg R. J., Ehlers M. D., and Philpot B. D. (2009) Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krumm N., O'Roak B. J., Shendure J., and Eichler E. E. (2014) A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 37, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Rubeis S., He X., Goldberg A. P., Poultney C. S., Samocha K., Cicek A. E., Kou Y., Liu L., Fromer M., Walker S., Singh T., Klei L., Kosmicki J., Shih-Chen F., Aleksic B., et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ernst C. (2016) Proliferation and differentiation deficits are a major convergence point for neurodevelopmental disorders. Trends Neurosci. 39, 290–299 [DOI] [PubMed] [Google Scholar]
- 17. Packer A. (2016) Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci. Biobehav. Rev. 64, 185–195 [DOI] [PubMed] [Google Scholar]
- 18. Polakis P. (2012) Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 4, a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tucci V., Kleefstra T., Hardy A., Heise I., Maggi S., Willemsen M. H., Hilton H., Esapa C., Simon M., Buenavista M. T., McGuffin L. J., Vizor L., Dodero L., Tsaftaris S., Romero R., et al. (2014) Dominant beta-catenin mutations cause intellectual disability with recognizable syndromic features. J. Clin. Investig. 124, 1468–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anastas J. N., and Moon R. T. (2013) WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13, 11–26 [DOI] [PubMed] [Google Scholar]
- 21. MacDonald B. T., Tamai K., and He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim S. E., Huang H., Zhao M., Zhang X., Zhang A., Semonov M. V., MacDonald B. T., Zhang X., Garcia Abreu J., Peng L., and He X. (2013) Wnt stabilization of β-catenin reveals principles for morphogen receptor-scaffold assemblies. Science 340, 867–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tai H. C., Besche H., Goldberg A. L., and Schuman E. M. (2010) Characterization of the brain 26S proteasome and its interacting proteins. Front. Mol. Neurosci. 3, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martínez-Noël G., Galligan J. T., Sowa M. E., Arndt V., Overton T. M., Harper J. W., and Howley P. M. (2012) Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol. Cell. Biol. 32, 3095–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jacobson A. D., MacFadden A., Wu Z., Peng J., and Liu C. W. (2014) Autoregulation of the 26S proteasome by in situ ubiquitination. Mol. Biol. Cell 25, 1824–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Besche H. C., Sha Z., Kukushkin N. V., Peth A., Hock E. M., Kim W., Gygi S., Gutierrez J. A., Liao H., Dick L., and Goldberg A. L. (2014) Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J. 33, 1159–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomaić V., and Banks L. (2015) Angelman syndrome-associated ubiquitin ligase UBE3A/E6AP mutants interfere with the proteolytic activity of the proteasome. Cell Death Dis. 6, e1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar S., Kao W. H., and Howley P. M. (1997) Physical interaction between specific E2 and Hect E3 enzymes determines functional cooperativity. J. Biol. Chem. 272, 13548–13554 [DOI] [PubMed] [Google Scholar]
- 29. Uchiki T., Kim H. T., Zhai B., Gygi S. P., Johnston J. A., O'Bryan J. P., and Goldberg A. L. (2009) The ubiquitin-interacting motif protein, S5a, is ubiquitinated by all types of ubiquitin ligases by a mechanism different from typical substrate recognition. J. Biol. Chem. 284, 12622–12632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zaaroor-Regev D., de Bie P., Scheffner M., Noy T., Shemer R., Heled M., Stein I., Pikarsky E., and Ciechanover A. (2010) Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 6788–6793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kleijnen M. F., Shih A. H., Zhou P., Kumar S., Soccio R. E., Kedersha N. L., Gill G., and Howley P. M. (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 6, 409–419 [DOI] [PubMed] [Google Scholar]
- 32. Hein M. Y., Hubner N. C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I. A., Weisswange I., Mansfeld J., Buchholz F., Hyman A. A., and Mann M. (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723 [DOI] [PubMed] [Google Scholar]
- 33. Mishra A., Godavarthi S. K., Maheshwari M., Goswami A., and Jana N. R. (2009) The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 284, 10537–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee S. Y., Ramirez J., Franco M., Lectez B., Gonzalez M., Barrio R., and Mayor U. (2014) Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell. Mol. Life Sci. 71, 2747–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Al-Shami A., Jhaver K. G., Vogel P., Wilkins C., Humphries J., Davis J. J., Xu N., Potter D. G., Gerhardt B., Mullinax R., Shirley C. R., Anderson S. J., and Oravecz T. (2010) Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One 5, e13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Puram S. V., Kim A. H., Park H. Y., Anckar J., and Bonni A. (2013) The ubiquitin receptor S5a/Rpn10 links centrosomal proteasomes with dendrite development in the mammalian brain. Cell Rep. 4, 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li V. S., Ng S. S., Boersema P. J., Low T. Y., Karthaus W. R., Gerlach J. P., Mohammed S., Heck A. J., Maurice M. M., Mahmoudi T., and Clevers H. (2012) Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell 149, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 38. Seeling J. M., Miller J. R., Gil R., Moon R. T., White R., and Virshup D. M. (1999) Regulation of β-catenin signaling by the B56 subunit of protein phosphatase 2A. Science 283, 2089–2091 [DOI] [PubMed] [Google Scholar]
- 39. Madan B., Walker M. P., Young R., Quick L., Orgel K. A., Ryan M., Gupta P., Henrich I. C., Ferrer M., Marine S., Roberts B. S., Arthur W. T., Berndt J. D., Oliveira A. M., Moon R. T., et al. (2016) USP6 oncogene promotes Wnt signaling by deubiquitylating Frizzleds. Proc. Natl. Acad. Sci. U.S.A. 113, E2945–E2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sominsky S., Kuslansky Y., Shapiro B., Jackman A., Haupt Y., Rosin-Arbesfeld R., and Sherman L. (2014) HPV16 E6 and E6AP differentially cooperate to stimulate or augment Wnt signaling. Virology 468–470, 510–523 [DOI] [PubMed] [Google Scholar]
- 41. Lichtig H., Gilboa D. A., Jackman A., Gonen P., Levav-Cohen Y., Haupt Y., and Sherman L. (2010) HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology 396, 47–58 [DOI] [PubMed] [Google Scholar]
- 42. Kuslansky Y., Sominsky S., Jackman A., Gamell C., Monahan B. J., Haupt Y., Rosin-Arbesfeld R., and Sherman L. (2016) Ubiquitin ligase E6AP mediates nonproteolytic polyubiquitylation of β-catenin independent of the E6 oncoprotein. J. Gen. Virol. 97, 3313–3330 [DOI] [PubMed] [Google Scholar]
- 43. Major M. B., Camp N. D., Berndt J. D., Yi X., Goldenberg S. J., Hubbert C., Biechele T. L., Gingras A. C., Zheng N., Maccoss M. J., Angers S., and Moon R. T. (2007) Wilms tumor suppressor WTX negatively regulates WNT/β-catenin signaling. Science 316, 1043–1046 [DOI] [PubMed] [Google Scholar]
- 44. Huang X., Luan B., Wu J., and Shi Y. (2016) An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 23, 778–785 [DOI] [PubMed] [Google Scholar]
- 45. Xu G., Paige J. S., and Jaffrey S. R. (2010) Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 28, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 47. Iossifov I., O'Roak B. J., Sanders S. J., Ronemus M., Krumm N., Levy D., Stessman H. A., Witherspoon K. T., Vives L., Patterson K. E., Smith J. D., Paeper B., Nickerson D. A., Dea J., Dong S., et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Elsasser S., Gali R. R., Schwickart M., Larsen C. N., Leggett D. S., Müller B., Feng M. T., Tübing F., Dittmar G. A., and Finley D. (2002) Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat. Cell Biol. 4, 725–730 [DOI] [PubMed] [Google Scholar]
- 49. Kumar S., Talis A. L., and Howley P. M. (1999) Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 274, 18785–18792 [DOI] [PubMed] [Google Scholar]
- 50. Kühne C., and Banks L. (1998) E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J. Biol. Chem. 273, 34302–34309 [DOI] [PubMed] [Google Scholar]
- 51. Havugimana P. C., Hart G. T., Nepusz T., Yang H., Turinsky A. L., Li Z., Wang P. I., Boutz D. R., Fong V., Phanse S., Babu M., Craig S. A., Hu P., Wan C., Vlasblom J., et al. (2012) A census of human soluble protein complexes. Cell 150, 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dantuma N. P., Lindsten K., Glas R., Jellne M., and Masucci M. G. (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 18, 538–543 [DOI] [PubMed] [Google Scholar]
- 53. Urraca N., Cleary J., Brewer V., Pivnick E. K., McVicar K., Thibert R. L., Schanen N. C., Esmer C., Lamport D., and Reiter L. T. (2013) The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 6, 268–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kühnle S., Mothes B., Matentzoglu K., and Scheffner M. (2013) Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc. Natl. Acad. Sci. U.S.A. 110, 8888–8893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Christensen J., Grønborg T. K., Sørensen M. J., Schendel D., Parner E. T., Pedersen L. H., and Vestergaard M. (2013) Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309, 1696–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wiltse J. (2005) Mode of action: inhibition of histone deacetylase, altering WNT-dependent gene expression, and regulation of β-catenin—developmental effects of valproic acid. Crit. Rev. Toxicol. 35, 727–738 [DOI] [PubMed] [Google Scholar]
- 57. Chenn A., and Walsh C. A. (2003) Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in β-catenin overexpressing transgenic mice. Cereb. Cortex 13, 599–606 [DOI] [PubMed] [Google Scholar]
- 58. Kim W. Y., Wang X., Wu Y., Doble B. W., Patel S., Woodgett J. R., and Snider W. D. (2009) GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sokol S. Y. (2011) Maintaining embryonic stem cell pluripotency with Wnt signaling. Development 138, 4341–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lien W. H., and Fuchs E. (2014) Wnt some lose some: transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 28, 1517–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Judson M. C., Sosa-Pagan J. O., Del Cid W. A., Han J. E., and Philpot B. D. (2014) Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol. 522, 1874–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ferrer-Vaquer A., Piliszek A., Tian G., Aho R. J., Dufort D., and Hadjantonakis A. K. (2010) A sensitive and bright single-cell resolution live imaging reporter of Wnt/β-catenin signaling in the mouse. BMC Dev. Biol. 10, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mutch C. A., Schulte J. D., Olson E., and Chenn A. (2010) β-Catenin signaling negatively regulates intermediate progenitor population numbers in the developing cortex. PLoS One 5, e12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim H., Vick P., Hedtke J., Ploper D., and De Robertis E. M. (2015) Wnt Signaling translocates Lys48-linked polyubiquitinated proteins to the lysosomal pathway. Cell Rep. 11, 1151–1159 [DOI] [PubMed] [Google Scholar]
- 65. Bandyopadhyay S., Chiang C. Y., Srivastava J., Gersten M., White S., Bell R., Kurschner C., Martin C., Smoot M., Sahasrabudhe S., Barber D. L., Chanda S. K., and Ideker T. (2010) A human MAP kinase interactome. Nat. Methods 7, 801–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Walker M. P., Stopford C. M., Cederlund M., Fang F., Jahn C., Rabinowitz A. D., Goldfarb D., Graham D. M., Yan F., Deal A. M., Fedoriw Y., Richards K. L., Davis I. J., Weidinger G., Damania B., et al. (2015) FOXP1 potentiates Wnt/β-catenin signaling in diffuse large B cell lymphoma. Sci. Signal. 8, ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mabb A. M., Je H. S., Wall M. J., Robinson C. G., Larsen R. S., Qiang Y., Corrêa S. A., and Ehlers M. D. (2014) Triad3A regulates synaptic strength by ubiquitination of Arc. Neuron 82, 1299–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Willert K., Brown J. D., Danenberg E., Duncan A. W., Weissman I. L., Reya T., Yates J. R. 3rd, and Nusse R. (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448–452 [DOI] [PubMed] [Google Scholar]
- 69. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.