

Abstract
Copper is an essential micronutrient for both pathogens and the animal hosts they infect. However, copper can also be toxic in cells due to its redox properties and ability to disrupt active sites of metalloproteins, such as Fe-S enzymes. Through these toxic properties, copper is an effective antimicrobial agent and an emerging concept in innate immunity is that the animal host intentionally exploits copper toxicity in antimicrobial weaponry. In particular, macrophages can attack invading microbes with high copper and this metal is also elevated at sites of lung infection. In addition, copper levels in serum rise during infection with a wide array of pathogens. To defend against this toxic copper, the microbial intruder is equipped with a battery of copper detoxification defenses that promote survival in the host, including copper exporting ATPases and copper binding metallothioneins. However, it is important to remember that copper is also an essential nutrient for microbial pathogens and serves as important cofactor for enzymes such as cytochrome c oxidase for respiration, superoxide dismutase for anti-oxidant defense and multi-copper oxidases that act on metals and organic substrates. We therefore posit that the animal host can also thwart pathogen growth by limiting their copper nutrients, similar to the well-documented nutritional immunity effects for starving microbes of essential zinc, manganese and iron micronutrients. This review provides both sides of the copper story and evaluates how the host can exploit either copper-the-toxin or copper-the-nutrient in antimicrobial tactics at the host-pathogen battleground.
Keywords: Copper, immunity, infection, metallothioneins, CTR1
Introduction
Copper serves as an essential micronutrient for virtually all forms of life. As a redox active metal, copper is the ideal cofactor for enzymes involved in electron transfer and oxygen chemistry and at least 30 metalloproteins have been classified as cuproenzymes [1]. Functions range from photosynthesis (plastocyanin) to respiration (cytochrome c oxidase; COX) to free radical detoxification (superoxide dismutases; SOD). Cuproenzymes are involved in oxidizing metals and organic substrates and produce a wide array of metabolites, neuropeptides, pigments and many other biologically active compounds [1]. However, the same redox properties that make copper an excellent enzymatic cofactor also make this element potentially deleterious. It has long been thought that cuprous copper can react with hydrogen peroxide in Fenton-like chemistry to produce the extremely reactive hydroxyl radical. More recent studies have shown that the reaction of cuprous copper with hydrogen peroxide produces a high oxidation Cu(III) product that may damage cellular components [2]. Even in the absence of hydrogen peroxide, copper can displace iron in Fe-S clusters, inactivating various essential Fe-S containing enzymes [3]. Given the potentially detrimental effects of this essential metal ion, maintaining copper homeostasis is crucial.
Humans are particularly skilled at controlling copper physiology and a number of homeostatic mechanisms ensure sufficient copper acquisition to meet dietary needs whilst minimizing side effects of copper overload. Instances of dietary copper starvation or copper toxicity are rare in humans except with cases of genetic disorders of copper metabolism. These disorders can result in copper overload, as in Wilson’s disease, or in copper deficiency, as in the fatal Menkes disease. Many excellent reviews have been written on the topic of mammalian copper homeostasis [4–9]. In contrast to mammals, microbial pathogens face key challenges when it comes to the bioavailability of copper as well as other essential and toxic metals.
In addition to copper, microbes require iron, manganese and zinc to activate a wide assortment of metalloenzymes and the animal host takes advantage of the microbial addiction to these metals through a tactic known as “nutritional immunity”, a host response designed to starve pathogens of essential metals [10, 11]. There is a system wide shut-down of available iron [10, 12–15] and manganese and zinc (as possibly iron) are also withheld at local sites of infection through extracellular S100 metal binding proteins, such as calprotectin, that bind these metals with nanomolar affinity [16, 17]. However, there have been no similar reports of nutritional immunity for copper, and in fact the opposite appears true. Rather than copper starvation, the current dogma is that the animal host elevates copper during infection to attack microbes with copper toxicity. A large number of reviews have been written on the topic of toxic copper as an antimicrobial weapon during infection [18–25]. We posit that the situation is far more complex and that depending on the niche in the host, copper can become either toxic or very limiting for the microbial intruder. This yin and yang of copper at the host-pathogen interface is the topic of this review.
The heightened copper response of the host during infection
Copper toxicity as an effective biocide
Dating back to the time of ancient Egyptian and Roman civilizations, copper containing compounds have been used as antimicrobials [26]. Currently, copper alloy surfaces are being utilized in hospitals to prevent hospital-transmitted infections [27–29] and copper-based compounds are being developed to treat human fungal infections [30, 31] as well as to protect crops against the damage of both fungal and bacterial pathogens [32]. These copper containing compounds are effective either through redox properties involving reactive oxygen species (ROS) or through copper mediated disruption of Fe-S clusters as described above [3, 20, 21]. In addition to these commercial uses of copper antimicrobials, animals also have built-in mechanisms for exploiting copper as a weapon to fight microbes. Here, we describe three sites within the animal host where copper is elevated in response to invading microbes.
The macrophage copper burst
When macrophages encounter pathogens, they can engulf and contain the microbe within a compartment known as the phagolysosome. This extremely hostile environment contains high ROS and reactive nitrogen, low pH and proteases, which are all designed to kill the microbe. With certain infectious agents, the phagolysosome can also accumulate high copper which, together with phagolysosomal ROS, can attack microbes through Fenton chemistry or through other pathways as described above. The high copper of macrophage phagolysosomes was first described for infection with Mycobacterium species including the M. tuberculosis pathogen for tuberculosis [33]. Since then, macrophage copper has been shown to be important in killing E. coli [34] and inducing copper toxicity stress for Salmonella [35, 36]. Even fungi such as Candida albicans show symptoms of high copper exposure during encounters with macrophages [37]. The mechanism by which macrophages accumulate this high copper is believed to involve a combination of increasing copper uptake by the high affinity copper transporter CTR1 and by activating the copper ATPase ATP7A, which can directly pump copper into the phagolysosome [34, 36, 38].
The heightened copper response in the lung – macrophages at work?
Studies conducted with three very different pathogens have provided evidence for elevated copper in the lung during microbial invasion. Infection with M. tuberculosis produces granulomas in animal lungs with significantly higher copper than the surrounding tissue, and copper detoxification mechanisms including a putative copper channel and copper transcriptional sensors in M. tuberculosis are essential for lung pathogenesis [39–41]. With Streptococcus pneumoniae, a leading cause of pneumonia, bacterial colonization of the lung was greatly diminished in S. pneumoniae mutants unable to export copper by the CopA copper transporting ATPase [42]. Even the human fungal pathogen Cryptococcus neoformans shows strong markers of fungal copper toxicity stress as this microbe invades the lung, including the induction of copper chelating metallothioneins (MT) for copper detoxification and lung pathogenesis [38]. Collectively, these studies point to elevated copper as antimicrobial armament for the lung as illustrated in Fig. 1. One likely source of this lung copper is the macrophage as described above. Pulmonary macrophages are a major constituent of tuberculosis granulomas and are known to densely populate sites of C. neoformans and S. pneumoniae infections in the lung. In recent experiments by Johnson et al, the copper toxicity stress of S. pneumoniae in the lung was ameliorated upon depletion of pulmonary macrophages [42], strongly implicating macrophages in this heightened copper response. As described above, the mechanism for increased copper in pulmonary macrophages may involve a combination of increasing copper uptake by the macrophage CTR1 copper permease and activation of the copper ATPase ATP7A in the phagolysosome [34, 36, 38]. The source of copper for pulmonary macrophages is unclear but may include extracellular cuproproteins (e.g., ceruloplasmin, see below) or non-proteinaceous copper binding ligands that are taken up by macrophages.
Fig. 1. Sites of copper excess or copper limitation for invading microbes.
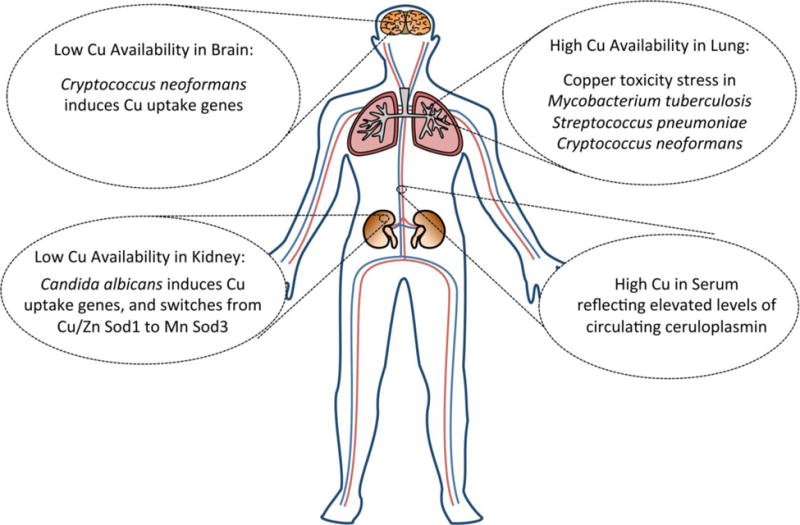
Illustrated are four sites in the animal host where copper is either elevated during infection or where the metal can become limiting for invading microbes. C. neoformans infection of the brain is associated with strong fungal markers of copper limitation stress including the activation of fungal copper uptake genes CTR1 and CTR4. As evidence for high copper in the lung, pulmonary pathogens with defects in copper detoxification pathways are poorly virulent, including M. tuberculosis mutants defective in copper sensing and copper transport [39–41], P. pneumonia mutants lacking the CopA copper exporting ATPase [42] and C. neoformans mutants lacking copper chelating MTs [38]. Invasion of the kidney with the fungal pathogen C. albicans stimulates a drop in total kidney copper during late stages of infection and the yeast responds by inducing copper uptake through fungal CTR1 and by switching from expressing Cu/Zn Sod1 to Mn Sod3 [45]. In serum, levels of the cuproprotein ceruloplasmin progressively rise during infection, causing a marked elevation in total serum copper. Ceruloplasmin is a multi-copper oxidase involved in iron homeostasis [38, 43–46, 53, 54].
Elevated serum copper and ceruloplasmin
A common hallmark of infection irrespective of the agent (viral, bacterial, fungal) is a marked and progressive rise in serum copper [38, 43–46]. This elevation in copper may all be attributed to a single source, namely the cuproprotein ceruloplasmin as illustrated in Fig. 1.
Ceruloplasmin is a multicopper oxidase [47] that is secreted from the liver and accounts for 95% of the copper content of the serum [48]. Its main physiological role is to oxidize Fe2+ to Fe3+, promoting both Fe mobilization from tissues and loading of ferric iron into serum transferrin [49–51]. Interestingly, ceruloplasmin is also an acute phase protein induced in response to inflammation, trauma, or infection [52], and levels of this protein are induced during infection with bacteria [44], viruses [53] and protozoans [54]. Since one molecule of ceruloplasmin binds 6 atoms of copper, even a modest increase in ceruloplasmin during infection can account for a substantial elevation in serum copper.
What is the purpose of elevating this multi-copper oxidase during infection? One possibility may be that ceruloplasmin helps deliver copper to sites of infection for attacking pathogens with copper toxicity. It has been suggested that certain tissues as well as lymphocytes contain cell surface receptors for ceruloplasmin [55, 56]. However, copper binds to ceruloplasmin with extraordinarily high affinity [57], and studies of mammalian ceruloplasmin deficiency have established a role for this cuproprotein in the homeostasis of iron, not copper [50, 51]. As a ferroxidase, ceruloplasmin helps mobilize iron from tissues and could therefore help starve invading pathogens of their essential iron nutrients as part of the nutritional immunity response. Thus, we posit that the marked elevation in serum copper during infection is not part of the toxic copper armament of the host, but rather reflects the production of ceruloplasmin for the purpose of depleting tissue iron. It is possible that the toxic copper response of host immunity is a very specialized case involving the macrophage phagolysosome.
Copper limitation during infection
While the role of copper toxicity in innate immunity is widely accepted, the less appreciated side of copper involves its role as a micronutrient for host and pathogen alike. Should copper become limiting during infection, a tug of war for this essential element could ensue between animal and microbe.
Copper as a nutrient for bacterial versus eukaryotic pathogens
When considering copper as a nutrient for infectious microbes, it is important to distinguish bacterial and eukaryotic pathogens. Bacteria for the most part seem very adverse to copper and go to great lengths to avoid any copper accumulation in the intracellular/cytoplasmic compartment. Virtually all the copper taken up by a typical Gram negative or Gram positive bacterial cell is actively exported by copper transporting ATPases into the periplasmic/extracellular space [58, 59]. Accordingly, bacterial cuproenzymes are generally extracellular/periplasmic, not cytoplasmic, including Cu/Zn SOD, COX and multi-copper oxidases [60, 61]. By comparison, eukaryotes have evolved with an extensive requirement for copper. Cuproenzymes can be found in practically every location inside and out of the cell and even extracellular cuproenzymes acquire their copper inside the cell via the secretory pathway. As a result, eukaryotes are equipped with sophisticated systems for acquiring copper and using copper chaperones to deliver the metal to various locations, including the mitochondria to activate COX, the secretory pathway to activate extracellular cuproenzymes such as Cu/Zn SOD and ceruloplasmin, and to sites in the cytosol for activating a cytosolic Cu/Zn SOD [62–69]. With such a thirst for copper, eukaryotic cells should be vulnerable to copper limitation. Nevertheless, eukaryotic pathogens have evolved to withstand droughts and swells of copper availability by inducing appropriate homeostatic responses. This is best understood with fungi.
Pathogenic fungi are designed to survive extreme highs and lows of copper
The molecular mechanisms by which fungi respond to copper were originally uncovered in the baker’s yeast Saccharomyces cerevisiae and many of the same principles apply to pathogenic fungi. This yeast corrects states of copper overload or copper deficiency by altering transcription of genes for copper homeostasis. The transcription factors that mediate these responses directly bind copper through cuprous-thiolate clusters [70] and therefore act as both sensors for copper and as transcriptional regulators. Two distinct copper regulators sense copper in bakers’ yeast: Cup2 (also known as Ace1) senses high copper and activates genes for MTs that chelate and detoxify copper [71], while copper deprivation is sensed by the Mac1 regulator that induces genes for copper uptake, including the fungal copper transporter gene CTR1 [72, 73] (Fig. 2).
Fig. 2. Fungal pathways for sensing and responding to Cu.
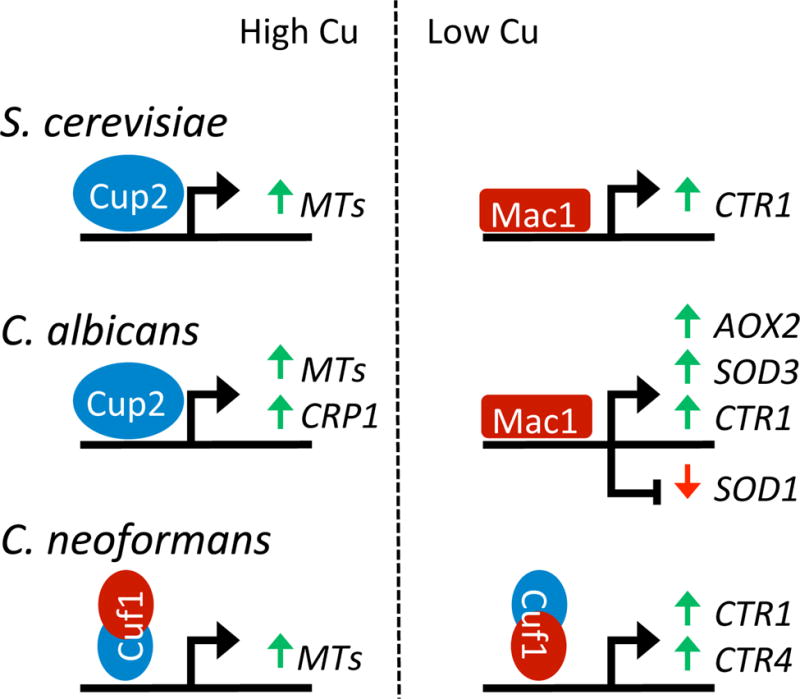
In pathogenic (C. neoformans and C. albicans) as well as non-pathogenic yeast (S. cerevisiae), copper sensing is mediated through transcription factors that bind copper through cuprous-thiolate clusters [70]. Shown are the transcription factors and protein domains that sense high (blue) versus low copper (red) and the target genes they regulate in italics. Gene activation and repression are indicated by green and red arrows respectively. In bakers’ yeast S. cerevisiae, separate Cup2 and Mac1 sensors respond to high and low copper by activating expression of copper chelating MTs and the copper uptake transporter CTR1, respectively [70–73]. Variations on this theme are seen in pathogenic fungi, presumably to accommodate challenges in copper at the host-pathogen interface. The Cup2 and Mac1 regulons in C. albicans have been expanded to include a copper exporting ATPase Crp1 induced with high copper [74, 75] and the induction of non-copper alternatives for enzymes involved in mitochondrial respiration (AOX2) and cytosolic anti-oxidant protection (SOD3) during times of low copper [76, 77, 79]. C. albicans Mac1 can also repress the gene encoding Cu/Zn Sod1, helping to spare copper under cases of copper limitation [45]. In C. neoformans, the Cuf1 copper sensor appears as a hybrid copper regulator and contains Cup1-like copper binding features at the N-terminus for responding to high copper and Mac1-like features at the C-terminus for responding to low copper [80–83].
Like bakers’ yeast, C. albicans uses separate Mac1 and Cup2 systems for responding to copper, but the respective copper regulons are more extensive. With high Cu, C. albicans Cup2 not only induces MTs but also a cell surface copper exporting ATPase (Crp1) to extrude copper from the cell, analogous to the copper elimination response of bacteria [74, 75]. Additionally, the C. albicans Mac1 sensor for low copper induces copper uptake as well as genes that control iron metabolism and modulate utilization of copper as an enzymatic cofactor [76, 77]. Regarding the latter, C. albicans Mac1 induces non-copper alternatives for the cuproenzymes COX and Cu/Zn SOD. Most eukaryotes (including bakers’ yeast) rely wholly on COX for mitochondrial respiration and a cytosolic Cu/Zn Sod1 for antioxidant protection, but C. albicans can express non-copper alternatives for these including an iron-requiring alternative oxidase (Aox2) [78] and a cytosolic Mn-Sod3 [79]. AOX2 and SOD3 expression are both induced by Mac1, while the Cu/Zn SOD1 is repressed by Mac1 during copper starvation [45, 77] (Fig. 2). In fact, the SOD3/SOD1 ratio is an excellent indicator of fungal copper status. By inducing non-copper alternatives for cuproenzymes and by repressing Cu/Zn Sod1 as a major consumer of cellular Cu, C. albicans can efficiently spare copper during times of copper limitation [45].
Interestingly, the pulmonary fungal pathogen C. neoformans has only a single factor for sensing copper, namely Cuf1, and this regulator can respond to both high and low copper. The N-terminus of Cuf1 is similar to the high copper sensor Cup2 while the C-terminus is similar to the low copper sensor Mac1 [80, 81] (Fig. 2). With high copper, Cuf1 transcriptionally activates a pair of unusually large MTs with a heightened capacity for chelating copper [82]. During copper deprivation, this same factor activates several copper uptake genes including CTR1 and CTR4, enabling substantial copper influx [80, 82, 83] (Fig. 2).
The aforementioned fungal responses to high copper make sense based on the dogma for copper toxicity of host innate immunity. Yet if copper is always high in the host, then why have these fungi retained copper starvation responses? One might argue that these responses function under copper limitation conditions outside the animal host, e.g., when C. neoformans exists in avian guano or plants. However, the animal is the only natural habitat for many pathogenic fungi, including C. albicans, suggesting there must certainly be copper limitation conditions inside the host.
Symptoms of copper starvation during brain dissemination of C. neoformans
C. neoformans can infect humans through inhalation of spores and the lungs are the initial target of infection. In the lung, the host attacks C. neoformans with elevated copper presumably through pulmonary macrophages as described above [38]. In response, C. neoformans mounts a copper detoxification response by inducing MTs, and accordingly, mutants of C. neoformans lacking MTs are poorly infectious in the lung [38]. Over prolonged infections, C. neoformans can enter the blood stream and disseminate to the brain causing meningoencephalitis. In stark contrast to the lung, C. neoformans exhibits symptoms of copper deprivation in the brain and induces fungal copper uptake transporters, but not the MTs [80, 84]. Thus C. neoformans encounters environments of both high and low copper bioavailability during infection and can adapt accordingly using its copper stress responses involving Cuf1 [21].
Copper starvation stress for Candida albicans in the kidney
C. albicans can thrive in many niches inside an animal host, but when it disseminates through the blood stream the major target tissue is the kidney. We investigated whether this pathogenic yeast encounters states of high or low copper in the kidney using the ratio of fungal Mn Sod3 to Cu/Zn Sod1 as an indicator, as described above. We observed that with early stages of kidney infection, C. albicans solely expressed Cu/Zn Sod1 and not Mn Sod3, indicative of abundant copper availability for the yeast. However, as infection progressed, the yeast shut down its production of the copper-enzyme and switched to expressing the non-copper alternative Mn-Sod3 [45]. This appearance of Mn-Sod3 was accompanied by induction of the fungal copper transport gene CTR1, a hallmark of C. albicans copper deficiency [45, 77]. Moreover, the switch from Cu/Zn Sod1 to Mn-Sod3 tracked temporarily with key changes in kidney copper. Kidney copper was initially high at early stages of infection, consistent with fungal expression of Cu/Zn Sod1, but as infection progressed, kidney copper levels dropped and C. albicans switched to expressing Mn Sod3 [45]. Thus, at least in the kidney, the host response is to limit copper for C. albicans, similar to the situation described above for C. neoformans invasion of brain [80, 84]. Despite these copper limiting environments of the animal host, the copper starvation stress responses in pathogenic fungi allows these microbes to thrive.
Is there a nutritional immunity response for copper?
The aforementioned studies with C. neoformans and C. albicans demonstrate that copper can be limiting in certain niches of the animal host, but is this a bonafide nutritional immunity response, similar to that described for iron, manganese and zinc? In other words, does the host deliberately withhold copper from the invading pathogen and if so, what are the potential mechanisms? Currently nothing is known about pathways for host withholding of copper during infection, but some proposals have been put forth. For example, it has been postulated that the brain can sequester copper from invading C. neoformans using either mammalian copper binding MTs or neuronal copper transporting ATPases that pump copper away from the microbe intruder [21]. The same could be true for the kidney during C. albicans infection. As another possibility, copper may be withheld from microbial pathogens using metal binding extracellular S100 proteins, similar to how the S100 protein calprotectin withholds manganese and zinc during nutritional immunity [11]. In fact certain metal binding S100 proteins have the ability to coordinate copper [85, 86] and may be involved in host withholding of copper. The precise mechanisms underlying host limitation for copper are to be determined.
Looking to the future
The findings summarized here likely represent the tip-of-the iceberg of copper effects at the host-pathogen interface. One size will not fit all pathogens when it comes to copper and depending on the site and course of infection, the invading microbe may be subject to either toxic copper overload or deprivation of this essential nutrient. It is important to note that the examples of copper limitation provided here are with fungal pathogens, yet eukaryotic parasitic microbes may also be vulnerable to copper limitation inside the host, as has been reported for Plasmodium falciparum [87]. Lastly, it is important to remember that although bacterial cells are classically known for their copper avoidance behavior, they still require this metal to activate cuproenzymes in the periplasmic or extracellular environment such as COX for respiration, Cu/Zn SOD to remove superoxide free radicals and multi-copper oxidases for copper detoxification [25, 60, 61]. It is therefore conceivable that under certain conditions, the host may thwart bacterial growth and survival through copper limitation, not copper overload.
Acknowledgments
We thank Dr. Ryan Petersen for critical review of this manuscript. The preparation of this review was supported by NIH RO1 grants AI 119949 and GM 50016 to VCC. A. Besold is supported by T32 CA009110.
Abbreviations
- ROS
reactive oxygen species
- COX
cytochrome oxidase
- SOD
superoxide dismutase
- MT
metallothioneins
References
- 1.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pham AN, Xing GW, Miller CJ, Waite TD. J Catal. 2013;301:54–64. doi: 10.1016/j.jcat.2013.01.025. [DOI] [Google Scholar]
- 3.Macomber L, Imlay JA. Proc Natl Acad Sci U S A. 2009;106:8344–8349. doi: 10.1073/pnas.0812808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barry AN, Shinde U, Lutsenko S. J Biol Inorg Chem. 2010;15:47–59. doi: 10.1007/s00775-009-0595-4. [DOI] [PubMed] [Google Scholar]
- 5.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. DOI 87/3/1011 [pii] 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan JH, Lutsenko S. J Biol Chem. 2009;284:25461–25465. doi: 10.1074/jbc.R109.031286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madsen E, Gitlin JD. Curr Opin Gastroenterol. 2007;23:187–192. doi: 10.1097/MOG.0b013e32801421bb. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki M, Gitlin JD. Pediatr Int. 1999;41:436–442. doi: 10.1046/j.1442-200x.1999.01090.x. [DOI] [PubMed] [Google Scholar]
- 9.Scheiber I, Dringen R, Mercer JF. Met Ions Life Sci. 2013;13:359–387. doi: 10.1007/978-94-007-7500-8_11. [DOI] [PubMed] [Google Scholar]
- 10.Weinberg ED. JAMA. 1975;231:39–41. doi: 10.1001/jama.231.1.39. [DOI] [PubMed] [Google Scholar]
- 11.Kehl-Fie TE, Skaar EP. Curr Opin Chem Biol. 2010;14:218–224. doi: 10.1016/j.cbpa.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cassat JE, Skaar EP. Cell Host Microbe. 2013;13:509–519. doi: 10.1016/j.chom.2013.04.010. S1931-3128 (13) 00152-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan J, Ward DM, De Domenico I. Int J Hematol. 2011;93:14–20. doi: 10.1007/s12185-010-0760-0. [DOI] [PubMed] [Google Scholar]
- 14.Correnti C, Strong RK. J Biol Chem. 2012;287:13524–13531. doi: 10.1074/jbc.R111.311829. DOI R111.311829 [pii] 10.1074/jbc.R111.311829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clifton MC, Corrent C, Strong RK. Biometals. 2009;22:557–564. doi: 10.1007/s10534-009-9207-6. [DOI] [PubMed] [Google Scholar]
- 16.Damo SM, Kehl-Fie TE, Sugitani N, Holt ME, Rathi S, Murphy WJ, Zhang Y, Betz C, Hench L, Fritz G, Skaar EP, Chazin WJ. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1220341110. DOI 1220341110 [pii] 10.1073/pnas.1220341110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakashige TG, Zhang B, Krebs C, Nolan EM. Nat Chem Biol. 2015;11:765–771. doi: 10.1038/nchembio.1891. DOI 10.1038/nchembio.1891 nchembio.1891 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stafford SL, Bokil NJ, Achard ME, Kapetanovic R, Schembri MA, McEwan AG, Sweet MJ. Biosci Rep. 2013;33 doi: 10.1042/BSR20130014. DOI 10.1042/BSR20130014 e00049 [pii] BSR20130014 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaturvedi KS, Henderson JP. Front Cell Infect Microbiol. 2014;4:3. doi: 10.3389/fcimb.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Festa RA, Thiele DJ. PLoS Pathog. 2012;8:e1002887. doi: 10.1371/journal.ppat.1002887. DOI 10.1371/journal.ppat.1002887 PPATHOGENS-D-12-01461[pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Santamarina S, Thiele DJ. J Biol Chem. 2015 doi: 10.1074/jbc.R115.649129. DOI jbc.R115.649129 [pii] R115.649129 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodgkinson VL, Petris MJ. J Biol Chem. 2012 doi: 10.1074/jbc.R111.316406. DOI R111.316406 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ladomersky E, Petris MJ. Metallomics. 2015;7:957–964. doi: 10.1039/c4mt00327f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samanovic MI, Ding C, Thiele DJ, Darwin KH. Cell Host Microbe. 2012;11:106–115. doi: 10.1016/j.chom.2012.01.009. DOI 10.1016/j.chom.2012.01.009 S1931-3128(12)00032-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu Y, Chang FM, Giedroc DP. Acc Chem Res. 2014 doi: 10.1021/ar500300n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dollwet HHA, Sorenson JRJ. Trace Elem Med. 1985;2:80–87. [Google Scholar]
- 27.Casey AL, Adams D, Karpanen TJ, Lambert PA, Cookson BD, Nightingale P, Miruszenko L, Shillam R, Christian P, Elliott TS. J Hosp Infect. 2010;74:72–77. doi: 10.1016/j.jhin.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 28.Michels HT, Keevil CW, Salgado CD, Schmidt MG. HERD. 2015;9:64–79. doi: 10.1177/1937586715592650. DOI 10.1177/1937586715592650 1937586715592650 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grass G, Rensing C, Solioz M. Appl Environ Microbiol. 2011;77:1541–1547. doi: 10.1128/AEM.02766-10. DOI 10.1128/AEM.02766-10 AEM.02766-10 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Festa RA, Helsel ME, Franz KJ, Thiele DJ. Chem Biol. 2014;21:977–987. doi: 10.1016/j.chembiol.2014.06.009. DOI 10.1016/j.chembiol.2014.06.009 S1074-5521(14)00214-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavet JS. Chem Biol. 2014;21:921–922. doi: 10.1016/j.chembiol.2014.07.011. DOI 10.1016/j.chembiol.2014.07.011 S1074-5521(14)00240-3[pii] [DOI] [PubMed] [Google Scholar]
- 32.Cha J, Cooksey SA. Proc Natl Acad Sci USA. 1991;88:8915–8519. doi: 10.1073/pnas.88.20.8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner D, Maser J, Lai B, Cai Z, Barry CE, 3rd, Honer Zu Bentrup K, Russell DG, Bermudez LE. J Immunol. 2005;174:1491–1500. doi: 10.4049/jimmunol.174.3.1491. [DOI] [PubMed] [Google Scholar]
- 34.White C, Lee J, Kambe T, Fritsche K, Petris MJ. J Biol Chem. 2009;284:33949–33956. doi: 10.1074/jbc.M109.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osman D, Waldron KJ, Denton H, Taylor CM, Grant AJ, Mastroeni P, Robinson NJ, Cavet JS. J Biol Chem. 2010;285:25259–25268. doi: 10.1074/jbc.M110.145953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Achard ME, Stafford SL, Bokil NJ, Chartres J, Bernhardt PV, Schembri MA, Sweet MJ, McEwan AG. Biochem J. 2012 doi: 10.1042/BJ20112180. DOI BJ20112180 [pii] 10.1042/BJ20112180. [DOI] [PubMed] [Google Scholar]
- 37.Douglas LM, Wang HX, Keppler-Ross S, Dean N, Konopka JB. MBio. 2012;3 doi: 10.1128/mBio.00254-11. DOI 10.1128/mBio.00254-11 e00254-11 [pii] mBio. 00254-11 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding C, Festa RA, Chen YL, Espart A, Palacios O, Espin J, Capdevila M, Atrian S, Heitman J, Thiele DJ. Cell Host Microbe. 2013;13:265–276. doi: 10.1016/j.chom.2013.02.002. DOI 10.1016/j.chom.2013.02.002 S1931-3128(13)00068-1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. Proc Natl Acad Sci U S A. 2011;108:1621–1626. doi: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darwin KH. J Biol Chem. 2015;290:18962–18966. doi: 10.1074/jbc.R115.640193. DOI 10.1074/jbc.R115.640193 R115.640193 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi X, Festa RA, Ioerger TR, Butler-Wu S, Sacchettini JC, Darwin KH, Samanovic MI. MBio. 2014;5 doi: 10.1128/mBio.00876-13. DOI 10.1128/mBio.00876-13 e00876-13 [pii] mBio. 00876-13 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson MD, Kehl-Fie TE, Klein R, Kelly J, Burnham C, Mann B, Rosch JW. Infect Immun. 2015;(83):1684–1694. doi: 10.1128/IAI.03015-14. DOI 10.1128/IAI.03015-14 IAI.03015-14 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ilback NG, Frisk P, Tallkvist J, Gadhasson IL, Blomberg J, Friman G. J Trace Elem Med Biol. 2008;22:120–130. doi: 10.1016/j.jtemb.2007.12.001. DOI 10.1016/j.jtemb.2007.12.001 S0946-672X(07)00173-3 [pii] [DOI] [PubMed] [Google Scholar]
- 44.Cernat RI, Mihaescu T, Vornicu M, Vione D, Olariu RI, Arsene C. Int J Tuberc Lung Dis. 2011;15:1239–1245. doi: 10.5588/ijtld.10.0445. [DOI] [PubMed] [Google Scholar]
- 45.Li CX, Gleason JE, Zhang SX, Bruno VM, Cormack BP, Culotta VC. Proc Natl Acad Sci U S A. 2015;112:E5336–5342. doi: 10.1073/pnas.1513447112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milanino R, Buchner V. Rev Environ Health. 2006;21:153–215. doi: 10.1515/reveh.2006.21.3.153. [DOI] [PubMed] [Google Scholar]
- 47.Kosman DJ. J Biol Inorg Chem. 2010;15:15–28. doi: 10.1007/s00775-009-0590-9. [DOI] [PubMed] [Google Scholar]
- 48.Hellman NE, Gitlin JD. Annu Rev Nutr. 2002;22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- 49.Roeser HP, Lee GR, Nacht S, Cartwright GE. J Clin Invest. 1970;49:2408–2417. doi: 10.1172/JCI106460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nittis T, Gitlin JD. Semin Hematol. 2002;39:282–289. doi: 10.1053/shem.2002.35633. DOI S0037196302500718 [pii] [DOI] [PubMed] [Google Scholar]
- 51.Harris ZL, Durley AP, Man TK, Gitlin JD. Proc Natl Acad Sci U S A. 1999;96:10812–10817. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eckersall PD, Saini PK, McComb C. Vet Immunol Immunopathol. 1996;51:377–385. doi: 10.1016/0165-2427(95)05527-4. [DOI] [PubMed] [Google Scholar]
- 53.Novikova I, Zlotnikova M. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2011;155:361–366. doi: 10.5507/bp.2011.051. [DOI] [PubMed] [Google Scholar]
- 54.Kocyigit A, Erel O, Gurel MS, Avci S, Aktepe N. Biol Trace Elem Res. 1998;65:271–281. doi: 10.1007/BF02789102. [DOI] [PubMed] [Google Scholar]
- 55.Lee SH, Lancey R, Montaser A, Madani N, Linder MC. Proc Soc Exp Biol Med. 1993;203:428–439. doi: 10.3181/00379727-203-43619. [DOI] [PubMed] [Google Scholar]
- 56.Kataoka M, Tavassoli M. Exp Hematol. 1985;13:806–810. [PubMed] [Google Scholar]
- 57.Hellman NE, Kono S, Mancini GM, Hoogeboom AJ, De Jong GJ, Gitlin JD. J Biol Chem. 2002;277:46632–46638. doi: 10.1074/jbc.M206246200. DOI 10.1074/jbc.M206246200 M206246200 [pii] [DOI] [PubMed] [Google Scholar]
- 58.Solioz M, Abicht HK, Mermod M, Mancini S. J Biol Inorg Chem. 2010;15:3–14. doi: 10.1007/s00775-009-0588-3. 10.1007/s00775-009-0588-3. [DOI] [PubMed] [Google Scholar]
- 59.Smith AT, Smith KP, Rosenzweig AC. J Biol Inorg Chem. 2014;19:947–960. doi: 10.1007/s00775-014-1129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rensing C, McDevitt SF. Metal Ions Life Sci. 2013;12:417–450. doi: 10.1007/978-94-007-5561-1_12. [DOI] [PubMed] [Google Scholar]
- 61.Nies DH, Herzberg M. Mol Microbiol. 2013;87:447–454. doi: 10.1111/mmi.12123. [DOI] [PubMed] [Google Scholar]
- 62.Harrison MD, Jones CE, Dameron CT. J Biol Inorg Chem. 1999;4:145–153. doi: 10.1007/s007750050297. [DOI] [PubMed] [Google Scholar]
- 63.O’Halloran TV, Culotta VC. J Biol Chem. 2000;275:25057–25060. doi: 10.1074/jbc.R000006200. [DOI] [PubMed] [Google Scholar]
- 64.Nevitt T, Ohrvik H, Thiele DJ. Biochim Biophys Acta. 2012;1823:1580–1593. doi: 10.1016/j.bbamcr.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lutsenko S. Curr Opin Chem Biol. 2010;14:211–217. doi: 10.1016/j.cbpa.2010.01.003. DOI 10.1016/j.cbpa.2010.01.003 S1367-5931(10)00002-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boal AK, Rosenzweig AC. Chem Rev. 2009;109:4760–4779. doi: 10.1021/cr900104z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luk E, Jensen LT, Culotta VC. J Biol Inorg Chem. 2003;8:803–809. doi: 10.1007/s00775-003-0482-3. [DOI] [PubMed] [Google Scholar]
- 68.Elam JS, Thomas ST, Holloway SP, Taylor AB, Hart PJ. Adv Protein Chem. 2002;60:151–219. doi: 10.1016/s0065-3233(02)60054-3. [DOI] [PubMed] [Google Scholar]
- 69.Robinson NJ, Winge DR. Annu Rev Biochem. 2010 doi: 10.1146/annurev-biochem-030409-143539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown KR, Keller GL, Pickering IJ, Harris HH, George GN, Winge DR. Biochemistry. 2002;41:6469–6476. doi: 10.1021/bi0160664. [DOI] [PubMed] [Google Scholar]
- 71.Palacios O, Atrian S, Capdevila M. J Biol Inorg Chem. 2011;16:991–1009. doi: 10.1007/s00775-011-0827-2. [DOI] [PubMed] [Google Scholar]
- 72.Keller G, Bird A, Winge DR. Eukaryot Cell. 2005;4:1863–1871. doi: 10.1128/EC.4.11.1863-1871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gross C, Kelleher M, Iyer VR, Brown PO, Winge DR. J Biol Chem. 2000;275:32310–32316. doi: 10.1074/jbc.M005946200. [DOI] [PubMed] [Google Scholar]
- 74.Weissman Z, Berdicevsky I, Cavari BZ, Kornitzer D. Proc Natl Acad Sci U S A. 2000;97:3520–3525. doi: 10.1073/pnas.97.7.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schwartz JA, Olarte KT, Michalek JL, Jandu GS, Michel SL, Bruno VM. Eukaryot Cell. 2013;12:954–961. doi: 10.1128/EC.00344-12. DOI 10.1128/EC.00344-12 EC.00344-12 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marvin ME, Mason RP, Cashmore AM. Microbiology. 2004;150:2197–2208. doi: 10.1099/mic.0.27004-0. DOI 10.1099/mic.0.27004-0 150/7/2197 [pii] [DOI] [PubMed] [Google Scholar]
- 77.Woodacre A, Mason RP, Jeeves RE, Cashmore AM. Microbiology. 2008;154:1502–1512. doi: 10.1099/mic.0.2007/013441-0. DOI 10.1099/mic.0.2007/013441-0 154/5/1502 [pii] [DOI] [PubMed] [Google Scholar]
- 78.Huh WK, Kang SO. Biochem J. 2001;356:595–604. doi: 10.1042/0264-6021:3560595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lamarre C, LeMay JD, Deslauriers N, Bourbonnais Y. J Biol Chem. 2001;276:43784–43791. doi: 10.1074/jbc.M108095200. [DOI] [PubMed] [Google Scholar]
- 80.Waterman SR, Hacham M, Hu G, Zhu X, Park YD, Shin S, Panepinto J, Valyi-Nagy T, Beam C, Husain S, Singh N, Williamson PR. J Clin Invest. 2007;117:794–802. doi: 10.1172/JCI30006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Waterman SR, Park YD, Raja M, Qiu J, Hammoud DA, O’Halloran TV, Williamson PR. MBio. 2012;3 doi: 10.1128/mBio.00285-12. DOI 10.1128/mBio.00285-12 e00285-12 [pii] mBio.00285-12 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ding C, Yin J, Tovar EM, Fitzpatrick DA, Higgins DG, Thiele DJ. Mol Microbiol. 2011;81:1560–1576. doi: 10.1111/j.1365-2958.2011.07794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raja MR, Waterman SR, Qiu J, Bleher R, Williamson PR, O’Halloran TV. Metallomics. 2013;5:363–371. doi: 10.1039/c3mt20220h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun TS, Ju X, Gao HL, Wang T, Thiele DJ, Li JY, Wang ZY, Ding C. Nat Commun. 2014;5:5550. doi: 10.1038/ncomms6550. DOI 10.1038/ncomms6550 ncomms6550 [pii] [DOI] [PubMed] [Google Scholar]
- 85.Moroz OV, Antson AA, Grist SJ, Maitland NJ, Dodson GG, Wilson KS, Lukanidin E, Bronstein IB. Acta Crystallogr D Biol Crystallogr. 2003;59:859–867. doi: 10.1107/s0907444903004700. DOI S0907444903004700 [pii] [DOI] [PubMed] [Google Scholar]
- 86.Kerkhoff C, Vogl T, Nacken W, Sopalla C, Sorg C. FEBS Lett. 1999;460:134–138. doi: 10.1016/s0014-5793(99)01322-8. DOI S0014-5793(99)01322-8 [pii] [DOI] [PubMed] [Google Scholar]
- 87.Asahi H, Tolba ME, Tanabe M, Sugano S, Abe K, Kawamoto F. BMC Microbiol. 2014;14:167. doi: 10.1186/1471-2180-14-167. DOI 10.1186/1471-2180-14-167 1471-2180-14-167 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]