

Abstract
Background
The duplication of genes can occur through various mechanisms and is thought to make a major contribution to the evolutionary diversification of organisms. There is increasing evidence for a large-scale duplication of genes in some chelicerate lineages including two rounds of whole genome duplication (WGD) in horseshoe crabs. To investigate this further, we sequenced and analyzed the genome of the common house spider Parasteatoda tepidariorum.
Results
We found pervasive duplication of both coding and non-coding genes in this spider, including two clusters of Hox genes. Analysis of synteny conservation across the P. tepidariorum genome suggests that there has been an ancient WGD in spiders. Comparison with the genomes of other chelicerates, including that of the newly sequenced bark scorpion Centruroides sculpturatus, suggests that this event occurred in the common ancestor of spiders and scorpions, and is probably independent of the WGDs in horseshoe crabs. Furthermore, characterization of the sequence and expression of the Hox paralogs in P. tepidariorum suggests that many have been subject to neo-functionalization and/or sub-functionalization since their duplication.
Conclusions
Our results reveal that spiders and scorpions are likely the descendants of a polyploid ancestor that lived more than 450 MYA. Given the extensive morphological diversity and ecological adaptations found among these animals, rivaling those of vertebrates, our study of the ancient WGD event in Arachnopulmonata provides a new comparative platform to explore common and divergent evolutionary outcomes of polyploidization events across eukaryotes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-017-0399-x) contains supplementary material, which is available to authorized users.
Keywords: Parasteatoda tepidariorum, Genome, Centruroides sculpturatus, Gene duplication, Evolution, Hox genes
Background
Gene duplication plays an important role in the evolutionary diversification of organisms [1, 2]. Unequal crossing-over commonly results in one or a few tandemly duplicated genes, but larger scale events, including whole genome duplications (WGDs) can also occur. Tandem duplication has been shown to underlie the evolution of many genes in both plants and animals, for example, of up to 32% of genes in the centipede Strigamia maritima [3, 4]. WGD is arguably the most sudden and massive change that a genome can experience in a single evolutionary event. The occurrence of WGDs across a wide variety of eukaryotic groups, including plants [5, 6], fungi [7, 8], ciliates [9], oomycetes [10], and animals [11–17], attests to the major impact that polyploidization events have had in reshaping the genomes of many different organisms.
Although most of the duplicated genes resulting from tandem duplication or WGD are subsequently lost, it is thought that these events provide new genetic material for some paralogous genes to undergo sub-functionalization or neo-functionalization and thus contribute to the rewiring of gene regulatory networks, morphological innovations and, ultimately, organismal diversification [2, 7, 18–24]. Comparisons of independent paleopolyploidization events across different eukaryotes, such as plants, yeast, and vertebrates [5, 8, 11, 13, 14, 24], have led to the development of models to elucidate genome-wide evolutionary patterns of differential gene loss and retention compared to smaller-scale events [2, 25]. However, the enormous differences between these disparate eukaryotic lineages in terms of genome structure, morphological and developmental organization, and ecology have impeded a critical assessment of the potential selective advantages and actual evolutionary consequences of WGDs. Thus, the extent to which WGDs may have contributed to taxonomic “explosions” and evolutionary novelties remains controversial, especially in the case of vertebrates [26–28]. For example, the two WGDs shared by all vertebrates have given rise to four clusters of Hox genes, providing new genetic material that may underlie the evolutionary success and innovations among these animals [24, 29, 30]. However, only three WGD events have been demonstrated in animals other than vertebrates, namely one in bdelloid rotifers and possibly two in horseshoe crabs [11, 14, 31], and these events are not associated with any bursts of diversification [32, 33]. It is clear, therefore, that documenting additional examples of WGD in metazoans would significantly increase our understanding of the genomic and morphological consequences of these events.
Intriguingly, there is increasing evidence for extensive gene duplication among chelicerates other than horseshoe crabs, particularly in spiders and scorpions [34–44], indicating that large-scale gene duplications occurred during the evolution of these arachnids. However, although the genomes of some arachnids have been sequenced, including the tick Ixodes scapularis [45, 46], the mite Tetranychus urticae [47], the Chinese scorpion Mesobuthus martensii [48], and three spiders (the velvet spider Stegodyphus mimosarum [49], the Brazilian whiteknee tarantula Acanthoscurria geniculata [49], and the golden orb-weaver Nephila clavipes [50]), a systematic analysis of genome evolution among these diverse animals has yet to be performed (Fig. 1) [51].
Fig. 1.
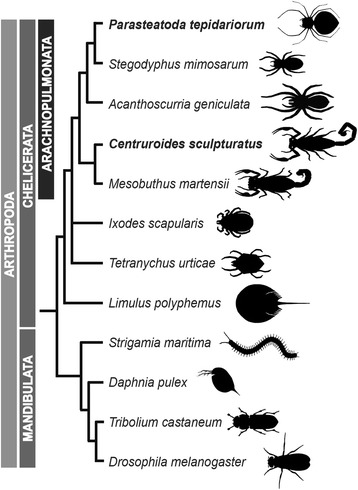
The relationships of Parasteatoda tepidariorum to select arthropods. Representatives of spiders (Araneae) with sequenced genomes (P. tepidariorum, Stegodyphus mimosarum, and Acanthoscurria geniculata) are shown with respect to other chelicerates with sequenced genomes including scorpions (Centruroides sculpturatus and Mesobuthus martensii), a tick (Ixodes scapularis), a mite (Tetranychus urticae), and a horseshoe crab (Limulus polyphemus) as well as representatives of Myriapoda (Strigamia maritima), Crustacea (Daphnia pulex), and Insecta (Drosophila melanogaster). Topology is based on Sharma et al. [53]
As a step towards this goal, we herein report the sequencing and analysis of the genomes of the common house spider Parasteatoda tepidariorum (C. L. Koch, 1841; formerly Achaearanea tepidariorum) [52] and the bark scorpion Centruroides sculpturatus (Wood, 1863) (Fig. 1), together with comparative genomic analyses of other available chelicerate genomes. We found that the genome of P. tepidariorum contains many paralogous genes, including two Hox gene clusters, which is also the case in other spiders and in scorpions (this work; [36]). These similar patterns of gene duplication between spiders and scorpions are consistent with recent molecular phylogenies, which support a much closer phylogenetic relationship of spiders and scorpions than previously thought, in a clade known collectively as Arachnopulmonata [53] (Fig. 1). We also document extensive divergence in the timing and location of expression of each pair of Hox gene paralogs, suggesting there may be far reaching functional consequences. Furthermore, an analysis of synteny among paralogs across the P. tepidariorum genome is consistent with a WGD. Comparison with other chelicerates suggests that this WGD took place in the common ancestor of the Arachnopulmonata and is probably independent of the WGDs in the horseshoe crab lineage.
Results
P. tepidariorum has many duplicated genes
The final P. tepidariorum genome assembly has a size of 1443.9 Mb. The number of predicted protein-coding genes in P. tepidariorum (27,990) is consistent with those of another spider, S. mimosarum (27,235) [49], as are the numbers of predicted genes of the two scorpions M. martensii (32,016) [48] and C. sculpturatus (30,456) (this study). Spiders and scorpions have significantly higher numbers of predicted genes than other arachnids such as the mite Tetranychus urticae (18,414) [47]. We evaluated the completeness of the P. tepidariorum gene set and assessed the extent of gene duplication using 1427 benchmarked universal single-copy ortholog (BUSCO) groups of arthropod genes [54], with input datasets ranging from 2806 (Strigamia maritima) to 3031 (Tribolium castaneum) putatively single-copy orthologs. For P. tepidariorum, the HMMER3 homology search revealed 91% complete single-copy orthologs (C), 41% complete duplicated orthologs (D), and 6.5% fragmented orthologs (F). Only 2% of conserved BUSCO groups from the universal ortholog arthropods database were missing (M) from the assembly. The number of duplicated orthologs was very high compared to Drosophila melanogaster (C: 99%, D: 3.7%, F: 0.2%, M: 0.0%, 13,918 genes in total) or Caenorhabditis elegans (C: 90%, D: 11%, F: 1.7%, M: 7.5%, 20,447 genes in total).
We then undertook a different approach to further investigate the extent of gene duplication, by estimating the ratios of orthologs in arachnopulmonate and non-arachnopulmonate genomes. Specifically, we compared the P. tepidariorum and C. sculpturatus genomes to the genomes of four other arthropods with a single Hox cluster and no evidence of large-scale gene duplication (“1X genomes”), including another chelicerate (the tick Ixodes scapularis) and three mandibulates (the red flour beetle T. castaneum, the crustacean Daphnia pulex, and the centipede S. maritima). The Orthologous Matrix (OMA) [55] algorithm was used to identify orthologs after pairwise mapping of genomes. The orthology mapping indicated that, depending upon the 1X genome used for comparison, between 7.5% and 20.5% of spider genes that could be mapped to a single mandibulate or tick ortholog had undergone duplication (Additional file 1: Table S1). Using the well-annotated T. castaneum genome as the reference, we found that 14.6% (523) of the P. tepidariorum genes with a single T. castaneum ortholog had undergone duplication (Additional file 1: Table S1). We obtained similar results when comparing the genome of the scorpion C. sculpturatus with that of T. castaneum (10.1%, 290 genes). However, only 4.9% (175) of I. scapularis genes had been duplicated since its divergence from T. castaneum (Additional file 1: Table S1). Moreover, higher numbers of 1:1 orthologs were found among 1X genomes than in comparisons that included either the spider or the scorpion genome, which is consistent with a greater degree of paralogy in the spider and scorpion genomes. The highest proportion of duplicated genes in a 1X genome, with reference to T. castaneum, was found in D. pulex (7.8%), which is known to have a large number of tandemly duplicated gene clusters [56] (Additional file 1: Table S1).
Most of the spider and scorpion duplicates occurred in 1:2 paralogy (i.e., two copies in spiders/scorpions for a given mandibulate or tick homolog) (Fig. 2, Additional file 1: Table S1), whereas duplicates in other arthropods showed no particular enrichment for this category. Two-copy duplicates accounted for 5.9–10.9% of the total spider duplicated genes, and 7.4–13.5% of the total scorpion duplicated genes (depending on the mandibulate or tick genome used for comparison). In both cases, these proportions were significantly higher than those of other arthropod genomes (P = 6.67 × 10–4) (Fig. 2a). Intriguingly, 11.8% of the two-copy duplicates were shared between spiders and scorpions. Inversely, comparing either P. tepidariorum or C. sculpturatus to mandibulate or tick genomes recovered a much lower proportion of single-copy orthologs (i.e., 1:1) relative to comparisons of any two species of mandibulate or tick. The number of duplicated genes was significantly higher in scorpions and spiders relative to comparing mandibulate or ticks among themselves, and particularly so for the 1:2 paralog bin (two-sample t-test; P = 3.75 × 10–4) (Fig. 2b, Additional file 1: Table S1). We found very similar profiles of paralog distributions using a more conservative approach comparing the spider and scorpion genes to a benchmarked set of 2806–3031 single-copy genes common to arthropods (the BUSCO-Ar database of the OrthoDB project) (Fig. 2c, d). Even within this database of genes with no reported cases of duplication in all other studied arthropods, a considerable fraction of genes was found in two copies in both the P. tepidariorum and C. sculpturatus genomes (63–78 genes) when compared to the mandibulate or tick datasets (Fig. 2c, d, Additional file 1: Table S1).
Fig. 2.
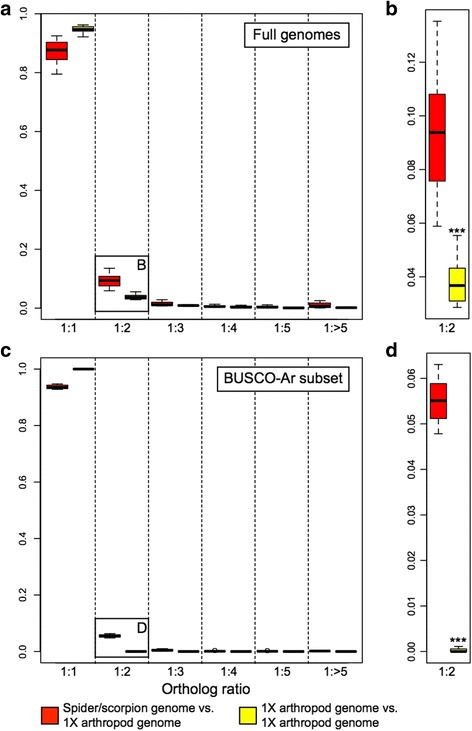
Orthology inference suggests substantial duplication in spiders and scorpions. a Distribution of orthology ratios from Orthologous Matrix analysis of full genomes. Comparisons of an arachnopulmonate genome to a 1X genome are shown in red and comparisons among 1X genomes are shown in yellow. A significantly higher number of 1:1 orthologs is recovered in pairwise comparisons within the non-arachnopulmonate genomes (P = 1.46 × 10–3). b Magnification of the 1:2 ortholog ratio category in (a) shows a significantly higher number of duplicated genes in comparisons of spider or scorpion genomes to a 1X genome (P = 6.67 × 10–4). c Distribution of orthology ratios for a subset of genes benchmarked as putatively single copy across Arthropoda (BUSCO-Ar). As before, a significantly higher number of 1:1 orthologs is recovered within the 1X genome group (P = 3.43 × 10–8). d Magnification of the 1:2 ortholog ratio category in (c) shows a significantly higher number of duplicated genes in spiders and scorpions (P = 7.28 × 10–9)
Dispersed and tandem gene duplicates abound in spiders and scorpions
We carried out systematic analysis of the frequency and synteny of duplicated genes in P. tepidariorum compared to C. sculpturatus and the horseshoe crab Limulus polyphemus. The genome of P. tepidariorum is characterized by an elevated number of tandem (3726 vs. 1717 and 2066 in C. sculpturatus and L. polyphemus, respectively) and proximal duplicates (2233 vs. 1114 and 97), i.e., consecutive duplicates and duplicates found at most 10 genes away from their paralog (Additional file 2: Figure S1, Additional file 3: Figure S2, Additional file 4: Figure S3). However, the most salient aspect in all three genomes was the very high number of dispersed duplicates, i.e., genes for which paralogous gene models were detected more than 10 genes apart or on different scaffolds, which amounted to approximately 14,700 genes in each species (Additional file 2: Figure S1, Additional file 3: Figure S2, Additional file 4: Figure S3).
To better understand the patterns of gene duplication in P. tepidariorum, we next investigated the duplication level and colinearity of specific coding and non-coding genes. We identified 80 homeobox gene families in P. tepidariorum (Additional file 5: Table S2) of which 58% were duplicated, giving a total of 145 genes (Fig. 3). Note that a very similar repertoire was also observed in C. sculpturatus, where 59% of homeobox gene families were duplicated (156 genes representing 82 gene families (Additional file 6: Table S3)). Of the 46 and 48 homeobox gene families with multiple gene copies in P. tepidariorum and C. sculpturatus, respectively, 38 were common to both species. In addition, 23 families were represented by a single gene in both the spider and scorpion genomes (Fig. 3). The few remaining families contained duplicates in only one of these two species or were only found in one species (Fig. 3). In addition, one family, Dmbx, had two copies in P. tepidariorum but was missing in C. sculpturatus.
Fig. 3.
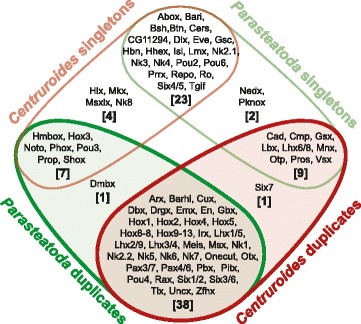
Homeobox-containing genes are frequently duplicated in P. tepidariorum and C. sculpturatus. Many duplicated homeobox gene families (overlap of red and green shading) are shared between P. tepidariorum (indicated in green) and C. sculpturatus (indicated in red). Single copy families are the next largest group shared, then families that are single copy in one species but duplicated in the other. There are also a few families that were only found in one species
The duplication of Hox gene clusters in vertebrates was among the first clues that led to the discovery of ancient WGDs in this group [13]. Therefore, we assessed the repertoire and organization of Hox genes in P. tepidariorum in comparison to three other spider genomes (L. hesperus, S. mimosarum, and A. geniculata [49]), two scorpion genomes (C. sculpturatus and M. martensii [48], this study), and the tick genome (I. scapularis [45, 46]).
We identified and manually annotated orthologs of all ten arthropod Hox gene classes (labial (lab), proboscipedia (pb), Hox3, Deformed (Dfd), Sex combs reduced (Scr), fushi tarazu (ftz), Antennapedia (Antp), Ultrabithorax (Ubx), abdominal-A (abdA), and Abdominal-B (AbdB)) in all genomes surveyed (Fig. 4, Additional file 7: Figure S4, Additional file 8: Figure S5, Additional file 9: Table S4). Whereas the tick genome contains only one copy of each Hox gene, nearly all Hox genes are found in two copies in the spider and scorpion genomes (Fig. 4, Additional file 8: Figure S5, Additional file 9: Table S4). The only Hox gene not found in duplicate is ftz in P. tepidariorum (Fig. 4, Additional file 8: Figure S5, Additional file 9: Table S4).
Fig. 4.
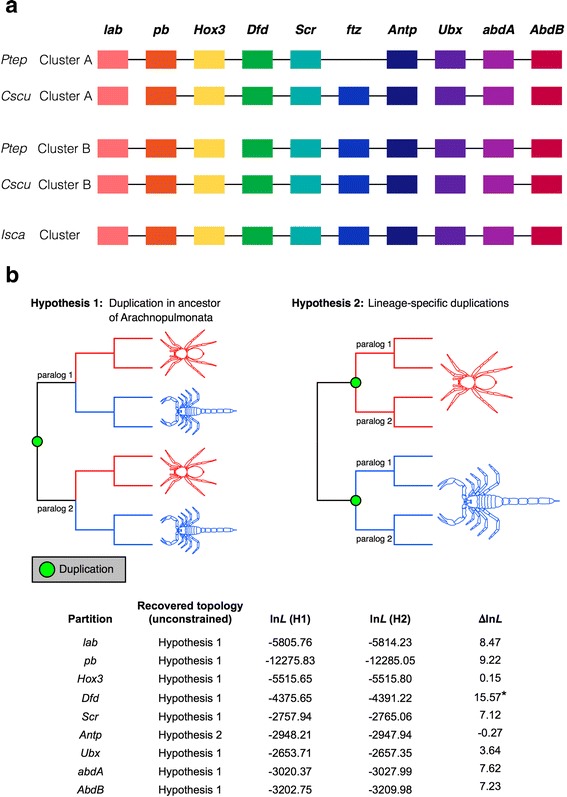
Hox gene complement and hypothetical Hox clusters in chelicerate genomes. Hox gene clusters in the spider Parasteatoda tepidariorum, the scorpion Centruroides sculpturatus, and in the tick (a). For details, see Additional file 9: Table S4. Transcription for all genes is in the reverse direction. Genes (or fragments thereof, see Additional file 9: Table S4) that are found on the same scaffold are joined by black horizontal lines. Abbreviations: Ptep Parasteatoda tepidariorum, Cscu Centruroides sculpturatus, Isca Ixodes scapularis. b Gene tree analysis of individual Hox genes support a shared duplication event in the common ancestor of spiders and scorpions in all cases except Antennapedia
Interestingly, none of the Hox paralogs present in spiders and scorpions were found as tandem duplicates. Instead, in P. tepidariorum, the species with the most complete assembly in this genomic region, it was clear that the entire Hox cluster had been duplicated. We found one P. tepidariorum Hox cluster copy in a single scaffold, lacking only a ftz copy, as is probably the case for this particular cluster (cluster A) in all spiders (Fig. 4, Additional file 8: Figure S5, Additional file 9: Table S4). The second Hox cluster (cluster B) was split between two scaffolds, which could be due to the incomplete assembly of this region due to there not being enough sequence downstream of Dfd (~70 kb) and upstream of Hox3 (~320 kb) to cover the paralogous ~840 kb between Dfd and Hox3 on Cluster A in P. tepidariorum or even the ~490 kb between Dfd and Hox3 in I. scapularis (Fig. 4, Additional file 8: Figure S5, Additional file 9: Table S4). Note that for clarity and to be consistent with the vertebrate nomenclature, we have named the P. tepidariorum Hox paralogs after the cluster that they are found in, for example, pb-A, pb-B, etc. (Additional file 8: Figure S5, Additional file 9: Table S4).
In addition to the Hox genes, the clusters also contained microRNAs, including a single copy of mir-10 in cluster B. Two copies of microRNAs iab4/8 were identified in both clusters, between abdA and AbdB (Additional file 8: Figure S5, Additional file 10: Table S5). Furthermore, mir-993b-1 was found in cluster B, but the other two P. tepidariorum mir-993 paralogs [44] were located in non-Hox containing scaffolds. In addition to these microRNAs, 98 other putative/predicted coding and non-coding genes were also found in the P. tepidariorum Hox clusters (Additional file 8: Figure S5, Additional file 10: Table S5). However, none of these other genes were present as duplicates in both clusters in the same syntenic arrangement.
It was also recently reported that approximately 36% of annotated microRNAs in P. tepidariorum are present as two or more copies [44]. Analysis of the synteny of the paralogous P. tepidariorum microRNAs shows that only 8 out of 30 are found on the same scaffold. Furthermore, nearly all of the tandemly duplicated microRNAs in P. tepidariorum are microRNAs largely specific to this spider (e.g., mir-3971 paralogs) or clustered in arthropods (e.g., mir-2 from the mir-71/mir-2 cluster) (Additional file 11: Table S6) [44]. These findings suggest that the majority of duplicated microRNAs were not generated by tandem duplication.
Comparative analyses suggest that other key developmental genes are also commonly duplicated in P. tepidariorum. A synteny analysis of these previously reported duplications showed that only the two Pax6 paralogs were located on the same scaffold (Additional file 12: Table S7), suggesting that they arose through tandem duplication. The paralogs of other duplicated developmental genes examined were found on different scaffolds (Additional file 12: Table S7), including retinal differentiation (dachshund and sine oculis), head patterning (six3, orthodenticle, collier) [57, 58], Wnt pathway genes (Wnt7, Wnt11, frizzled 4) [37, 59], and appendage formation genes (homothorax, extradenticle, Lim1, spineless, trachealess, and clawless) (Prpic et al., unpublished data).
Classification of duplicated genes in spiders and scorpions shows that tandem and especially dispersed duplications abound in these genomes. The observation that most of the duplicated genes are found on different scaffolds is suggestive of large-scale duplication, with the caveat that the scaffolds do not represent chromosomes, and therefore the frequency of tandem duplications could be underestimated. Taken together, these results, and the finding that the Hox cluster has also been duplicated, could be indicative of a WGD.
Conservation of synteny among P. tepidariorum scaffolds supports the hypothesis of a WGD event
To further test the hypothesis that a WGD event had occurred in an ancestor of P. tepidariorum, we next searched for conserved synteny among the genomic scaffolds of this spider using Satsuma [60] (note that this approach was not possible in C. sculpturatus because of the assembly quality of the genome of this scorpion). This analysis revealed signatures of large segmental duplications suggestive of a WGD followed by numerous rearrangements (inversions, translocations, tandem duplications) (Fig. 5a). These signatures were observed among many of the larger scaffolds (Fig. 5, Additional file 13: Figure S6), but were particularly strong and clear between scaffolds 1 and 7, between scaffolds 9 and 30, and among scaffolds 60, 78, and 103 (Fig. 5b). These results are comparable to findings from a similar analysis of the genome of the fish Tetraodon nigroviridis [17] and are consistent with an ancient WGD event in an ancestor of this spider.
Fig. 5.
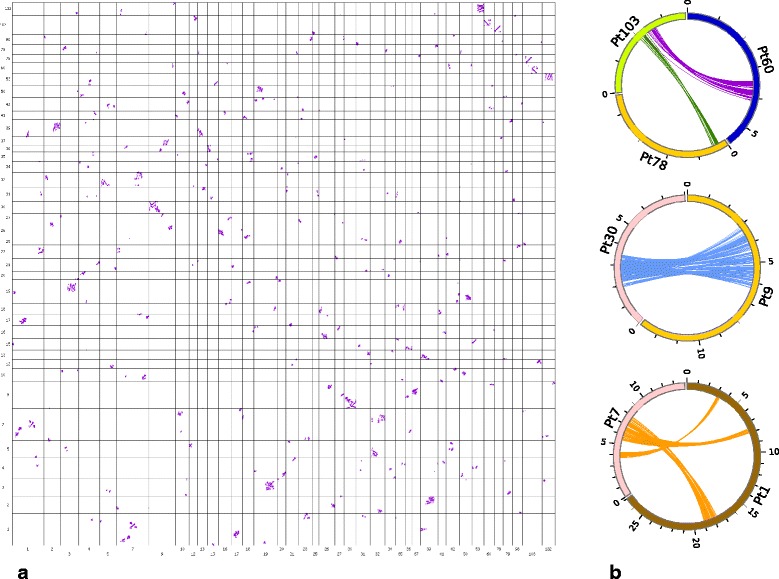
Genome-scale conservation of synteny among P. tepidariorum scaffolds reveals signatures of an ancient WGD. a Oxford grid displaying the colinearity detected by SatsumaSynteny among the 39 scaffolds presenting the greatest numbers of hits on one another. On this grid (not drawn to scale), each point represents a pair of identical or nearly identical 4096-bp regions. Alignments of points reveal large segmental duplications suggestive of a whole-genome duplication event along with other rearrangements such as inversions, translocations and tandem duplications. b Circos close-ups of some of the colinearity relationships revealed by the Oxford grid
When did WGD occur in chelicerates?
To determine the timing of duplication relative to species divergence within a broader taxonomic sampling of arachnids than analyzed thus far, we grouped the protein-coding genes of 30 arachnid species into gene families with either P. tepidariorum or C. sculpturatus translated genes used as a seed plus L. polyphemus and S. maritima as outgroups (Additional file 14: Table S8) [61]. This method resulted in 2734 unique P. tepidariorum-seeded gene families (Additional file 15: Figure S7). Note that seeding gene families with C. sculpturatus resulted in fewer families (1777) but similar patterns of gene duplication (not shown); we thus focused on the results of P. tepidariorum-seeded families.
To analyze the timing of the putative WGD event, we calculated molecular distances between paralog pairs by averaging the maximum likelihood branch lengths estimated under the HKY model of evolution [62] within gene trees from the duplication node to all descendant within-species paralogs. We fit the molecular distances of duplication nodes with HKY > 0.01 (avoid inferring alleles as paralogs) and HKY < 2.0 (minimize mutational saturation) to five distribution models. The results show that P. tepidariorum duplication nodes best fit three Gaussian distributions (four other distributions were rejected by the Kolmogorov–Smirnoff goodness-of-fit test, see Additional file 16: Table S9). The first Gaussian distribution, with an average genetic distance of μ = 0.038 likely represents recent individual gene duplications. The second (μ = 0.491) and third (μ = 1.301) distributions of genetic distance among paralogs are consistent with two ancient large-scale duplication events (Fig. 6a) [11, 63]. We observed a similar distribution of paralog molecular distances in five deeply sequenced spider species and C. sculpturatus (Additional file 17: Figure S8, Additional file 18: Table S10), but not T. urticae and I. scapularis. The shift in distribution patterns between the scorpion and the mite is consistent with a shared WGD in spiders and scorpions that was not experienced by the more distantly related arachnid species. It is also possible that spiders and scorpions experienced independent duplication events shortly after their divergence, but this is unlikely given the shared retention of paralogs from this analysis (see below) and from the BUSCO-Ar and OMA gene sets (see above).
Fig. 6.
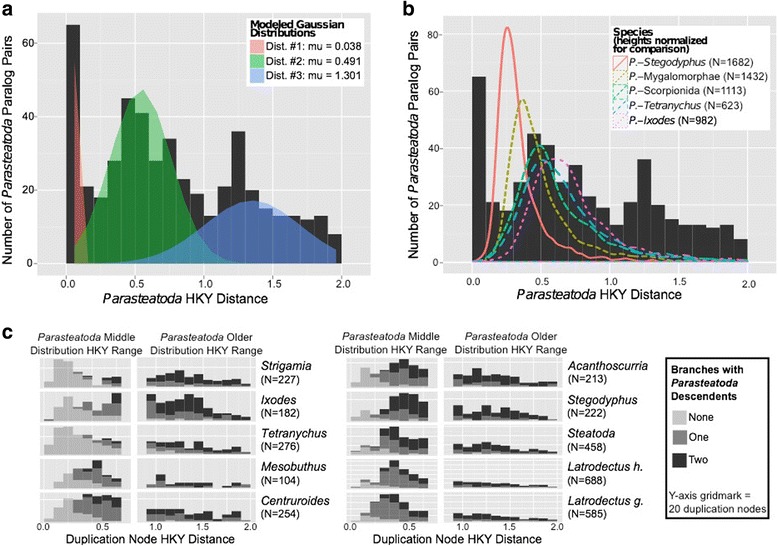
Molecular distance distributions of P. tepidariorum paralogs and speciation nodes. The distribution of mean HKY distances from P. tepidariorum duplication nodes to P. tepidariorum descendants reveals three distributions shown in different colors in (a). Comparing the distribution of HKY distances from speciation nodes to P. tepidariorum (lines in b) reveals that distribution #1 (red in a) is restricted to the P. tepidariorum branch, distribution #2 (green in a) is similar to pre-spider and post-tick speciation nodes, and distribution #3 (blue in a) is older than the P. tepidariorum-tick speciation event. N = number of speciation nodes in (b). Comparing the number of duplication nodes in non-P. tepidariorum species (c) that are either partially or fully retained in P. tepidariorum reveals that the duplication nodes with HYK distances in the range of the oldest P. tepidariorum distribution (blue in a) are retained at a similar rate across all species (right sub-columns in c), but that those duplication nodes with HKY distances in the range of the middle P. tepidariorum distribution (green in a) are only retained in scorpions or more closely related species (left sub-columns in c)
The possibility that a WGD occurred prior to the divergence of spiders and scorpions and after the divergence of spiders from mites is additionally supported by comparison of the distributions of HKY distances of the duplication nodes to speciation nodes, with an almost identical pattern found for the paralog distances and the spider–scorpion distances (Fig. 6b, Additional file 19: Figure S9, Additional file 20: Table S11). Shared paralog retention is also high for spiders and scorpions, but not between spiders and ticks or mites, further supporting a shared WGD in the spider and scorpion common ancestor (Fig. 6c, Additional file 21: Table S12). Furthermore, the tandem duplication nodes identified above formed the majority of the duplication nodes in the younger Gaussian distribution (71%), and minorities of the second (24%) and third distributions (9%) (Additional file 22: Figure S10). This is the opposite of what is seen with the duplication nodes containing dispersed duplications (younger: 29%, second: 62%, and third: 50%). Additionally, a slight majority of the older tandem duplication nodes showed evidence of being shared with other arachnids (57%), but mostly with other species in the same family as P. tepidariorum (44%). This suggests that an ancient WGD was followed by pervasive lineage-specific tandem duplications, especially in spiders.
Analysis of the gene families containing a duplication pair from the middle and oldest Gaussian distributions (Fig. 6a), excluding tandem duplicates, showed that they are enriched in several GO terms compared to gene families without duplication pairs, including several terms associated with transcription and metabolism (Additional file 23: Table S13). The same GO terms are also enriched in these gene families compared to the families with tandem duplications, but the difference is not significant. However, the gene families with tandem duplication pairs are depleted in GO terms relating to translation.
Gene trees support the common duplication of genes in Arachnopulmonata
The results of our analysis of duplicated genes in P. tepidariorum and other arachnids from the OMA and BUSCO gene sets, as well as our dating of the divergence in gene families, strongly suggest that there was a WGD in the ancestor of spiders and scorpions. To further explore whether the duplicated genes in spiders and scorpions were the result of duplication in the most recent common ancestor of these arachnopulmonates (Hypothesis 1) or lineage-specific duplications (Hypothesis 2), we applied a phylogenetic approach to examine P. tepidariorum and C. sculpturatus genes (Fig. 7, Additional file 24: Table S14, Additional file 25: Table S15). Of the 116 informative gene trees (see Methods) of orthogroups, wherein exactly two P. tepidariorum paralogs were present for a single T. castaneum ortholog, 67 (58%; henceforth Tree Set 1) were consistent with a common duplication (Hypothesis 1) and 49 (42%) were consistent with lineage specific duplications (Hypothesis 2) (Fig. 7, Additional file 24: Table S14, Additional file 25: Table S15). Of the 67 tree topologies supporting a common duplication, 18 were fully congruent with the idealized Hypothesis 1 tree topology and 49 were partially congruent with Hypothesis 1 (i.e., the two spider paralogs formed a clade with respect to a single scorpion ortholog) (Fig. 7, Additional file 24: Table S14, Additional file 25: Table S15).
Fig. 7.
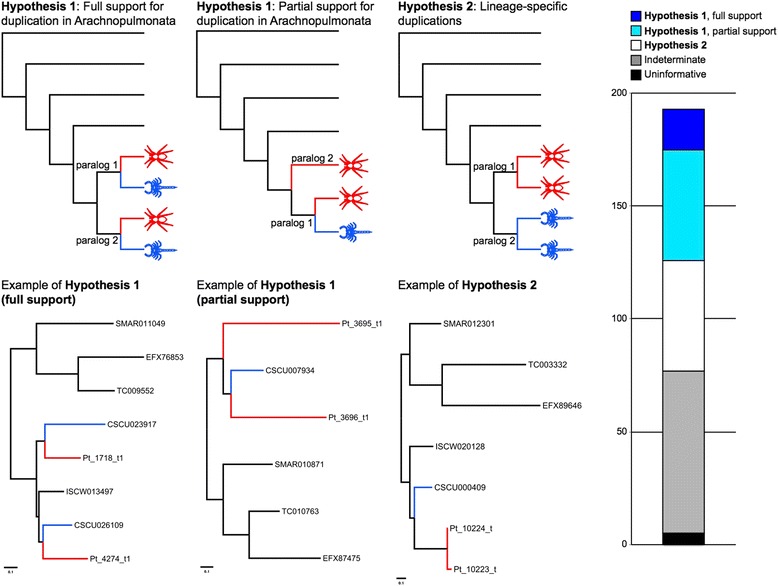
Gene trees support the common duplication of genes in Arachnopulmonata. Analysis of gene trees inferred from six arthropod genomes was conducted, with the gene trees binned by topology. Trees corresponding to a shared duplication event were binned as Hypothesis 1, and trees corresponding to lineage-specific duplication events as Hypothesis 2. Gene trees with spider paralogs forming a clade with respect to a single scorpion paralog were treated as partially consistent with Hypothesis 1. Top row of panels shows hypothetical tree topologies; bottom row of panels shows empirical examples. Right panel shows distribution of gene trees as a function of bin frequency
If the gene trees in Tree Set 1 were the result of large-scale duplication events or WGD as opposed to tandem duplication, we would expect each resulting copy to occupy two different scaffolds. Of the 18 P. tepidariorum paralog pairs from gene trees fully consistent with Hypothesis 1, 15 were found to occupy different P. tepidariorum scaffolds; of the 49 paralog pairs from gene trees partially congruent with Hypothesis 1, all but ten pairs were found to occupy different P. tepidariorum scaffolds (Additional file 26: Table S16). In addition, of the 18 C. sculpturatus paralog pairs that were fully consistent with Hypothesis 1, all 18 were found on different scaffolds. To test whether P. tepidariorum paralog pairs located on different scaffolds compared to the three paralog pairs found on the same scaffolds was simply a consequence of differences in assembly quality, we examined the length of the scaffolds for these two groups. We found the lengths of the scaffolds were statistically indistinguishable between the two groups (Additional file 26: Table S16; Wilcoxon rank sum test: W = 358, P = 0.9179). This analysis was not required for the 18 scorpion paralog pairs because, in all cases, each member of the scorpion paralog pair was distributed on a different scaffold.
The occurrence of two clusters of Hox genes in both the spider and scorpion genomes could also be consistent with either of these alternative hypotheses (Fig. 4b). However, only in the case of Antp was a tree topology consistent with Hypothesis 2 recovered and the difference in log likelihood between the two hypotheses was negligible (lnL = –0.27) (Fig. 4b). Higher statistical support for the Hypothesis 1 topology was generally obtained for data partitions with a large number of available sequences (e.g., Dfd, pb) (Fig. 4b). The sum of the Hox gene tree data is therefore consistent with the synteny analysis, and supports a shared duplication in the common ancestor of Arachnopulmonata.
WGD in Xiphosura is probably unrelated to the duplication of genes in Arachnopulmonata
The recent report of WGD and multiple Hox clusters in an analysis of horseshoe crabs (Order Xiphosura [31]) raises the possibility of two alternative interpretations, namely (1) a single WGD at the base of Chelicerata, with losses of duplicated genes in lineages like mites and ticks, or (2) separate WGD events in the horseshoe crab ancestor and in the arachnopulmonate ancestor. To discern whether the WGD event(s) recently reported in Xiphosura constitute separate (Hypothesis 3) or common (Hypothesis 4) evolutionary events from the duplication of genes in Arachnopulmonata, we added the three published horseshoe crab genomes to our dataset and reran OMA (Fig. 8). If the duplications reported here in spiders and scorpions were caused by the same event that drove the genome duplications in horseshoe crabs, we would expect to find paralog clusters that included members of all Euchelicerata (Xiphosura + Arachnida). This expected pattern is comparable to the case of whole genome duplications in the vertebrate ancestor [30], which resulted in the same sets of paralogs for all major vertebrate lineages, to the exclusion of non-vertebrate deuterostomes and the protostomes (e.g., the Sp gene family [64]). By contrast, if the duplications in spiders and scorpions were distinct from the duplications in horseshoe crabs, we would expect to observe a pattern where (1) horseshoe crab paralogs clustered together, (2) arachnopulmonate paralogs clustered together, and (3) all other arachnid orthologs would not be duplicated at all and fell somewhere in between horseshoe crabs and arachnopulmonates (Fig. 1) [53]. We thus examined gene trees recovered by OMA to discern which of these two scenarios was supported by the comparison of the nine full genomes.
Fig. 8.
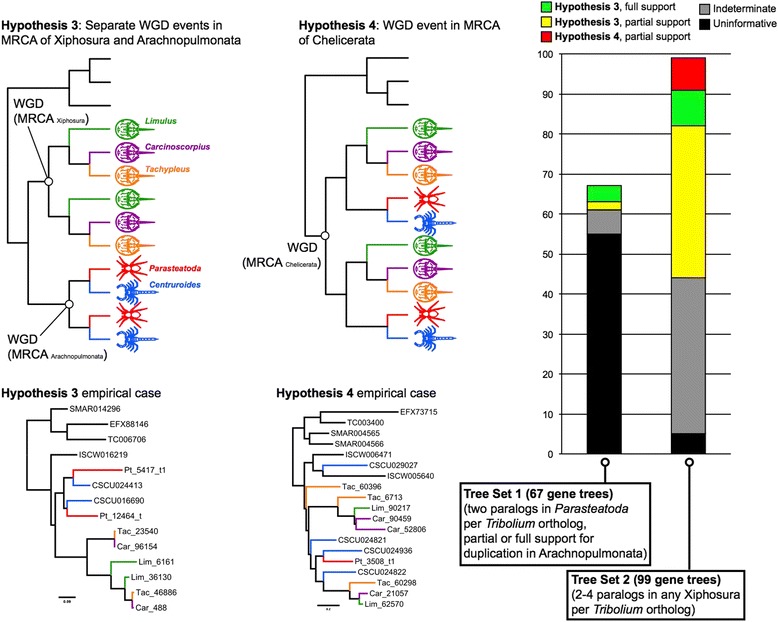
WGD in Xiphosura is probably unrelated to the duplication of genes in Arachnopulmonata. Analysis of gene trees inferred from nine arthropod genomes was conducted, with the gene trees binned by topology. Trees corresponding to two separate duplication events in the most recent common ancestor (MRCA) of Xiphosura and Arachnopulmonata were binned as Hypothesis 3, and trees corresponding to a single duplication event in the MRCA of Chelicerata as Hypothesis 4. Top row of panels shows hypothetical tree topologies; bottom row of panels shows empirical examples. Right panel shows distribution of gene trees as a function of bin frequency, for two different tree sets (i.e., gene trees retrieved under two alternate filtering criteria). Note the limited support for Hypothesis 4, with empirical gene trees poorly matching the expected tree topology (contra empirical cases supporting Hypothesis 3)
We first examined the orthogroups corresponding to Tree Set 1, after addition of horseshoe crab orthologs (Fig. 8). However, we found that 55 of the 67 gene trees constituting Tree Set 1 could not distinguish between Hypothesis 3 and Hypothesis 4 (i.e., no horseshoe crab paralogs were recovered in those orthogroups with duplicated spider genes).
We assembled a second tree set (henceforth, Tree Set 2) using the filtering criterion of orthogroups where 2–4 xiphosuran paralogs were recovered for a single T. castaneum ortholog. We thus recovered 99 gene trees in Tree Set 2 (Fig. 8). Of these, 44 were indeterminate (non-monophyletic outgroup) or uninformative (either missing all arachnopulmonates or missing all xiphosuran paralogs). A further 47 were consistent with Hypothesis 3, with nine gene trees completely congruent with Hypothesis 3 (i.e., multiple paralog clusters within both arachnopulmonates and horseshoe crabs, monophyly of Arachnopulmonata and Xiphosura, and monophyly of the mandibulate outgroup) (Fig. 8). The last eight gene trees in Tree Set 2 were scored as partially consistent with Hypothesis 4, but as shown in one empirical case (Fig. 8), these gene trees did not correspond well to the scenario of a common WGD at the base of Chelicerata, and may stem from algorithmic error in phylogenetic reconstruction (e.g., model misspecification). To be conservative, we treated these eight trees as consistent with our alternative hypothesis.
The sum of our gene tree analyses thus indicates support for Hypothesis 3 – the independent origins of arachnopulmonate and xiphosuran duplications. We found very little support for a shared duplication event at the base of Chelicerata (Hypothesis 4); no gene tree could be found where multiple paralogous groups each included exemplars of Xiphosura and Arachnopulmonata. Taken together, these results suggest that the duplication of genes in spiders and scorpions was probably independent of the proposed WGD events in horseshoe crabs.
Hox gene paralogs in P. tepidariorum show considerable divergence in temporal and spatial expression during embryogenesis
Alteration of the temporal and/or spatial expression can underlie the neo- or sub-functionalization of duplicated genes. To test whether the Hox gene paralogs in chelicerates have divergent expression patterns, we assayed the expression of all Hox genes throughout P. tepidariorum embryogenesis (for lab-A and lab-B expression see [65, 66]). For each pair of Hox paralogs, we found remarkable differences in spatial and temporal expression patterns (Fig. 9, Additional file 27: Figure S11, Additional file 28: Figure S12, Additional file 29: Figure S13, Additional file 30: Figure S14, Additional file 31: Figure S15, Additional file 32: Figure S16, Additional file 33: Figure S17, Additional file 34: Figure S18, Additional file 35: Figure S19, Additional file 36: Figure S20, Additional file 37: Figure S21, Additional file 38: Figure S22, Additional file 39: Figure S23, Additional file 40: Figure S24, Additional file 41: Figure S25, Additional file 42: Figure S26, Additional file 43: Figure S27, Additional file 44: Supplementary File1).
Fig. 9.
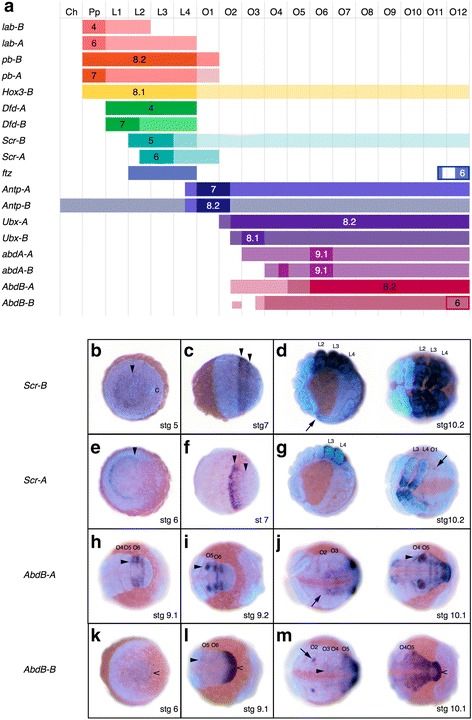
Expression of Hox paralogs in P. tepidariorum. a Summary of Hox gene expression domains and expression timing in P. tepidariorum embryos. Columns represent segments from anterior to posterior. Bars represent the extent of a gene’s expression domain with respect to the segments. The darkest color for each gene is used for the initial expression domain of each gene when it first appears, which usually coincides with a genes’ strongest expression. The next lighter color is used for the expanded domain, and the lightest color is used for further late expansions of the expression domains, which usually tends to be only in the nervous system. The stage at which a gene’s expression first appears is depicted by the stage number in the domain of first expression. ftz, in addition to its Hox domain, is expressed dynamically (i.e., budding off stripes) in the SAZ, and AbdB-B is continuously expressed in the SAZ after its formation at stage 6. These SAZ expression patterns are indicated by rectangular outlines in what is otherwise the O12 segment. Note that, since we did not detect clear expression boundaries for Hox3-A, the expression of this gene is not represented. b–m Two examples of Hox gene expression differences between paralogs of Scr (b–g) and AbdB (h–m). For detailed descriptions of expression patterns, see Additional file 44: Supplementary File 1 and the legends of Additional file 33: Figure S17, Additional file 34: Figure S18, Additional file 42: Figure S26, Additional file 43: Figure S27. All images are overlays of a bright-field images depicting the expression pattern and a fluorescent DAPI nuclear staining. Abbreviations: Ch cheliceral segment, Pp Pedipalpal segment, L–L4 walking leg segments 1–4, O1–12 opisthosomal segments 1–12
The expression of the paralogs of each Hox gene never appears at the same time during development; the expression of one paralog often precedes the other by at least 10 hours (e.g., lab, Scr, Ubx, and abdA) [65, 66] (Fig. 9b–g), if not 15 to 20 hours (pb, Dfd, Antp), or even 30 hours as in the case of AbdB (Fig. 9a, h–m). The expression domains of paralogs also differ significantly in their anterior and/or posterior borders. Scr, Ubx, abdA, and AbdB paralogs exhibit anterior borders that are shifted by half a segment or more, and several Hox gene paralogs expressed in the prosoma show shifts in their posterior expression borders by one or more segments (Fig. 9a). While the borders of the strongest expression domain are identical in the case of the paralogs of lab, Antp, and abdA, they differ substantially in all other paralogs (Fig. 9, Additional file 27: Figure S11, Additional file 28: Figure S12, Additional file 29: Figure S13, Additional file 30: Figure S14, Additional file 31: Figure S15, Additional file 32: Figure S16, Additional file 33: Figure S17, Additional file 34: Figure S18, Additional file 35: Figure S19, Additional file 36: Figure S20, Additional file 37: Figure S21, Additional file 38: Figure S22, Additional file 39: Figure S23, Additional file 40: Figure S24, Additional file 41: Figure S25, Additional file 42: Figure S26, Additional file 43: Figure S27), but note that the expression boundaries detected for Hox3-A were somewhat unclear (Additional file 29: Figure S13).
Most Hox gene paralogs also exhibit differences in the tissues and cell types they are expressed in (e.g., mesodermal vs. ectodermal expression, or groups of neuroectodermal cells that a paralog is expressed in), which hints at the possible neo-functionalization of one of the paralogs. For example, in the case of the AbdB paralogs (Fig. 9h–m), only AbdB-B, is expressed in the segment addition zone where it has a dynamic anterior expression border until a more Hox-like expression domain appears at stage 9.
While most Hox gene paralogs in P. tepidariorum follow spatial colinearity rules, i.e., genes at the beginning of the Hox cluster are expressed more anteriorly than genes at the end of the Hox cluster, a few Hox genes in P. tepidariorum do not adhere to these rules (Fig. 9a). Except for AbdB-B, all of the earliest expression domains are strictly spatially colinear; however, later during development, expression domains of a few genes extend beyond the expected spatial domains (ftz, Antp-A, AbdB-A, and -B).
Temporal colinearity rules, however, are not always followed by P. tepidariorum Hox genes. While genes at the beginning of the clusters are generally expressed earlier than the ones at the end of the clusters, there are many genes that do not adhere to temporal colinearity rules. Additionally, there is no temporal colinearity of expression initiation within either cluster A or B.
Taken together, we have observed considerable differences in the spatial and temporal expression between each of the P. tepidariorum Hox gene paralogs (Fig. 9). These differences likely reflect changes in function between the paralogs that have evolved in the time since the cluster was duplicated.
Discussion
Signatures of an ancient WGD in the last common ancestor of spiders and scorpions
Our study of the assembly and annotation of the P. tepidariorum genome revealed a high number of duplicated genes in accordance with previous observations [34–44]. This finding is further supported by our detection of a colinearity signal across many of the largest P. tepidariorum scaffolds. The fact that we find many smaller synteny blocks across scaffolds suggests that the WGD event occurred early during spider evolution and was followed by extensive disruption of previously larger blocks, for instance, by recombination or the activity of transposable elements. Intriguingly, the comparison of the gene content of the P. tepidariorum genome with other chelicerates and other arthropods suggests that a WGD likely occurred in the lineage leading to spiders and scorpions. Our dating efforts indeed confirmed that this WGD most likely occurred after the divergence of the common ancestor of spiders and scorpions from other arachnid lineages (mites, ticks, and harvestmen) prior to 430 MYA [67, 68] (Fig. 1). Furthermore, our results suggest that this event was independent of the apparent WGDs shared by all extant horseshoe crabs [31].
Divergence in gene function after duplication
It is thought that typically large-scale duplication events such as WGD are followed by a period of gene loss (for example, only 12% of paralogs have been retained after 100 MY in Saccharomyces cerevisiae [7, 23]), in concert with major genomic rearrangements, and that those duplicated genes that are subsequently retained are enriched in developmental genes such as those encoding transcription factors and other proteins that often act in multiprotein complexes [2, 18, 24, 25, 69]. Our GO term enrichment analysis partially confirms a similar trend for P. tepidariorum, since we find, for instance, proteins related to transcriptional regulation enriched in the group of duplicates. Indeed, it is striking that vertebrates, horseshoe crabs, and arachnopulmonates have retained duplicated Hox clusters and appear to be enriched in other paralogs that encode other transcription factors, suggesting that this retention pattern after WGDs is a general trend in animals.
Our study provides evidence for possible subsequent sub-functionalization and neo-functionalization among ohnologs [19–22, 69], most likely as a result of evolutionary changes in their regulatory sequences as has been observed in the case of other WGD events [70]. This is exemplified by the diversity in the temporal and spatial expression of the P. tepidariorum Hox gene paralogs during embryogenesis (e.g., Fig. 9). Divergence in the expression patterns of duplicated Hox genes has been previously reported for the genes Dfd, Scr, and Ubx in spiders [38, 71, 72] and for the posterior Hox genes Antp, Ubx, abdA, and AbdB in the scorpion C. sculpturatus [40]. However, these previous studies only investigated a few Hox gene families and analysis of the spatial expression of these genes was limited to later developmental stages after the appearance of limb buds. Divergence in gene expression has also been previously observed for duplicated Wnt ligand genes in P. tepidariorum [37]. In addition, a recent study of the two dachshund paralogs provided possible evidence for the neo-functionalization of a duplicated gene during the evolution of a morphological novelty in spiders [41].
Gene duplication and arachnid evolution
Our findings have profound implications for the evolution of chelicerates as a whole, a group whose internal phylogeny has proven extremely difficult to resolve [53]. Focal to understanding the evolution of terrestrialization in this group are the relationships of five arachnid orders possessing book lungs. The close relationship of four of these groups, namely spiders, amblypygids, thelyphonids, and schizomids, is generally not contested and both morphological and molecular trees place them together in a monophyletic clade, the Tetrapulmonata. The position of scorpions in the chelicerate tree, however, is much more controversial. It has been argued that their terrestrial adaptations, including the book lungs, evolved convergently to those of tetrapulmonates, whereas recent phylogenomic analyses have placed scorpions (possibly a sister group to Pseudoscorpiones) as the sister group to Tetrapulmonata [53, 73]. The shared paleopolyploidization event between spiders and scorpions provides further evidence that these two groups are more closely related to each other than they are to other apulmonate and non-duplicated arachnids (e.g., mites and ticks), which is in agreement with recent molecular phylogenies. This would imply a single origin of the arachnid book lungs as has been suggested previously based on detailed ultrastructural morphological analyses [74], raising the possibility that the ancient WGD identified here can be tested using new comparative genomic data and sampling such lineages as amblypygids, thelyphonids, and schizomids.
The age of the duplication event identified here must predate the most recent common ancestor of spiders and scorpions. Molecular clock approaches vary widely on the age of arachnids, and have suggested that arachnids diversified in the Ordovician [75, 76] or in the Silurian [77], with large confidence intervals on node age estimates that often span entire geological periods. However, the earliest stem-group spiders (the extinct order Uraraneida) date to the mid-Devonian (386 MYA [78]), whereas discoveries of Paleozoic scorpions have extended the stratigraphic range of scorpions into the Silurian (430 MYA [67]). The arachnid fossil record thus suggests the mid-Silurian is a conservative floor age of the duplication event. A Paleozoic age of the duplication event at the base of Arachnopulmonata would make this event approximately contemporaneous with the two-fold WGD in the ancestral vertebrate [30].
This reconstruction is consistent with the observation that few genes retain the ancient signal of shared duplication in both arachnopulmonates and vertebrates, and those that do often tend to be developmental patterning genes. For example, when compared to the Drosophila melanogaster genome, less than 5% of homologous vertebrate genes retain the 1:4 ortholog ratio expected from the vertebrate two-fold WGD event [30]. However, included among this minority are vertebrate orthologs of Hox genes, whose duplicates have been retained and deployed for various aspects of embryonic patterning. Thus, the patterns observed in arachnopulmonate arachnids are broadly consistent with counterparts in vertebrates.
Currently, it is not possible to address the question of whether the arachnopulmonate WGD facilitated the evolution of a terrestrial life-style and the development of book lungs. Taking advantage of the annotated spider genome sequences and the practical merits of P. tepidariorum, however, future functional studies in spiders could analyze paralog sub- and neo-functionalization and gene regulatory network rewiring after duplication to clarify these questions.
Conclusions
Much has been speculated about the long-term evolutionary consequences of genome duplications, including long-standing discussions on the evolution and origin of our own lineage, the vertebrates, and the complex body plan and diverse ecological adaptations that are hallmarks of this animal group [1, 2, 79–81]. However, it has been argued that there does not appear to be an association between genome duplication and teleost diversification [82]. Furthermore, other groups that have experienced WGD, such as horseshoe crabs and bdelloid rotifers, did not exhibit any apparent diversification or obvious increase in complexity following WGD, with the caveat that there might be changes in the complexity of their physiology, behavior and life history. This suggests that a putative link between WGD and increased diversification, as suggested in vertebrates, may not be generalizable to other taxa [11, 14, 32, 33].
To help address the contribution of WGD to animal diversification, analyzing the outcomes of those independent “experiments” that have naturally occurred during evolutionary time is of paramount importance. Recurrent and independent cases of paleopolyploidization should be studied systematically to reveal commonalities of evolutionary forces experienced across disparate lineages. Our discovery of an ancient genome duplication event preceding the origin of spiders and scorpions helps to fill a crucial gap in the comparative studies of WGDs. Previously reported cases of paleopolyploid lineages in different eukaryotes, including both unicellular and multicellular taxa, only allowed an extremely reduced set of core orthologous genes to be compared across lineages. However, the biology of vertebrates and arachnopulmonates is in many respects very similar, sharing the gene toolkit common to most animal species, highly conserved developmental pathways and even the general layout of the basic bilaterian body plan.
Thus, our results will open new research avenues, allowing the formulation of specific hypotheses about the impact of WGDs on developmental gene regulatory networks and morphological diversity by making direct comparisons and extrapolations with the vertebrate case. Moreover, since P. tepidariorum is arguably the primary chelicerate model system in the field of evolutionary development biology [51, 83–85], its genome sequence will provide an excellent resource to functionally test hypotheses based on genomic inferences.
Methods
Extraction of genomic DNA
Genomic DNA was extracted from four adult females and eight adult males of a genetically homogenous P. tepidariorum strain that was inbred for 15 generations and originally collected in Göttingen. All 12 animals were separated from the general stock before their final molt (to ensure that all specimens were virgin and did not contain genetic material from mating partners or developing embryos), and were starved for 2 weeks prior to DNA extraction (to minimize contamination from gut contents). Directly before DNA extraction, all animals were microscopically inspected to ensure they were free of external parasites (e.g., mites) and were macerated and digested in 80 mM EDTA (pH = 8.0), 100 mM Tris-HCl (pH = 8.0), 0.5% SDS, and 100 μg/mL proteinase K at 60 °C for 2 hours. Genomic DNA was isolated from this solution by salt-chloroform extraction, precipitated with ammonium acetate and ethanol, and dissolved in water. RNA contamination was removed with RNaseA. Purified genomic DNA was precipitated with sodium acetate, washed with ethanol, and dissolved in TE buffer (10 mM Tris-HCl (pH = 7.4), 1 mM EDTA) (pH = 8.0)).
For the bark scorpion C. sculpturatus, genomic DNA was extracted from four legs, a pedipalp patella and femur, and the fourth metasomal segment of an adult wild-caught female specimen (Tucson, Arizona, USA). Extraction was performed using the Animal Blood and Tissue protocol for a Qiagen DNeasy kit, with the addition of 16 μL of RNase A (25 mg/mL). Whole body RNA was extracted from the same adult female, an adult male, and a juvenile using one leg, the telson, the fifth metasomal segment, 1/3 of the abdomen (to avoid gut contamination), 1/2 of the cephalothorax, and a pedipalp patella. Total RNA was extracted using Trizol with the addition of glycogen.
Genome sequencing and assembly
The house spider and bark scorpion are two of 30 arthropod species sequenced as part of the pilot project for the i5K 5000 arthropod genomes project at the Baylor College of Medicine Human Genome Sequencing Center. For all of these species, an enhanced Illumina-ALLPATHS-LG sequencing and assembly strategy enabled multiple species to be approached in parallel at reduced costs. For the house spider, we sequenced five libraries of nominal insert sizes 180 bp, 500 bp, 2 kb, 3 kb, and 8 kb at genome coverages of 39.2x, 35.1x, 19.7x, 49.3x, and 19.3x, respectively (assuming a 1.5 Gb genome size [86]). These raw sequences have been deposited in the NCBI SRA: BioSample ID SAMN01932302. For the bark scorpion, we sequenced four libraries of nominal insert sizes 180 bp, 500 bp, 3 kb, and 8 kb at genome coverages of 102.1x, 25.6x, 35.2x, and 39.0x, respectively (assuming a 900 Mb genome size). These raw sequences have been deposited in the NCBI SRA: BioSample SAMN02617800.
To prepare the 180 bp and 500 bp libraries, we used a gel-cut paired-end library protocol. Briefly, 1 μg of the DNA was sheared using a Covaris S-2 system (Covaris, Inc. Woburn, MA) using the 180 bp or 500 bp program. Sheared DNA fragments were purified with Agencourt AMPure XP beads, end-repaired, dA-tailed, and ligated to Illumina universal adapters. After adapter ligation, DNA fragments were further size-selected on an agarose gel and PCR-amplified for 6 to 8 cycles using the Illumina P1 and Index primer pair and Phusion® High-Fidelity PCR Master Mix (New England Biolabs). The final library was purified using Agencourt AMPure XP beads and quality-assessed by Agilent Bioanalyzer 2100 (DNA 7500 kit) to determine library quantity and fragment size distribution before sequencing.
Long mate pair libraries with 2 kb, 3 kb, and 8 kb insert sizes were constructed according to the manufacturer’s protocol (Mate Pair Library v2 Sample Preparation Guide art # 15001464 Rev. A PILOT RELEASE). Briefly, 5 μg (for 2 and 3 kb gap size libraries) or 10 μg (8–10 kb gap size library) of genomic DNA was sheared to the desired size fragments by Hydroshear (Digilab, Marlborough, MA), then end-repaired and biotinylated. Fragment sizes between 1.8 and 2.5 kb (2 kb), 3 and 3.7 kb (3 kb), or 8 and 10 kb (8 kb) were purified from 1% low-melting agarose gel and then circularized by blunt-end ligation. These size-selected circular DNA fragments were then sheared to 400 bp (Covaris S-2), purified using Dynabeads M-280 Streptavidin Magnetic Beads, end-repaired, dA-tailed, and ligated to Illumina PE-sequencing adapters. DNA fragments with adapter molecules on both ends were amplified for 12 to 15 cycles with Illumina P1 and Index primers. Amplified DNA fragments were purified with Agencourt AMPure XP beads. Quantification and size distribution of the final library was determined before sequencing as described above.
Sequencing was performed using Illumina HiSeq2000 generating 100 bp paired-end reads. Reads were assembled using ALLPATHS-LG (v35218) [87] and further scaffolded and gap-filled using Atlas-Link (v.1.0) and Atlas gap-fill (v.2.2) [88]. For P. tepidariorum, this yielded an assembly size of 1443.9 Mb with 263,833 contigs with an N50 of 10.1 kb and, after scaffolding and gap closing, 31,445 scaffolds with an N50 of 465.5 kb. Approximately 2416 million reads (96.9x sequence coverage) are represented in this assembly of the P. tepidariorum genome. The assembly has been deposited in the NCBI: BioProject PRJNA167405 (Accession: AOMJ00000000).
For the C. sculpturatus this yielded an assembly size of 926.4 Mb with 214,941 contigs with an N50 of 5.1 kb and, after scaffolding and gap closing, 10,457 scaffolds with an N50 of 342.5 kb. The final assembly has been deposited in the NCBI: BioProject PRJNA168116.
Dovetail assembly
Chicago library preparation
To further improve the P. tepidariorum assembly we used in vitro contact genomics [89] based on the Chicago method (Dovetail Genomics, Santa Cruz, CA) [90]. A Chicago library was prepared as described previously [90]. Briefly, ≥ 0.5 μg of high molecular weight genomic DNA of ≥ 50 kb mean fragment size was extracted from a female P. tepidariorum, reconstituted into chromatin in vitro, and fixed with formaldehyde. Fixed chromatin was then digested with MboI or DpnII, the 5′ overhangs were filled in with biotinylated nucleotides, and the free blunt ends were then ligated. After ligation, crosslinks were reversed and the DNA was purified from protein. Purified DNA was treated to remove all biotin that was not internal to ligated fragments. The DNA was sheared to a mean fragment size of ~350 bp, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. Biotin-containing fragments were then isolated using streptavidin beads before PCR enrichment of the library.
Scaffolding the draft genome with HiRise
The P. tepidariorum draft genome in FASTA format (1443.9 Mb with a scaffold N50 of 465.5 kb), the shotgun sequences (from approximately 2416 million Illumina reads (see above)), and the Chicago library sequence (187 million read pairs from Illumina HiSeq 2500 2X100bp rapid run) in FASTQ format were used as input data for HiRise, a software pipeline designed specifically for using Chicago library sequence data to assemble genomes [90]. Shotgun and Chicago library sequences were aligned to the draft input assembly using a modified SNAP read mapper [91]. The separations of Chicago read pairs mapped within draft scaffolds were analyzed by HiRise to produce a likelihood model, and the resulting likelihood model was used to identify putative misjoins and score prospective joins. After scaffolding, shotgun sequences were used to close gaps between contigs. This resulted in 16,542 super-scaffolds with an N50 of 4050 kb.
Genome annotation
P. tepidariorum
The P. tepidariorum genome assembly (pre-Dovetail) was annotated using version 2.7 of AUGUSTUS [92]. AUGUSTUS constructs genes from evidence such as the RNA-Seq alignments – here called hints – but also uses statistical models for ab initio prediction. The parameters for the statistical models of P. tepidariorum genes were estimated on a training set of gene structures. Several steps of parameter estimation, prediction, visual quality control on a genome browser, and parameter tuning were performed.
P. tepidariorum transcript alignments were generated using available RNA-Seq libraries [86], namely 1,040,005 reads from 454-sequencing of P. tepidariorum embryonic stages, two RNA-Seq libraries from Illumina-sequencing of embryonic stages (333,435,949 and 602,430 reads), and two RNA-Seq libraries from Illumina-sequencing of post-embryonic stages (294,120,194 read and 317,853 reads). In addition, we downloaded all P. tepidariorum ESTs [93] and protein sequences available in GenBank. The assembly was repeat-masked using RepeatMasker (version 1.295) [94] and TandemRepeatFinder (version 4.07b) [95] based on a de novo repeat library compiled with RepeatScout (version 1.0.5) [96]; 46% of the bases were masked as repeats.
P. tepidariorum-specific parameters of AUGUSTUS were estimated iteratively. An initial training set of genes was generated with PASA (release 2012-06-25) [97] using the ESTs only. This yielded 851 genes that were used to estimate the first set of parameters of AUGUSTUS for the coding regions of genes. Additionally, eukaryotic core proteins were predicted in the masked assembly with CEGMA (version 2.4.010312) [98] and yielded 103 hints for CDS to AUGUSTUS, which were then used in the training stage predictions. With these initial parameters and integrating the evidence from transcriptome data, AUGUSTUS was used to annotate the masked assembly genome-wide. We then extracted another training gene set from the genome-wide prediction by mapping RNA-Seq reads from 454- and Illumina sequencing against predicted transcripts using GSNAP (version 2013-06-27) [99]; however, (1) only genes with 100% RNA-Seq alignment coverage were taken and (2) we mapped the proteins from the database UniRef50 (version UniProt Release 2013 06) [100] against predicted proteins using BLASTP (version 2.2.25) [101], keeping only fully covered transcripts. The genes in the intersection of both sets – that is, genes fulfilling constraints (1) and (2) simultaneously – were used for a second iteration of parameter training. The UTR parameters of AUGUSTUS were only trained once when other parameters had already become stable.
RNA-Seq reads from 454 and Illumina sequencing were mapped against the masked assembly using GSNAP (version 2013-06-27) [99]. The evidence from transcriptome data, protein homology and repeats was input to AUGUSTUS as a ‘hints’ file. The spliced alignments of the RNA-Seq reads using GSNAP resulted in 272,816 unique intron hints and further hints on exonic parts from transcribed regions. Furthermore, we obtained 97,785 hints from ESTs (not only for CDS) using BLAT (version v. 35x1) [102]. The roughly 2.1 million repeat-masked regions were used as ‘nonexonpart’ hints in the annotation, moderately penalizing the prediction of exons overlapping repeats. Consecutive gene sets were computed utilizing AUGUSTUS to stepwise improve prediction accuracy and reliability of the final gene set release referred to as aug3. All extrinsic hint data were incorporated into this last prediction. Allowing the occurrence of alternative transcripts in the results, the final gene set aug3 was then generated using the call:
augustus –species = parasteatoda –alternatives-from-evidence = true … --UTR = on --hintsfile = all.hints --extrinsicCfgFile = extrinsic.P.E.RM.cfg genome_masked.fa
The RNA-Seq data coverage was quantified using the transcript quantification tool eXpress [103], which estimates fragments per kb of transcript per million mapped reads at transcript level (FPKM) values, thereby quantifying the pooled abundances of the predicted transcripts in the RNA-Seq data.
The aug3 gene models were transferred to the Dovetail genome assembly using Exonerate v2.2 [104] with the command --model protein2genome --bestn 1 --showtargetgff YES. The resulting GFF files were converted into protein sets from the corresponding Dovetail genome fasta file.
The Trinotate annotation pipeline (Release 2.0.2) [105] was used for the functional annotation of the aug3 protein predictions following the standard procedure. Briefly, the predicted peptide sequences of the aug3 annotation were blasted against UniRef90 and SwissProt databases with E ≤ 0.05 and keeping only the best hit. HMMER (version 3.1b1) [106] was used to search the Pfam database to predict protein domains. All Blast searches were run in parallel on a high performance computer cluster utilizing the perl script HPC GridRunner (v1.0.2) [107]. The Blast and protein domain predictions were stored in a predefined sqlite (version 3.8.8.3) [108] database. Trinotate was used to export a final report that contains the best Blast hits, protein domain predictions, and GO categories extracted from the Blast result and the Pfam domain prediction for each of the aug3 predictions (Additional file 45: Table S17).
The final annotated gene set contained 27,990 genes and 31,186 transcripts; 85% of the predicted P. tepidariorum proteins had homology support derived from a BLASTP search against the UniRef50 data (E value ≤ 10–5). Transcript quantification from the RNA-Seq data (using estimates of FPKM values [103]) showed that 29,966 (93%) of predicted transcripts had transcriptome support at FPKM ≥ 0.034 and 26,381 (82%) of predicted transcripts had transcriptome support at FPKM ≥ 0.34. In the final gene set, only 1.1% of the predicted transcripts had neither homology nor transcriptome support at an FPKM threshold of less than 0.034. The annotated P. tepidariorum genome is available in JBrowse/Web Apollo Parasteatoda tepidariorum [109].
C. sculpturatus
The C. sculpturatus genome was annotated using MAKER [110] with RNA-Seq reads generated from a juvenile [111], an adult female [112], and adult males [113]. The annotated C. sculpturatus genome is available in the Centruroides Genome Browser [114].
Analysis of duplicated genes
Classification of duplicates using MCScanX
The data used to perform these analyses were, for P. tepidariorum, the aug3 version, and for C. sculpturatus, the 0.5.53 version of the MAKER annotation available at Centruroides sculpturatus MAKER annotation [115]. The same analysis was also performed on the Limulus polyphemus genome [116] as a comparison.
Out of the 32,949 gene models in the aug3 annotation of the P. tepidariorum genome (resulting from the transfer of the aug3 annotation on the Dovetail scaffolds), only the main transcript of each gene was retained, yielding a set of 28,746 gene models. This list was further shortened by removing all instances of 755 gene models that had become artifactually duplicated during the annotation transfer process from aug2 to aug3, resulting in a final set of 27,203 gene models. All of the 30,465 gene models in the C. sculpturatus annotation were retained for the synteny analyses. Finally, out of the 23,287 annotated proteins of L. polyphemus, 21,170 were retained for the synteny analyses after filtering out annotated isoforms of the same genes (based on their identical start and end positions).
Hits within and between gene sets were catalogued using BLASTP using an E value threshold of 10–10 and keeping only the five best hits as recommended in the instruction manual of MCScanX [117]. Then, MCScanX was used with default parameters to classify genes into five categories, namely singletons (i.e., genes without any duplicate), dispersed (duplicates occurring more than 10 genes apart or on different scaffolds), proximal (duplicates occurring on the same scaffold at most 10 genes apart), tandem (consecutive duplicates), and segmental (block of at least five collinear genes separated by less than 25 genes missing on one of the duplicated regions).
Orthology assessment of arthropod genomes
To investigate the extent of gene duplication in P. tepidariorum and C. sculpturatus, we compared these two genomes to those of four other arthropods with no demonstrable evidence of a WGD. These non-arachnopulmonate taxa were another chelicerate (the tick I. scapularis) and three mandibulates (the flour beetle Tribolium, the crustacean Daphnia pulex, and the centipede Strigamia maritima). Predicted peptide sets (aug3) were used as inputs, and redundancy reduction was performed with CD-HIT [118] to remove the variation in the coding regions of genomes attributed to allelic diversity R (>99% sequence similarity). Peptide sequences with all final candidate ORFs were retained as fasta files. We assigned predicted ORFs into orthologous groups across all samples using OMA stand-alone v.0.99u [119, 120] discarding sequences of less than 50 sites in length. All-by-all local alignments were parallelized across 400 CPUs. Orthology mapping of spider and scorpion genes that could be mapped to a mandibulate or tick counterpart was conducted using custom Python scripts on the OMA output.
To assess the possibility of incorrect orthology assessment stemming from algorithmic error, we identified the intersection of the OMA output (based on whole genomes) and a set of orthologs found to occur in single copy across Arthropoda, as benchmarked in the BUSCO-Ar database of OrthoDB [121]. The BUSCO-Ar set of the flour beetle T. castaneum was selected as the reference genome for the BUSCO set.
In a separate and subsequent analysis, three additional taxa (genomes of the horseshoe crabs L. polyphemus, Tachypleus gigas, and Carcinoscorpius rotundicauda) were added to the taxa in the principal OMA run, with all other procedures as specified above.
Analysis of gene tree topologies from six-genome dataset
From the output of the OMA analysis of six arthropod genomes, we extracted a subset of orthogroups wherein exactly two spider paralogs were present for one T. castaneum ortholog (i.e., 1:2 orthology). T. castaneum was chosen as the reference genome in comparative analyses both for the quality of its assembly and for its archetypal gene content among Arthropoda. Gene trees for this subset of orthogroups were inferred to examine the topological relationship between homologous sequences of arachnopulmonate and non-arachnopulmonate taxa. These orthogroups were aligned using MUSCLE v.3.8 [122] and ambiguously aligned regions were culled using GBlocks v.0.91b [123] using the commands –b3 = 8 (maximum of eight contiguous non-conserved positions), –b4 = 10 (minimum of ten positions in a block), and –b5 = h (gap positions allowed for a maximum of half the sequences). Maximum likelihood analyses were conducted using the LG + Γ model with four rate categories [124, 125] and 500 independent starts in RAxML v. 7.3.0 [126].
We characterized whether the resulting tree topologies corresponded to Hypothesis 1 (common duplication in the most recent common ancestor (MRCA) of spiders and scorpions), Hypothesis 2 (lineage-specific duplication events in each of spiders and scorpions), an indeterminate tree topology (corresponding to neither scenario, typically due to the non-monophyly of the outgroup taxa), or an uninformative tree topology (due to the lack of any scorpion paralogs). Cases where the two spider paralogs formed a grade with respect to a single scorpion paralog were additionally classified as partially congruent with Hypothesis 1. The set of gene trees either partially or fully congruent with Hypothesis 1 is henceforth termed “Tree Set 1”. Alignments and gene tree files are available on request.
Analysis of gene tree from nine-genome dataset
To infer the relationship between arachnopulmonate and xiphosuran paralogs, from the OMA analysis of nine genomes (the six genomes above, L. polyphemus, T. gigas, and C. rotundicauda) we separately extracted another subset of orthogroups, wherein two, three, or four horseshoe crab paralogs from any of the three horseshoe crab genomes were detected for one T. castaneum ortholog (i.e., 1:2, 1:3, or 1:4 orthology). We inferred gene trees with the approach specified above. We again distinguished two scenarios, namely (1) separate WGD events in the MRCA of Arachnopulmonata and Xiphosura (Hypothesis 3), and (2) a common WGD event in the MRCA of all Chelicerata (Hypothesis 4). Cases where ancient paralogy was detected in Xiphosura alone (and not Arachnopulmonata) were classified as partially congruent with Hypothesis 3. The set of gene trees either partially or fully consistent with Hypothesis 3 was termed “Tree Set 2”. Alignments and gene tree files are available on request.
Identification of paralog pairs in P. tepidariorum and other chelicerates
Putative families of homologous protein-coding genes were identified for 31 chelicerate species and a myriapod (Additional file 14: Table S8). Protein sequences from the publically available translated coding sequences were also used. Otherwise, transcripts were translated with Transdecoder [97]. For translated sequences with > 95% identity, only the single longest protein was retained for further analyses. For transcripts assembled by Trinity [127], the longest transcript per “contig” was retained (Trinity often generates multiple transcripts associated with a single “contig”, thought to represent isoforms).
We grouped genes into families using a modified version of the method applied in the Phytozome project described by Goodstein et al. [61], with either P. tepidariorum or C. sculpturatus translated genes used as a seed. In short, homologous protein pairs were identified using all-versus-all BLASTP comparisons of the 32 arthropod species with an E cutoff value of < 1 × 10–3 [101]. A global alignment score was calculated for each homologous pair using the Needleman–Wunsch algorithm with the Blosum62 matrix. We then used the Needleman–Wunsch score between P. tepidariorum (or C. sculpturatus) protein sequences and the rest of the sequences to seed the gene families in a three-step process. First, for each non-P. tepidariorum protein, the P. tepidariorum protein with the highest Needleman–Wunch score was identified. Second, all the non-Parasteatoda proteins with the same best-scoring P. tepidariorum protein were grouped with the P. tepidariorum protein. Third, all the groups were combined that contained P. tepidariorum proteins determined to be homologous to each other based on a BLASTP alignment with an E value of < 1 × 10–3. The same three-step process was repeated to identify C. sculpturatus-seeded gene families.
For each gene family, the protein sequences were multiply aligned using MUSCLE [122]. The multiple alignments were trimmed by removing all the bounding alignment positions that added more gaps than sequence by a custom Perl script. Entire protein sequences were removed from the alignment if the sequence had gaps in more than 25% of the aligned positions. For the P. tepidariorum-seeded gene families, only those containing at least one P. tepidariorum protein and four additional sequences were retained for further analyses. Within the retained families, poorly aligned columns were removed using TrimAL under a “strict-plus” setting, which optimizes the signal to noise ratio in the multiple alignment [128]. The protein alignments were then used to guide nucleotide alignments by replacing the amino acids with their encoding transcript sequences.
Protein alignments were used to infer gene trees with TreeBeST [129]. TreeBeST searches for an optimal gene tree given a species tree (we used the phylogeny in Additional file 15: Figure S7) and identifies duplication and speciation events within the optimal tree. Branch lengths were calculated for the optimal TreeBeST tree using maximum likelihood (PhyML type search) with the HKY model of evolution [62]. Alignments and gene tree inferences were repeated for the C. sculpturatus-seeded gene families.
Molecular distance of duplication and speciation events
We estimated the molecular distance of a P. tepidariorum (or C. sculpturatus) duplication or speciation node in P. tepidariorum (or C. sculpturatus)-seeded families by averaging the branch lengths in TreeBeST trees from the node to all its P. tepidariorum (or C. sculpturatus) descendants. We similarly estimated the molecular distance of other species’ duplication nodes by averaging the branch length from the node to all of the descendants of the species of interest. Distributions of molecular distances were estimated and statistical tests for goodness-of-fit calculated in R.
Ascertaining GO Term enrichment in P. tepidariorum paralog pairs
GO Terms were imputed to the P. tepidariorum AUGUSTUS gene models (aug3) through comparisons to the UniRef50 protein set by BLASTP comparisons using a cut-off of 1 × 10–5. The GO Terms of its closest UniRef by E value with documented GO Terms were assigned to a gene model via a custom perl script, with GO Slim values derived using GOSlimViewer [130]. Enrichment of GO Terms within gene families was ascertained using Fisher’s exact test.
Synteny analyses
A genome-scale synteny analysis of the P. tepidariorum scaffolds was conducted using the program SatsumaSynteny [60]. This approach does not rely on the annotation and can detect weak, degraded signals of synteny such as signatures of ancient WGDs that were followed by numerous rearrangements. For visualization, we selected only the 100 scaffolds for which the number of hits detected by Satsuma was maximal; in a second round, this list was further reduced to the set of 39 scaffolds that exhibited the greatest number of hits with each other. An Oxford grid [131] was drawn using the tool orthodotter [132], and a circular plot was drawn using Circos [133].
For the synteny analysis of selected developmental genes, their nucleotide sequences were first downloaded from NCBI (Accession numbers are given in Additional file 12: Table S7). BLASTN searches against the Augustus 3 gene set were used to identify the best aug3 prediction and BLASTN searches against the Dovetail assembly (Assembly 2.0) were used to identify their respective scaffold.
All 148 precursor microRNA sequences for P. tepidariorum [44], with the inclusion of flanking sequences 20 bp up- and down-stream, were BLASTN-searched in the Dovetail assembly to identify scaffold ID and position from the best matches. The scaffolds and positions of C. sculpturatus microRNAs from Leite et al. [44] were used.
Homeobox and Hox gene annotation
To identify possible homeobox genes in P. tepidariorum and C. sculpturatus, the complete set of homeodomain sequences from HomeoDB [134, 135], those identified previously in the scorpion Mesobuthus martensii [36], and the P. tepidariorum gene prospero (Accession: BAE87100.1) were BLASTP-searched (version 2.4.0+) [136] against the P. tepidariorum AUGUSTUS (aug3) and C. sculpturatus MAKER protein predictions. All blast hits were scanned for the presence of homeodomains and other functional domains with the CDD search tool [137]. Hits that contained at least one homeodomain were manually checked for the completeness of this sequence. The homeobox genes were annotated and classified based on the work by Holland et al. [138].
To identify the location of Hox genes on genomic scaffolds of P. tepidariorum, Latrodectus hesperus, S. mimosarum, A. geniculata, C. sculpturatus, I. scapularis, and genomic contigs of M. martensii were searched for Hox genes with tblastx BLAST (version 2.2.28+) [136] using published chelicerate Hox gene sequences. Scaffolds or contigs containing blast hits to Hox genes were extracted and intron-exon boundaries were hand-annotated in Geneious (version 7) [139] with the help of sequenced transcriptomes, sequences obtained by RACE PCR experiments (in the case of P. tepidariorum), cloned Hox gene sequences (in the case of C. sculpturatus), or by comparison between the chelicerate sequences. In case of additional splice variants containing additional small exons, the shortest version consisting of only two exons was used for the analysis. Naming of Hox genes followed orthologies to already published Hox genes in C. salei and P. tepidariorum for the spider sequences or, in the case of the scorpions, orthologies to published C. sculpturatus sequences.
Hox gene alignments and topological tests of gene trees
Nine Hox class genes were used as test cases for distinguishing two scenarios, namely (1) common duplication in the MRCA of spiders and scorpions (Hypothesis 1) and (2) lineage-specific duplication events in each of spiders and scorpions (Hypothesis 2). The single remaining Hox class gene (fushi tarazu) did not possess the minimum requirement – inclusion of two paralogs each of a spider and a scorpion species – and thus was not dispositive in topological tests. Peptide sequence alignments were constructed using MUSCLE v. 3.8 [122] and alignment ends manually trimmed, such that either terminus of the alignment sampled at least half of each alignment’s terminals. Preliminary efforts using outgroup taxa have demonstrated little statistical power resulting from rooting trees due to large phylogenetic distances between arachnopulmonates and arachnid outgroups (e.g., harvestmen, pycnogonids [40]), as well as accelerated evolution in other potential outgroup taxa (e.g., mites, ticks [53]). Therefore, outgroup-free tests were conducted using spider and scorpion sequences only.
Maximum likelihood analyses were conducted using the LG + Γ model with four rate categories [124, 125] and 500 independent starts in RAxML v. 7.3.0 [126]. To compare tree likelihoods of unconstrained runs to Hypothesis 2, a constraint tree was imposed for each Hox class enforcing mutual monophyly of spider and scorpion sequences, and the best tree topology was selected from 500 independent starts under the scenario of lineage-specific duplications.
Embryos, in situ hybridization, and imaging
P. tepidariorum embryos were obtained from laboratory cultures in Oxford, UK, Cambridge, MA, USA, and Cologne, Germany. RNA was extracted from embryos of stages 1–14 using either Trizol (Life Technologies) or Qiazol (Qiagen) and cDNA was synthesized with SuperscriptIII (Life Technologies). Probe templates were either synthesized by PCR using TOPO pCR4 vectors containing cloned RACE fragments of Hox genes (RACE was performed with the Marathon RACE kit or SMART RACE cDNA kit (Clontech)), or they were generated by adding T7 binding sites to RT-PCR fragments as described previously [140]. Primer sequences used for the RT-PCR fragments were based on the P. tepidariorum transcriptome [86] and genome sequences. The origin of gene fragments and primers is available on request. Embryos were fixed and probe synthesis and in situ hybridizations were carried out as described previously [141, 142]. The anti-DIG antibody (Roche, 11093274910) was pre-absorbed overnight at 4 °C with mixed-stage embryos. Stained embryos were staged according to Mittmann and Wolff [143] and imaged using a Leica stereoscope fitted with a Zeiss AxioCam MRc. Images were processed in Photoshop CS4 or CS6.
Additional files
Duplicated genes in Parasteatoda and Centruroides in comparison to each of the single-copy arthropods. (XLSX 55 kb)
Duplicated genes: three gene blocks. (PDF 36 kb)
Duplicated genes: four gene blocks. (PDF 36 kb)
Duplicated genes: five gene blocks. (PDF 36 kb)
P. tepidariorum homeobox genes. (XLSX 56 kb)
C. sculpturatus homeobox genes. (XLSX 54 kb)
Gene tree analysis of individual Hox genes. Blue branches indicate scorpion sequences; red branches indicate spider sequences. Gene trees are shown for labial (A), proboscipedia (B), Deformed (C), Sex combs reduced (D), Antennapedia (E), Ultrabithorax (F), abdominal-A, (G), and Abdominal-B (H). Log likelihood values are provided in Fig. 4. Due to insufficient sequences for hypothesis testing, gene trees were not inferred for Hox3-B and fushi tarazu. Abbreviations: Pt Parasteatoda tepidariorum, Lh Latrodectus hesperus, Sm Stegodyphus mimosarum, Ag Acanthoscurria geniculata, Cs Centruroides sculpturatus, Mm Mesobuthus martensii. (PDF 156 kb)
{kind=link}
P. tepidariorum Hox clusters to scale. The distances of Hox genes in clusters A and B are very similar, except for the break in cluster B. Hox genes are colored so that paralogs match, non-Hox genes are shown in grey and microRNAs are represented as lines. Genes on the positive strand and negative strand are indicated above and below the black lines, respectively. (JPG 503 kb)
Spider, scorpion and mite Hox clusters A and B. (XLSX 533 kb)
microRNAs and other genes identified in the P. tepidariorum Hox clusters. (DOCX 106 kb)
Duplication of miRNAs by possible large-scale and tandem duplication events in Parasteatoda and Centruroides. (XLSX 52 kb)
Duplicated developmental genes in P. tepidariorum. (XLSX 50 kb)
Synteny of 39 P. tepidariorum scaffolds with greatest number of reciprocal hits. Circos plot of the subset of 39 scaffolds presenting the greatest numbers of hits on one another (as detected using SatsumaSynteny). One unit on the perimeter represents one Mbp. (PDF 819 kb)
Genomic and transcriptomic datasets used to build gene families. (DOCX 141 kb)
Phylogeny of 31 arthropod species used in comparison with P. tepidariorum. Species relationships are based on JC Regier, JW Shultz, A Zwick, A Hussey, B Ball, R Wetzer, JW Martin, and CW Cunningham [73], JE Bond, NL Garrison, CA Hamilton, RL Godwin, M Hedin, and I Agnarsson [144], PP Sharma and G Giribet [145]. The number of genes within P. tepidariorum-seeded gene families are shown parenthetically (n =), the number of speciation nodes observed in the gene families between P. tepidariorum and the other species (N =), and the median HKY distance between each speciation node and P. tepidariorum (HKY =) descendants are shown at the node. (DOCX 348 kb)
P values of Kolmogorov–Smirnov goodness-of-fit tests for five models of the HKY distance1 distribution of duplication nodes with non-P. tepidariorum descendants. Distributions unable to be rejected at an alpha level of 0.05 are in bold. Best fitting models are in italics. (DOCX 80 kb)
HKY distance distributions and Gaussian mixture models of duplication nodes from P. tepidariorum-seeded gene families for eight arachnid species. The HKY distances for duplication nodes were calculated as the mean HKY branch length from the duplication node to each of the descendent genes in the species of interest (A–H). In panel (I) the distribution is for all the duplication nodes with at least one P. tepidariorum descendant, using the mean HKY distance from the node to P. tepidariorum descendants. For each panel, the best match to one of five distributions (Uniform, exponential (G), or a Gaussian mixture model with 1, 2 (H), or 3 (A–F, I) distributions) is shown. The Gaussian mixture models were seeded with Gaussian mean and standard deviations estimated from the P. tepidariorum duplication nodes (Fig. 6a). (DOCX 430 kb)
Percent of duplication nodes assigned to three Gaussian distributions of HKY distances. The mean and standard deviation of the P. tepidariorum distributions (Fig. 6a) were used to estimate the other species’ Gaussian distributions. (DOCX 65 kb)
HKY distance distributions of all P. tepidariorum speciation nodes in P. tepidariorum-seeded gene families. The distributions are for mean HKY branch lengths from the P. tepidariorum speciation node to the descendant P. tepidariorum genes. (DOCX 244 kb)
Comparison of distributions for P. tepidariorum duplication and speciation nodes. Kolmogorov–Smirnov goodness-of-fit tests were used to compare the middle Gaussian distribution of HKY distances of duplication nodes (Fig. 6a, Additional file 13: Figure S6) to the log-normal distributions for speciation nodes. (DOCX 73 kb)
Shared paralog pair retention between P. tepidariorum and other arthropod species. Shaded species have complete genomes or deeply sequenced transcriptomes. (DOCX 92 kb)
Tandem duplications are abundant in young duplication events, but rare in older duplication events. HKY distance distributions and Gaussian mixture models of duplication nodes from P. tepidariorum-seeded gene families mirror those in Fig. 6a, but paralog pairs that are found in tandem (within five genes of each other on the same scaffold) are now shown in light grey, and dispersed paralogs in dark grey. (PDF 158 kb)
Analysis of the gene families containing a duplication pair from each of the Gaussian distributions (Fig. 6a), excluding tandem duplicates, show enrichment in several GO terms compared to gene families without duplication pairs. (XLSX 2339 kb)
Gene IDs of duplicated P. tepidariorum and C. sculpturatus genes compared to other arthropods. (XLSX 86 kb)
Frequency of tree topologies supporting different duplication scenarios. (XLSX 34 kb)
Orthology of P. tepidariorum genes giving full or partial support for WGD with respect to T. castaneum. (XLSX 19 kb)
{kind=link}
P. tepidariorum proboscipedia-A expression. pb-A is first expressed in a thin stripe in the anterior of the embryo at stage 7 (A and B). The stripe broadens and as segmentation commences, it is found in the pedipalpal segment, and additionally, four weaker stripes start to appear in L1–L4 (C). The expression in the pedipalpal segment remains strongest, when by the end of stage 8.1, another stripe of weak pb-A expression is found in O1 (arrow) (D and E). During stage 9 and 10, pb-A expression is found in the mesoderm of the outgrowing pedipalp and walking legs, but also ectodermally in the distal tip of the outgrowing appendages of Pp-L4 (caret) (F–H). In the nervous system, pb-A is expressed most strongly in the Pp segment, starting at stage 9 (arrowhead), but it is also present in a smaller lateral domain of L1–O1 (arrow) (F–H). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–H). Anterior is to the left. Abbreviations: Pp pedipalpal segment, L walking leg segments, O opisthosomal segments. (JPG 6345 kb)
{kind=link}
P. tepidariorum proboscipedia-B expression. Weak pb-B expression emerges at stage 8 in a domain spanning from Pp to L4 (A). The weak expression is located in the mesoderm, and at stage 8.2 also visible in O1 (arrow) (B). At stages 9 and 10, pb-B is expressed in the mesoderm of the outgrowing appendages of Pp–L4, as well as in dorsolateral parts of the neuroectoderm of segments Pp–O1 (arrows) (C and D). Each panel (except A) shows the same embryo, viewed laterally (left) and ventrally (center, right). Anterior is to the left. Abbreviations: see Additional file 27: Figure S11. (JPG 143 kb)
{kind=link}
P. tepidariorum Hox3-A expression. Hox3-A expression appears first at stage 7 (not shown). At stage 8.1, the expression can be found in segmental stripes in the pedipalpal and the walking leg segments, and weak expression can also be seen in O1. The strongest expression is found in the first walking leg segment (A). At stage 8.2 and 9.1, Hox3-A is expressed in the mesoderm of the developing limb buds (B, C) and in the mesoderm of posterior segments. Note that Hox3-A expression is very weak and due to the high background in the head segments as well as in the opisthosomal segments, it is difficult to define anterior/posterior boundaries. Expression of Hox3-A vanishes after stage 9 (not shown). Panels show flat-mounted embryos, brightfield image above, nuclear stain of the same embryo below. Anterior is to the left. Abbreviations: see Additional file 27: Figure S11. (JPG 214 kb)
{kind=link}
P. tepidariorum Hox3-B expression. Hox3-B is not expressed before stage 8, when expression starts in broad segmental stripes from Pp to L4, most likely in the mesoderm (A). Expression is strongest in L1 and L4 (A and B). In stage 9 and 10 embryos, Hox3-B is expressed in the mesoderm of the outgrowing limb buds, except for the pedipalps, which show a broad ring of expression (white arrow in C) that later refines into rings and additional expression at the tip of the pedipalp (D–F). Additionally, expression extends anterior-ventral to the limb buds in a triangular shape at stage 9 (arrowhead in C). This expression vanishes by stage 10. Instead, Hox3-B is now also expressed in the pedipalpal and walking leg segments in segmental groups of cells in the medial neuroectoderm (arrowheads in D–F). In stage 11 embryos, Hox3-B is additionally expressed in dots in the ventral neuroectoderm of every opisthosomal segment (carets in F). Furthermore, a dot of expression can be found in the first opisthosomal limb bud (arrow in F). Embryos in A and B are shown laterally, embryos C–E are shown laterally on the left and ventrally on the right. F shows ventral views of the head region (left) and the opisthosoma (right) of an embryo at a similar stage as the embryo in E. Anterior is to the left. Abbreviations: see Additional file 27: Figure S11. (JPG 4203 kb)
{kind=link}
P. tepidariorum Deformed-A expression. Dfd-A is first expressed at stage 4, in almost all cells but the rim (arrowhead) of the germ disc (A). During stage 6, cells not expressing Dfd-A at the outer rim, the future anterior, have multiplied (arrowheads) (B). Later during stage 6, the expression clears from the posterior (former center of the germ disc, arrow) and the expression starts to form a broad stripe (bracket) (C). This uniform stripe of expression first subdivides into two stripes with lower expression between these stripes during stage 7. Then, the posterior domain splits into two more stripes and, in between the anterior and posterior domains, a new stripe gets inserted such that there are now four stripes of Dfd-A expression (asterisks) (D). These four stripes are later located in the four walking leg segments at stage 8 (E). In stage 9–11 embryos, Dfd-A is strongly expressed in the outgrowing walking legs with strongest expression in the tips of the legs (white carets) (F–H) and in the neuroectoderm (arrowheads in G and H). Each panel (except for A, B, C) shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left in all panels. Abbreviations: See Additional file 27: Figure S11. (JPG 6387 kb)
{kind=link}
P. tepidariorum Deformed-B expression. Dfd-B expression is first detected in a weak, broad stripe in the developing germ band (bracket) (A). This stripe can be allocated to the first two walking leg segments at stage 8 (B). The anterior of the segments is stained stronger than the posterior. Additional, but much weaker expression can be seen in the L3 and L4 segments. At stage 9, Dfd-B is predominantly expressed in the ventral neuroectoderm (C). The anterior and posterior expression domain is marked by arrowheads in B and C, respectively. Each panel shows the same embryo viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 2272 kb)
{kind=link}
P. tepidariorum Sex combs reduced-A expression. A stripe of Scr-A expression first appears at stage 6 (A). The stripe broadens during stage 7 (B), at the end of which it starts to split into two stripes, and a third new stripe appears posterior to the initial stripes (borders of expression marked by arrowheads) (C). At stage 8.1, there are four stripes located in the posterior parts of L2, L3, and L4 and the newest stripe appears in O1 (white arrow) (D and E). In limb bud stages, only the L3 and L4 limb buds carry Scr-A expression, mostly in their distal tips. Expression in L2 is restricted to the neuroectoderm (not shown), and expression in O1 is restricted to the posterior part of each hemisegment as well (F). At later stages, Scr-A expression in L3 and L4 refines to several rings in the distal part of the legs. Expression in L2 becomes undetectable. (G–I). At stage 10.1, Scr-A is visible in a neuroectodermal patch in L4 and a dot in O1 (arrow) (H). Each panel shows the same embryo viewed laterally (left) and ventrally (center and right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 266 kb)
{kind=link}
P. tepidariorum Sex combs reduced-B expression. Scr-B is first expressed in a cap-like domain (arrowhead) in the center of the germ disc at stage 5. The position of the cumulus is marked by a ‘c’ (A). The expression widens during stage 6. Scr-B now forms an open ring, localized roughly halfway between anterior rim of the opening germ disc and the future posterior center of the germ disc. The cells posterior to the ring also express Scr-B, but at a much lower level (B). At stage 7, the ring splits up into two stripes (arrowheads) (C) and, later, two new stripes appear (carets) (D). The most anterior stripe of expression lies between L1 and L2 at stage 8, the second anterior stripe lies between L2 and L3. The two posterior stripes cover L3 and L4. The weak expression of Scr-B continues posterior to these stripes (E and F). Scr-B is predominantly expressed in the limb buds and the ventral neuroectoderm of stage 9.1 embryos (G). The anterior expression border is directly posterior to the L1 limb bud. Expression of Scr-B continues in the opisthosoma, but is much weaker, except for a domain in the SAZ (arrow) (G). This expression in the SAZ continues to the end of segmentation (H and I). No more expression is visible at the posterior end of the embryo at stage 11 (white caret) (J). Scr-B expression forms multiple rings in the legs (H–J), but it is much more strongly expressed in L3 compared to L2 and L4. At late stage 9, the expression of Scr-B in the neuroectoderm can also be found in every segment of the opisthosoma (arrowhead in H). Each panel shows the same embryo viewed laterally (left) and ventrally (center and right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 393 kb)
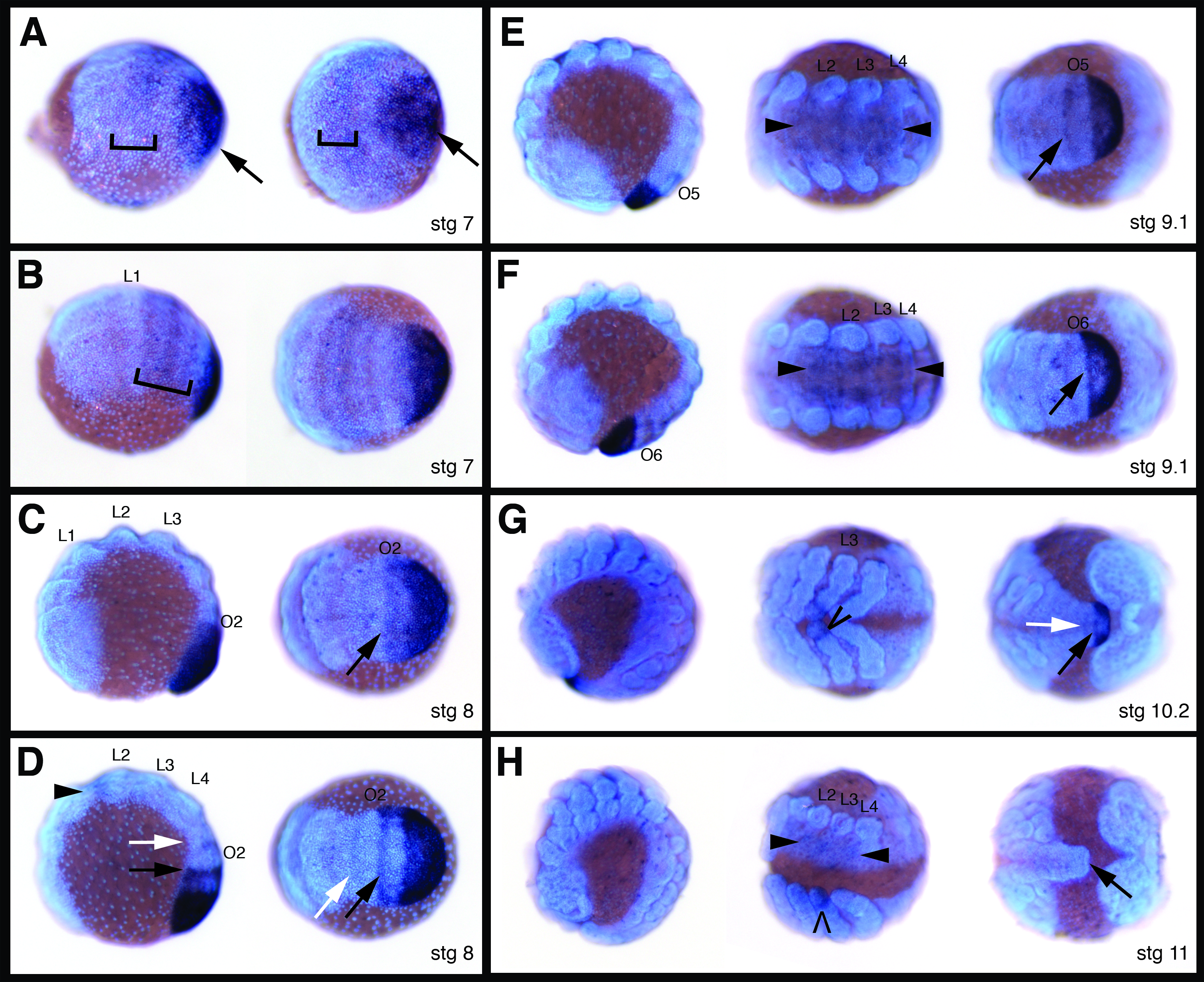{kind=link}
P. tepidariorum fushi tarazu expression. ftz is first expressed at stage 6 in a semicircle at the posterior end where the SAZ is forming (arrow) (A). The bracket marks an anterior broad stripe of ftz expression. This domain of expression gets weaker during stages 7 and 8, where mostly only the anterior border of the domain anterior to L2 (arrowhead in D) is visible. The expression is almost invisible when it is viewed ventrally. The expression in the SAZ persists, and does not clear from the posterior end (A–G). It emanates stripes at the anterior end of the SAZ (arrow in C–G). Stripes stay visible at the anterior border of the last formed segment (white arrow in D and G) until the next stripe of ftz expression forms. Only after segmentation finishes does the expression at the posterior disappear (arrow in H). Meanwhile, the anterior border of the Hox expression domain stretches from L2 to L4 and is predominantly found ventrally (black arrowheads in F and H). The legs on one side of a stage 11 embryo were removed to show the ventral Hox domain, which has concentrated to one spot per hemisegment (black arrowheads) within the L2–L4 domain (H). At stage 10, ftz expression appears in a ring near the distal tip of L3 (carets in G–H). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–H). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 7681 kb)
{kind=link}
P. tepidariorum Antennapedia-A expression. Antp-A expression first develops in the SAZ at stage 7 (A). This expression transforms into a stripe and is followed by cells that show only weak expression of Antp-A (B), before new expression appears at the posterior end of the SAZ (C). The first two stripes can be allocated to the first two opisthosomal segments at stage 8 (D). New stripes keep appearing from the SAZ (arrows, E–H) until the end of segmentation (G). At stage 9, the anterior border of Antp-A expression reaches into the posterior half of L4 (arrowhead, E). Expression is now also seen in the ventral neuroectoderm, where it forms longitudinal rows in each hemisegment (white arrowheads, E–G), and rings in the L4 appendage (caret, F and G). Throughout development, Antp-A expression is always strongest in O1 and the anterior half of O2. Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–G). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 6657 kb)
{kind=link}
P. tepidariorum Antennapedia-B expression. Antp-B first emerges in the first two opisthosomal segments at stage 8.2 (arrows) (A). The Antp-B domain expands anteriorly into the posterior part of L4 during stage 9 (white arrowheads) (B–D). Additionally, Antp-B forms rings of expression in the L4 appendage (carets in C and D). While initially the very posterior part of O2 is free of Antp-B expression (the O2/O3 border is demarcated by a black vertical line in D–F), during stage 10, expression can also be found at the O2/O3 border (arrows) (F). Furthermore, during stage 10, the expression pattern refines, in that the L4 and O1 Antp-B expression is mostly in the ventral neuroectoderm and not found dorsally, while the O2 expression is excluded from the most ventral region (E–G). Starting at stage 10, Antp-B is also expressed in one dot each in the opisthosomal limb buds on O4 and O5 (arrowheads) (E–G). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–G). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 5223 kb)
{kind=link}
P. tepidariorum Ultrabithorax-A expression. Ubx-A is first expressed at stage 8.1 in the SAZ and in a weak stripe anterior to that (arrow) (A). This stripe gets stronger and the posterior domain enlarges (B and C) so that all new tissue added posteriorly expresses Ubx-A at equal levels (D–F). The anterior border is located in O2, while the very anterior part of O2 initially does not express Ubx-A (segmental boundaries in C–F indicated by black vertical lines) until the end of stage 9.2, when it is expressed dorsally and in the neuroectoderm also in the anterior part of O2 (white arrow in E). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 4678 kb)
{kind=link}
P. tepidariorum Ultrabithorax-B expression. Ubx-B is first expressed at stage 8.2, in the SAZ, as well as in two weaker stripes in the O3 and O4 segments (arrows) (A). Expression is initially restricted to the posterior part of O3 (B), but eventually the anterior expression border extends ventrally into the posterior part of O2 (C–F), and is also found in the posterior part of the O2 limb bud (caret in E and F). Expression is strongest in O3, but otherwise is fairly uniform in all segments posterior to this one (D–F). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 4129 kb)
{kind=link}
P. tepidariorum abdominalA-A expression. abdA-A expression starts in the SAZ during stage 9.1, posterior to O5 (A and B). By stage 10.1, the anterior border of abdA-A expression has extended into the posterior part of O3 (arrowhead, C). While the dorsal border of abdA-A expression remains there, in the ventral neuroectoderm, the expression expands anteriorly into the posterior half of O2 (arrowhead, D). These borders of expression persist during later development (E to G). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 5068 kb)
{kind=link}
P. tepidariorum abdominalA-B expression. abdA-B expression first appears at stage 9.1 in the SAZ (A). Slightly later, shortly before the opisthosomal limb buds appear, abdA-B additionally emerges in the posterior part of O4 (B). From stage 9.2 onwards, abdA-B is expressed in the entire O4 segment and all posterior segments with strong expression in the opisthosomal limb buds (C–F). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 4092 kb)
{kind=link}
P. tepidariorum AbdominalB-A expression. AbdB-A is first expressed at stage 9.1 in O6 (arrowhead) and in a stripe in the anterior-most portion of the SAZ (A). The anterior expression border subsequently shifts anteriorly, first into the posterior part of O5 (arrowhead) (B and C), and the dorsal part of the AbdB-A domain then shifts into the posterior portion of O4 (arrowhead) (D and E). From the beginning of opisthosomal limb bud development, it is strongly expressed in the O5 limb buds. At stage 9.2, the ventral part of the AbdB-A domain expands into the posterior half of O2 (arrow). These anterior borders remain the same throughout the rest of embryonic development (E–G). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in D–G). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 245 kb)
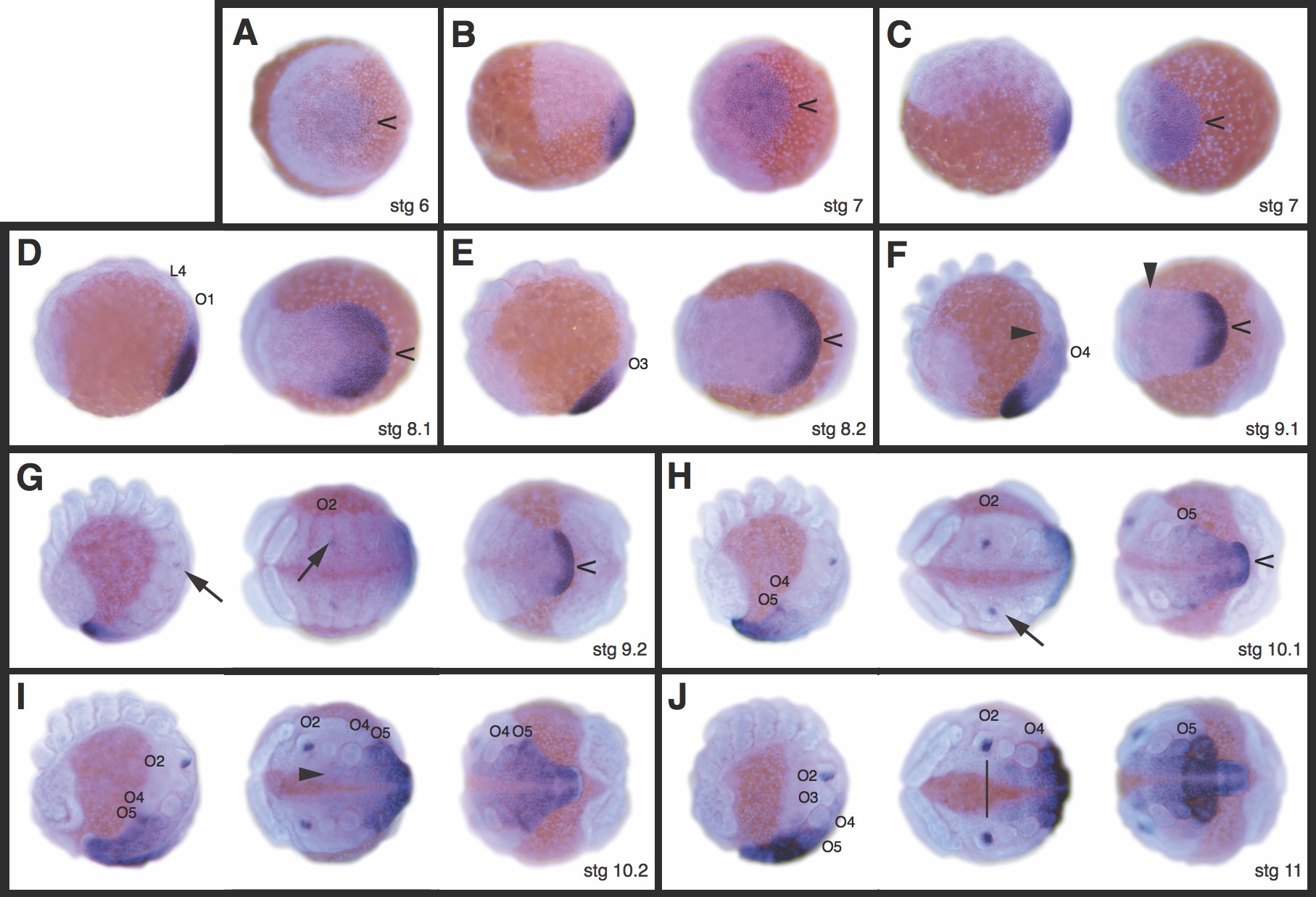{kind=link}
P. tepidariorum AbdominalB-B expression. AbdB-B expression emerges with the appearance of the SAZ at stage 6 (A). It is continuously expressed in the SAZ until the end of segmentation (caret) (B–H). Additionally, from stage 9.1 on, weak AbdB-B expression is found posterior to the O3/O4 border (arrowhead) (F). Slightly later, additional expression appears in the O2 limb buds, in the prospective genital opening (arrow) (G and H). Dorsally, the AbdB-B expression domain extends from O5 to the posterior end (H–J), but in the ventral neuroectoderm, the AbdB-B expression border is located in the posterior half of O3 (arrowhead in I). The vertical line delineates the O2/O3 border, which the AbB-B expression does not reach in the neuroectoderm (J). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in G–J). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 332 kb)
Hox gene expression in P. tepidariorum. Detailed description and comparison of the expression patterns of the Hox gene paralogs. (DOCX 143 kb)
Trinotate report of the best Blast hits, protein domain predictions and GO categories extracted from the Blast result, and the Pfam domain prediction for each of the aug3 predictions. (XLSX 7616 kb)
Acknowledgements
We thank Wim Damen for all his support and mentoring, and for many productive discussions about spider genetics and development.
Funding
This work was supported by NIH grant NHGRI U54 HG003273 to RAG, the National Science Foundation (IOS-0951886 to NAA and DEB-1257053 to JHW), a Leverhulme visiting fellowship for EES (VF-2012-016), funding and PhD studentships (DJL, LG and AS) from Oxford Brookes University, and a Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) scholarship to CLBP. N-MP was funded by the Deutsche Forschungsgemeinschaft (grant numbers PR 1109/4-1, PR 1109/7-1 and PR 1109/6-1 to N-MP). Additional financial backing has been received from the Göttingen Graduate School for Neurosciences, Biophysics and Molecular Biosciences (GGNB), the Göttingen Center for Molecular Biosciences (GZMB), and the University of Göttingen (GAU). NT is supported by a Christiane-Nüsslein-Volhard-Foundation fellowship and a “Women in Science” Award by L'Oréal Deutschland and the Deutsche UNESCO-Kommission. NP has been funded by the Volkswagen Foundation (project number: 85 983) and the Emmy Noether Programme of the Deutsche Forschungsgemeinschaft (grant number: PO 1648/3-1). Funding to RJ was provided by the Swedish Research Council VR grant 621-2011-4703.
Availability of data and material
The raw sequences for P. tepidariorum have been deposited in the NCBI SRA: BioSample ID SAMN01932302. For C. sculpturatus, the raw sequences have been deposited in the NCBI SRA: BioSample SAMN02617800. For P. tepidariorum the assembly has been deposited in the NCBI: BioProject PRJNA167405 (Accession: AOMJ00000000). For C. sculpturatus the final assembly has been deposited in the NCBI: BioProject PRJNA168116.
The annotated P. tepidariorum genome is available at https://i5k.nal.usda.gov/JBrowse-partep.
The C. sculpturatus RNA-Seq reads from a juvenile, adult female and adult males, respectively, are available at http://www.ncbi.nlm.nih.gov/sra/SRX911075; http://www.ncbi.nlm.nih.gov/sra/SRX911064; http://www.ncbi.nlm.nih.gov/sra/SRX911079.
The annotated C. sculpturatus genome is available at https://apollo.nal.usda.gov/cenexi/jbrowse.
Authors’ contributions
APM, NP, N-MP, HO, JC, and SR conceived the project. Library construction, genome sequencing and assembly: N-MP SR, SD, SLL, HC, HVD, HD, YH, JQ, SCM, DSTH, KCW, DMM, RAG, VLG, and JC. Gene duplication analyses: TC, PPS, NAA, and JFF. Hox gene analysis: EES, PPS, DJL, and CS. Parasteatoda genome data was analyzed by all authors. The manuscript was written by APM, EES, PPS, TC, IM, NAA, MS, TW, NP, N-MP, JFF, and SR with the help of all other authors. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-017-0399-x) contains supplementary material, which is available to authorized users.
Contributor Information
Evelyn E. Schwager, Email: evelyn_schwager@uml.edu
Prashant P. Sharma, Email: prashant.sharma@wisc.edu
Thomas Clarke, Email: toby.h.clarke@gmail.com.
Daniel J. Leite, Email: danielleite@brookes.ac.uk
Torsten Wierschin, Email: torsten.wierschin@gmail.com.
Matthias Pechmann, Email: pechmanm@uni-koeln.de.
Yasuko Akiyama-Oda, Email: yasuko@brh.co.jp.
Lauren Esposito, Email: espositola@gmail.com.
Jesper Bechsgaard, Email: jesper.bechsgaard@bios.au.dk.
Trine Bilde, Email: trine.bilde@bios.au.dk.
Alexandra D. Buffry, Email: alex.buffry-2015@brookes.ac.uk
Hsu Chao, Email: hchao@bcm.edu.
Huyen Dinh, Email: hdinh@bcm.edu.
HarshaVardhan Doddapaneni, Email: doddapan@bcm.edu.
Shannon Dugan, Email: sdugan@bcm.edu.
Cornelius Eibner, Email: cornelius.eibner@uni-jena.de.
Cassandra G. Extavour, Email: extavour@oeb.harvard.edu
Peter Funch, Email: peter.funch@biology.au.dk.
Jessica Garb, Email: jessica_garb@uml.edu.
Luis B. Gonzalez, Email: luis.baudouin.gonzalez-2016@brookes.ac.uk
Vanessa L. Gonzalez, Email: gonzalezv@si.edu
Sam Griffiths-Jones, Email: sam.griffiths-jones@manchester.ac.uk.
Yi Han, Email: yhan@bcm.edu.
Cheryl Hayashi, Email: cheryl.hayashi@ucr.edu.
Maarten Hilbrant, Email: m.hilbrant@uni-koeln.de.
Daniel S. T. Hughes, Email: dsthughes@gmail.com
Ralf Janssen, Email: ralf.janssen@geo.uu.se.
Sandra L. Lee, Email: sllee@bcm.edu
Ignacio Maeso, Email: nacho.maeso@gmail.com.
Shwetha C. Murali, Email: shwethacm@gmail.com
Donna M. Muzny, Email: donnam@bcm.edu
Rodrigo Nunes da Fonseca, Email: rodrigo.nunes.da.fonseca@gmail.com.
Christian L. B. Paese, Email: 15054968@brookes.ac.uk
Jiaxin Qu, Email: jiaxinqu@gmail.com.
Matthew Ronshaugen, Email: matthew.ronshaugen@manchester.ac.uk.
Christoph Schomburg, Email: c.schomburg@stud.uni-goettingen.de.
Anna Schönauer, Email: aschonauer@brookes.ac.uk.
Angelika Stollewerk, Email: a.stollewerk@qmul.ac.uk.
Montserrat Torres-Oliva, Email: montserrat.torres-oliva@biologie.uni-goettingen.de.
Natascha Turetzek, Email: natascha.turetzek@biologie.uni-goettingen.de.
Bram Vanthournout, Email: bram.vanthournout@ugent.be.
John H. Werren, Email: werr@mail.rochester.edu
Carsten Wolff, Email: carsten.wolff.1@rz.hu-berlin.de.
Kim C. Worley, Email: kworley@bcm.edu
Gregor Bucher, Email: gbucher1@gwdg.de.
Richard A. Gibbs, Email: agibbs@bcm.edu
Jonathan Coddington, Email: coddington@si.edu.
Hiroki Oda, Email: hoda@brh.co.jp.
Mario Stanke, Email: mario.stanke@uni-greifswald.de.
Nadia A. Ayoub, Email: ayoubn@wlu.edu
Nikola-Michael Prpic, Email: nprpic@uni-goettingen.de.
Jean-François Flot, Email: jflot@ulb.ac.be.
Nico Posnien, Email: nico.posnien@biologie.uni-goettingen.de.
Stephen Richards, Email: stephenr@bcm.edu.
Alistair P. McGregor, Email: amcgregor@brookes.ac.uk
References
- 1.Ohno S. Evolution by Gene Duplication. Berlin: Springer; 1970. [Google Scholar]
- 2.Semon M, Wolfe KH. Consequences of genome duplication. Curr Opin Genet Dev. 2007;17(6):505–12. doi: 10.1016/j.gde.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Yun S, Furlong M, Sim M, Cho M, Park S, Cho EB, Reyes-Alcaraz A, Hwang JI, Kim J, Seong JY. Prevertebrate local gene duplication facilitated expansion of the neuropeptide GPCR superfamily. Mol Biol Evol. 2015;32(11):2803–17. doi: 10.1093/molbev/msv179. [DOI] [PubMed] [Google Scholar]
- 4.Chipman AD, Ferrier DE, Brena C, Qu J, Hughes DS, Schroder R, Torres-Oliva M, Znassi N, Jiang H, Almeida FC, et al. The first myriapod genome sequence reveals conservative arthropod gene content and genome organisation in the centipede Strigamia maritima. PLoS Biol. 2014;12(11) doi: 10.1371/journal.pbio.1002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fawcett JA, Maere S, Van de Peer Y. Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc Natl Acad Sci U S A. 2009;106(14):5737–42. doi: 10.1073/pnas.0900906106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z, Baniaga AE, Sessa EB, Scascitelli M, Graham SW, Rieseberg LH, Barker MS. Early genome duplications in conifers and other seed plants. Sci Adv. 2015;1(10) doi: 10.1126/sciadv.1501084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387(6634):708–13. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- 8.Ma LJ, Ibrahim AS, Skory C, Grabherr MG, Burger G, Butler M, Elias M, Idnurm A, Lang BF, Sone T, et al. Genomic analysis of the basal lineage fungus Rhizopus oryzae reveals a whole-genome duplication. PLoS Genet. 2009;5(7) doi: 10.1371/journal.pgen.1000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aury JM, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Segurens B, Daubin V, Anthouard V, Aiach N, et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444(7116):171–8. doi: 10.1038/nature05230. [DOI] [PubMed] [Google Scholar]
- 10.Martens C, Van de Peer Y. The hidden duplication past of the plant pathogen Phytophthora and its consequences for infection. BMC Genomics. 2010;11:353. doi: 10.1186/1471-2164-11-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nossa CW, Havlak P, Yue JX, Lv J, Vincent KY, Brockmann HJ, Putnam NH. Joint assembly and genetic mapping of the Atlantic horseshoe crab genome reveals ancient whole genome duplication. GigaScience. 2014;3:9. doi: 10.1186/2047-217X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bisbee CA, Baker MA, Wilson AC, Haji-Azimi I, Fischberg M. Albumin phylogeny for clawed frogs (Xenopus) Science. 1977;195(4280):785–7. doi: 10.1126/science.65013. [DOI] [PubMed] [Google Scholar]
- 13.Amores A, Force A, Yan YL, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang YL, et al. Zebrafish hox clusters and vertebrate genome evolution. Science. 1998;282(5394):1711–4. doi: 10.1126/science.282.5394.1711. [DOI] [PubMed] [Google Scholar]
- 14.Flot JF, Hespeels B, Li X, Noel B, Arkhipova I, Danchin EG, Hejnol A, Henrissat B, Koszul R, Aury JM, et al. Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature. 2013;500(7463):453–7. doi: 10.1038/nature12326. [DOI] [PubMed] [Google Scholar]
- 15.Edger PP, Pires JC. Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009;17(5):699–717. doi: 10.1007/s10577-009-9055-9. [DOI] [PubMed] [Google Scholar]
- 16.Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, Fukui A, Hikosaka A, Suzuki A, Kondo M, et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538(7625):336–43. doi: 10.1038/nature19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431(7011):946–57. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- 18.Davis JC, Petrov DA. Do disparate mechanisms of duplication add similar genes to the genome? Trends Genet. 2005;21(10):548–51. doi: 10.1016/j.tig.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151(4):1531–45. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154(1):459–73. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch M, O’Hely M, Walsh B, Force A. The probability of preservation of a newly arisen gene duplicate. Genetics. 2001;159(4):1789–804. doi: 10.1093/genetics/159.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–5. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 23.Kellis M, Birren BW, Lander ES. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428(6983):617–24. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- 24.Putnam NH, Butts T, Ferrier DE, Furlong RF, Hellsten U, Kawashima T, Robinson-Rechavi M, Shoguchi E, Terry A, Yu JK, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453(7198):1064–71. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- 25.Hakes L, Pinney JW, Lovell SC, Oliver SG, Robertson DL. All duplicates are not equal: the difference between small-scale and genome duplication. Genome Biol. 2007;8(10):R209. doi: 10.1186/gb-2007-8-10-r209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurley IA, Mueller RL, Dunn KA, Schmidt EJ, Friedman M, Ho RK, Prince VE, Yang Z, Thomas MG, Coates MI. A new time-scale for ray-finned fish evolution. Proc Biol Sci. 2007;274(1609):489–98. doi: 10.1098/rspb.2006.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furlong RF, Holland PW. Were vertebrates octoploid? Philos Trans R Soc Lond B Biol Sci. 2002;357(1420):531–44. doi: 10.1098/rstb.2001.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donoghue PC, Purnell MA. Genome duplication, extinction and vertebrate evolution. Trends Ecol Evol. 2005;20(6):312–9. doi: 10.1016/j.tree.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 29.McGinnis W, Krumlauf R. Homeobox genes and axial patterning. Cell. 1992;68(2):283–302. doi: 10.1016/0092-8674(92)90471-N. [DOI] [PubMed] [Google Scholar]
- 30.Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3(10) doi: 10.1371/journal.pbio.0030314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kenny NJ, Chan KW, Nong W, Qu Z, Maeso I, Yip HY, Chan TF, Kwan HS, Holland PW, Chu KH, et al. Ancestral whole-genome duplication in the marine chelicerate horseshoe crabs. Heredity. 2016;116(2):190–9. doi: 10.1038/hdy.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ricci CN. Ecology of bdelloids: how to be successful. Hydrobiologia. 1987;147:117–27. doi: 10.1007/BF00025734. [DOI] [Google Scholar]
- 33.Rudkin DM, Young GA. Horseshoe crabs – an ancient ancestry revealed. In: Tanacredi JT, Botton ML, Smith DR, editors. Biology and Conservation of Horseshoe Crabs. Boston: Springer; 2009. pp. 25–44. [Google Scholar]
- 34.Clarke TH, Garb JE, Hayashi CY, Arensburger P, Ayoub NA. Spider transcriptomes identify ancient large-scale gene duplication event potentially important in silk gland evolution. Genome Biol Evol. 2015;7(7):1856–70. doi: 10.1093/gbe/evv110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke TH, Garb JE, Hayashi CY, Haney RA, Lancaster AK, Corbett S, Ayoub NA. Multi-tissue transcriptomics of the black widow spider reveals expansions, co-options, and functional processes of the silk gland gene toolkit. BMC Genomics. 2014;15:365. doi: 10.1186/1471-2164-15-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Z, Yu Y, Wu Y, Hao P, He Y, Zhao H, Li Y, Zhao G, Li X, Li W, et al. Genome-wide analysis of homeobox genes from Mesobuthus martensii reveals Hox gene duplication in scorpions. Insect Biochem Mol Biol. 2015;61:25–33. doi: 10.1016/j.ibmb.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Janssen R, Le Gouar M, Pechmann M, Poulin F, Bolognesi R, Schwager EE, Hopfen C, Colbourne JK, Budd GE, Brown SJ, et al. Conservation, loss, and redeployment of Wnt ligands in protostomes: implications for understanding the evolution of segment formation. BMC Evol Biol. 2010;10:374. doi: 10.1186/1471-2148-10-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwager EE, Schoppmeier M, Pechmann M, Damen WG. Duplicated Hox genes in the spider Cupiennius salei. Front Zool. 2007;4:10. doi: 10.1186/1742-9994-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma PP, Santiago MA, Gonzalez-Santillan E, Monod L, Wheeler WC. Evidence of duplicated Hox genes in the most recent common ancestor of extant scorpions. Evol Dev. 2015;17(6):347–55. doi: 10.1111/ede.12166. [DOI] [PubMed] [Google Scholar]
- 40.Sharma PP, Schwager EE, Extavour CG, Wheeler WC. Hox gene duplications correlate with posterior heteronomy in scorpions. Proc Biol Sci. 2014;281(1792). [DOI] [PMC free article] [PubMed]
- 41.Turetzek N, Pechmann M, Schomburg C, Schneider J, Prpic NM. Neofunctionalization of a duplicate dachshund gene underlies the evolution of a novel leg segment in arachnids. Mol Biol Evol. 2016;33(1):109–21. doi: 10.1093/molbev/msv200. [DOI] [PubMed] [Google Scholar]
- 42.Fuzita FJ, Pinkse MW, Patane JS, Juliano MA, Verhaert PD, Lopes AR. Biochemical, transcriptomic and proteomic analyses of digestion in the scorpion Tityus serrulatus: insights into function and evolution of digestion in an ancient arthropod. PLoS One. 2015;10(4) doi: 10.1371/journal.pone.0123841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuzita FJ, Pinkse MW, Patane JS, Verhaert PD, Lopes AR. High throughput techniques to reveal the molecular physiology and evolution of digestion in spiders. BMC Genomics. 2016;17:716. doi: 10.1186/s12864-016-3048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leite DJ, Ninova M, Hilbrant M, Arif S, Griffiths-Jones S, Ronshaugen M, McGregor AP. Pervasive microRNA duplication in Chelicerates: insights from the embryonic microRNA repertoire of the spider Parasteatoda tepidariorum. Genome Biol Evol. 2016;8(7):2133–44. doi: 10.1093/gbe/evw143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawson D, Arensburger P, Atkinson P, Besansky NJ, Bruggner RV, Butler R, Campbell KS, Christophides GK, Christley S, Dialynas E, et al. VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Res. 2009;37(Database issue):D583–7. doi: 10.1093/nar/gkn857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gulia-Nuss M, Nuss AB, Meyer JM, Sonenshine DE, Roe RM, Waterhouse RM, Sattelle DB, de la Fuente J, Ribeiro JM, Megy K, et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat Commun. 2016;7:10507. doi: 10.1038/ncomms10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grbic M, Van Leeuwen T, Clark RM, Rombauts S, Rouze P, Grbic V, Osborne EJ, Dermauw W, Ngoc PC, Ortego F, et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature. 2011;479(7374):487–92. doi: 10.1038/nature10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao Z, Yu Y, Wu Y, Hao P, Di Z, He Y, Chen Z, Yang W, Shen Z, He X, et al. The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nat Commun. 2013;4:2602. doi: 10.1038/ncomms3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanggaard KW, Bechsgaard JS, Fang X, Duan J, Dyrlund TF, Gupta V, Jiang X, Cheng L, Fan D, Feng Y, et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat Commun. 2014;5:3765. doi: 10.1038/ncomms4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babb PL, Lahens NF, Correa-Garhwal SM, Nicholson DN, Kim EJ, Hogenesch JB, Kuntner M, Higgins L, Hayashi CY, Agnarsson I, et al. The Nephila clavipes genome highlights the diversity of spider silk genes and their complex expression. Nat Genet. 2017;49(6):895–903. doi: 10.1038/ng.3852. [DOI] [PubMed] [Google Scholar]
- 51.Schwager EE, Schoenauer A, Leite DJ, Sharma PP, McGregor AP. Chelicerata. In: Wanninger A, editor. Evolutionary Developmental Biology of Invertebrates. Berlin: Spinger; 2015. [Google Scholar]
- 52.Yoshida H. A revision of the genus Achaearanea (Araneae: Theridiidae) Acta Arachnologica. 2008;57(1):37–40. doi: 10.2476/asjaa.57.37. [DOI] [Google Scholar]
- 53.Sharma PP, Kaluziak ST, Pérez-Porro AR, González VL, Hormiga G, Wheeler WC, Giribet G. Phylogenomic interrogation of Arachnida reveals systemic conflicts in phylogenetic signal. Mol Biol Evol. 2014;31:2963–84. doi: 10.1093/molbev/msu235. [DOI] [PubMed] [Google Scholar]
- 54.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 55.Altenhoff AM, Skunca N, Glover N, Train CM, Sueki A, Pilizota I, Gori K, Tomiczek B, Muller S, Redestig H, et al. The OMA orthology database in 2015: function predictions, better plant support, synteny view and other improvements. Nucleic Acids Res. 2015;43(Database issue):D240–9. doi: 10.1093/nar/gku1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331(6017):555–61. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schomburg C, Turetzek N, Schacht MI, Schneider J, Kirfel P, Prpic NM, Posnien N. Molecular characterization and embryonic origin of the eyes in the common house spider Parasteatoda tepidariorum. EvoDevo. 2015;6:15. doi: 10.1186/s13227-015-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samadi L, Schmid A, Eriksson BJ. Differential expression of retinal determination genes in the principal and secondary eyes of Cupiennius salei Keyserling (1877) EvoDevo. 2015;6:16. doi: 10.1186/s13227-015-0010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janssen R, Schönauer A, Weber M, Turetzek N, Hogvall M, Goss GE, Patel NH, McGregor AP, Hilbrant M. The evolution and expression of panarthropod frizzled genes. Front Ecol Evol. 2015;3:96. doi: 10.3389/fevo.2015.00096. [DOI] [Google Scholar]
- 60.Grabherr MG, Russell P, Meyer M, Mauceli E, Alfoldi J, Di Palma F, Lindblad-Toh K. Genome-wide synteny through highly sensitive sequence alignment: Satsuma. Bioinformatics. 2010;26(9):1145–51. doi: 10.1093/bioinformatics/btq102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(Database issue):D1178–86. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22(2):160–74. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 63.Kagale S, Robinson SJ, Nixon J, Xiao R, Huebert T, Condie J, Kessler D, Clarke WE, Edger PP, Links MG, et al. Polyploid evolution of the Brassicaceae during the Cenozoic era. Plant Cell. 2014;26(7):2777–91. doi: 10.1105/tpc.114.126391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schaeper ND, Prpic NM, Wimmer EA. A clustered set of three Sp-family genes is ancestral in the Metazoa: evidence from sequence analysis, protein domain structure, developmental expression patterns and chromosomal location. BMC Evol Biol. 2010;10:88. doi: 10.1186/1471-2148-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akiyama-Oda Y, Oda H. Cell migration that orients the dorsoventral axis is coordinated with anteroposterior patterning mediated by Hedgehog signaling in the early spider embryo. Development. 2010;137(8):1263–73. doi: 10.1242/dev.045625. [DOI] [PubMed] [Google Scholar]
- 66.Pechmann M, Schwager EE, Turetzek N, Prpic NM. Regressive evolution of the arthropod tritocerebral segment linked to functional divergence of the Hox gene labial. Proc Biol Sci. 2015;282(1814):20151162. doi: 10.1098/rspb.2015.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Waddington J, Rudkin DM, Dunlop JA. A new mid-Silurian aquatic scorpion-one step closer to land? Biol Lett. 2015;11(1):20140815. doi: 10.1098/rsbl.2014.0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dunlop JA. Geological history and phylogeny of Chelicerata. Arthropod Struct Dev. 2010;39(2-3):124–42. doi: 10.1016/j.asd.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 69.Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424(6945):194–7. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- 70.Papp B, Pal C, Hurst LD. Evolution of cis-regulatory elements in duplicated genes of yeast. Trends Genet. 2003;19(8):417–22. doi: 10.1016/S0168-9525(03)00174-4. [DOI] [PubMed] [Google Scholar]
- 71.Abzhanov A, Popadic A, Kaufman TC. Chelicerate Hox genes and the homology of arthropod segments. Evol Dev. 1999;1(2):77–89. doi: 10.1046/j.1525-142x.1999.99014.x. [DOI] [PubMed] [Google Scholar]
- 72.Damen WG, Hausdorf M, Seyfarth EA, Tautz D. A conserved mode of head segmentation in arthropods revealed by the expression pattern of Hox genes in a spider. Proc Natl Acad Sci U S A. 1998;95(18):10665–70. doi: 10.1073/pnas.95.18.10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, Martin JW, Cunningham CW. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463(7284):1079–83. doi: 10.1038/nature08742. [DOI] [PubMed] [Google Scholar]
- 74.Scholtz G, Kamenz C. The book lungs of Scorpiones and Tetrapulmonata (Chelicerata, Arachnida): evidence for homology and a single terrestrialisation event of a common arachnid ancestor. Zoology. 2006;109(1):2–13. doi: 10.1016/j.zool.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 75.Rota-Stabelli O, Daley AC, Pisani D. Molecular timetrees reveal a Cambrian colonization of land and a new scenario for ecdysozoan evolution. Curr Biol. 2013;23(5):392–8. doi: 10.1016/j.cub.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 76.Lozano-Fernandez J, Carton R, Tanner AR, Puttick MN, Blaxter M, Vinther J, Olesen J, Giribet G, Edgecombe GD, Pisani D. A molecular palaeobiological exploration of arthropod terrestrialization. Philos Trans R Soc Lond B Biol Sci. 2016;371(1699): pii: 20150133. [DOI] [PMC free article] [PubMed]
- 77.Sharma PP, Wheeler WC. Cross-bracing uncalibrated nodes in molecular dating improves congruence of fossil and molecular age estimates. Front Zool. 2014;11:57. doi: 10.1186/s12983-014-0057-x. [DOI] [Google Scholar]
- 78.Selden PA, Shear WA, Sutton MD. Fossil evidence for the origin of spider spinnerets, and a proposed arachnid order. Proc Natl Acad Sci U S A. 2008;105(52):20781–5. doi: 10.1073/pnas.0809174106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Freeling M, Scanlon MJ, Fowler JE. Fractionation and subfunctionalization following genome duplications: mechanisms that drive gene content and their consequences. Curr Opin Genet Dev. 2015;35:110–8. doi: 10.1016/j.gde.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 80.Glasauer SM, Neuhauss SC. Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol Genet Genomics. 2014;289(6):1045–60. doi: 10.1007/s00438-014-0889-2. [DOI] [PubMed] [Google Scholar]
- 81.Holland LZ. Evolution of new characters after whole genome duplications: insights from amphioxus. Semin Cell Dev Biol. 2013;24(2):101–9. doi: 10.1016/j.semcdb.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 82.Clarke JT, Lloyd GT, Friedman M. Little evidence for enhanced phenotypic evolution in early teleosts relative to their living fossil sister group. Proc Natl Acad Sci U S A. 2016;113(41):11531–6. doi: 10.1073/pnas.1607237113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hilbrant M, Damen WG, McGregor AP. Evolutionary crossroads in developmental biology: the spider Parasteatoda tepidariorum. Development. 2012;139(15):2655–62. doi: 10.1242/dev.078204. [DOI] [PubMed] [Google Scholar]
- 84.McGregor AP, Hilbrant M, Pechmann M, Schwager EE, Prpic NM, Damen WG. Cupiennius salei and Achaearanea tepidariorum: Spider models for investigating evolution and development. Bioessays. 2008;30(5):487–98. doi: 10.1002/bies.20744. [DOI] [PubMed] [Google Scholar]
- 85.Oda H, Akiyama-Oda Y. Differing strategies for forming the arthropod body plan: lessons from Dpp, Sog and Delta in the fly Drosophila and spider Achaearanea. Develop Growth Differ. 2008;50(4):203–14. doi: 10.1111/j.1440-169X.2008.00998.x. [DOI] [PubMed] [Google Scholar]
- 86.Posnien N, Zeng V, Schwager EE, Pechmann M, Hilbrant M, Keefe JD, Damen WGM, Prpic N-M, McGregor AP, Extavour CG. A comprehensive reference transcriptome resource for the common house spider Parasteatoda tepidariorum. PLoS ONE. 2014;9(8) doi: 10.1371/journal.pone.0104885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108(4):1513–8. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.BCM-HGSC Software. https://www.hgsc.bcm.edu/software/.
- 89.Flot JF, Marie-Nelly H, Koszul R. Contact genomics: scaffolding and phasing (meta)genomes using chromosome 3D physical signatures. FEBS Lett. 2015;589(20 Pt A):2966–74. doi: 10.1016/j.febslet.2015.04.034. [DOI] [PubMed] [Google Scholar]
- 90.Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW, et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016;26(3):342–50. doi: 10.1101/gr.193474.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scalable Nucleotide Alignment Program. http://snap.cs.berkeley.edu.
- 92.Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24(5):637–44. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 93.Oda H, Nishimura O, Hirao Y, Tarui H, Agata K, Akiyama-Oda Y. Progressive activation of Delta-Notch signaling from around the blastopore is required to set up a functional caudal lobe in the spider Achaearanea tepidariorum. Development. 2007;134(12):2195–205. doi: 10.1242/dev.004598. [DOI] [PubMed] [Google Scholar]
- 94.Smit A, Hubley R, Green P. Repeatmasker open-3.0. 1996-2010. http://www.repeatmasker.org.
- 95.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21(Suppl 1):i351–8. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 97.Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9(1):R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 99.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26(7):873–81. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.UniProtConsortium Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42(Database issue):D191–8. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 102.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10(1):71–3. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Trinotate: Transcriptome Functional Annotation and Analysis. https://trinotate.github.io/.
- 106.HMMER: biosequence analysis using profile hidden Markov models. http://hmmer.org/.
- 107.HPC GridRunner. http://hpcgridrunner.github.io/.
- 108.SQLite. https://www.sqlite.org/.
- 109.JBrowse/Web Apollo Parasteatoda tepidariorum. https://i5k.nal.usda.gov/JBrowse-partep.
- 110.Campbell MS, Holt C, Moore B, Yandell M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr Protoc Bioinformatics. 2014;48:4.11.11–39. doi: 10.1002/0471250953.bi0411s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.RNA sequencing of Centruroides exilicauda whole organism sample CEXI.00-juvenile. http://www.ncbi.nlm.nih.gov/sra/SRX911075.
- 112.RNA sequencing of Centruroides exilicauda whole organism sample CEXI.00-female. http://www.ncbi.nlm.nih.gov/sra/SRX911064.
- 113.RNA sequencing of Centruroides exilicauda whole organism sample CEXI.00-male. http://www.ncbi.nlm.nih.gov/sra/SRX911079.
- 114.Centruroides Genome Browser. https://apollo.nal.usda.gov/cenexi/jbrowse.
- 115.Centruroides sculpturatus MAKER annotation. ftp://ftp.hgsc.bcm.edu/I5K-pilot/Bark_scorpion/.
- 116.Battelle BA, Ryan JF, Kempler KE, Saraf SR, Marten CE, Warren WC, Minx P, Montague MJ, Green PJ, Schmidt SA, Fulton L, Patel NH, Protas ME, Wilson RK, Porter ML. Opsin repertoire and expression patterns in horseshoe crabs: evidence from the genome of Limulus polyphemus (Arthropoda: Chelicerata). Genome Biol Evol. 2016;8(5):1571-89. doi:10.1093/gbe/evw100. [DOI] [PMC free article] [PubMed]
- 117.Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7) doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–2. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Altenhoff AM, Gil M, Gonnet GH, Dessimoz C. Inferring hierarchical orthologous groups from orthologous gene pairs. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0053786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39(Database issue):D289–94. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41(Database issue):D358–65. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 124.Le SQ, Dang CC, Gascuel O. Modeling protein evolution with several amino acid replacement matrices depending on site rates. Mol Biol Evol. 2012;29(10):2921–36. doi: 10.1093/molbev/mss112. [DOI] [PubMed] [Google Scholar]
- 125.Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol. 1996;11(9):367–72. doi: 10.1016/0169-5347(96)10041-0. [DOI] [PubMed] [Google Scholar]
- 126.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 127.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.TreeSoft: Softwares for Phylogenetic Trees. http://treesoft.sourceforge.net/treebest.shtml.
- 130.McCarthy FM, Wang N, Magee GB, Nanduri B, Lawrence ML, Camon EB, Barrell DG, Hill DP, Dolan ME, Williams WP, et al. AgBase: a functional genomics resource for agriculture. BMC Genomics. 2006;7:229. doi: 10.1186/1471-2164-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Edwards JH. The Oxford Grid. Ann Hum Genet. 1991;55(Pt 1):17–31. doi: 10.1111/j.1469-1809.1991.tb00394.x. [DOI] [PubMed] [Google Scholar]
- 132.Orthodotter. http://www.genoscope.cns.fr/orthodotter.
- 133.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhong Y-F, Butts T, Holland PWH. HomeoDB: a database of homeobox gene diversity. Evol Dev. 2008;10(5):516–8. doi: 10.1111/j.1525-142X.2008.00266.x. [DOI] [PubMed] [Google Scholar]
- 135.Zhong Y-F, Holland PWH. HomeoDB2: functional expansion of a comparative homeobox gene database for evolutionary developmental biology. Evol Dev. 2011;13(6):567–8. doi: 10.1111/j.1525-142X.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43(Database issue):D222–6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Holland PW, Booth HA, Bruford EA. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007;5:47. doi: 10.1186/1741-7007-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sharma PP, Schwager EE, Extavour CG, Giribet G. Hox gene expression in the harvestman Phalangium opilio reveals divergent patterning of the chelicerate opisthosoma. Evol Dev. 2012;14(5):450–63. doi: 10.1111/j.1525-142X.2012.00565.x. [DOI] [PubMed] [Google Scholar]
- 141.Akiyama-Oda Y, Oda H. Early patterning of the spider embryo: a cluster of mesenchymal cells at the cumulus produces Dpp signals received by germ disc epithelial cells. Development. 2003;130(9):1735–47. doi: 10.1242/dev.00390. [DOI] [PubMed] [Google Scholar]
- 142.Prpic NM, Schoppmeier M, Damen WG. The American Wandering Spider Cupiennius salei. Cold Spring Harb Protoc. 2008 doi: 10.1101/pdb.emo103. [DOI] [PubMed] [Google Scholar]
- 143.Mittmann B, Wolff C. Embryonic development and staging of the cobweb spider Parasteatoda tepidariorum C. L. Koch, 1841 (syn.: Achaearanea tepidariorum; Araneomorphae; Theridiidae) Dev Genes Evol. 2012;222(4):189–216. doi: 10.1007/s00427-012-0401-0. [DOI] [PubMed] [Google Scholar]
- 144.Bond JE, Garrison NL, Hamilton CA, Godwin RL, Hedin M, Agnarsson I. Phylogenomics resolves a spider backbone phylogeny and rejects a prevailing paradigm for orb web evolution. Curr Biol. 2014;24(15):1765–71. doi: 10.1016/j.cub.2014.06.034. [DOI] [PubMed] [Google Scholar]
- 145.Sharma PP, Giribet G. A revised dated phylogeny of the arachnid order Opiliones. Front Genet. 2014;5:255. doi: 10.3389/fgene.2014.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Duplicated genes in Parasteatoda and Centruroides in comparison to each of the single-copy arthropods. (XLSX 55 kb)
Duplicated genes: three gene blocks. (PDF 36 kb)
Duplicated genes: four gene blocks. (PDF 36 kb)
Duplicated genes: five gene blocks. (PDF 36 kb)
P. tepidariorum homeobox genes. (XLSX 56 kb)
C. sculpturatus homeobox genes. (XLSX 54 kb)
Gene tree analysis of individual Hox genes. Blue branches indicate scorpion sequences; red branches indicate spider sequences. Gene trees are shown for labial (A), proboscipedia (B), Deformed (C), Sex combs reduced (D), Antennapedia (E), Ultrabithorax (F), abdominal-A, (G), and Abdominal-B (H). Log likelihood values are provided in Fig. 4. Due to insufficient sequences for hypothesis testing, gene trees were not inferred for Hox3-B and fushi tarazu. Abbreviations: Pt Parasteatoda tepidariorum, Lh Latrodectus hesperus, Sm Stegodyphus mimosarum, Ag Acanthoscurria geniculata, Cs Centruroides sculpturatus, Mm Mesobuthus martensii. (PDF 156 kb)
P. tepidariorum Hox clusters to scale. The distances of Hox genes in clusters A and B are very similar, except for the break in cluster B. Hox genes are colored so that paralogs match, non-Hox genes are shown in grey and microRNAs are represented as lines. Genes on the positive strand and negative strand are indicated above and below the black lines, respectively. (JPG 503 kb)
Spider, scorpion and mite Hox clusters A and B. (XLSX 533 kb)
microRNAs and other genes identified in the P. tepidariorum Hox clusters. (DOCX 106 kb)
Duplication of miRNAs by possible large-scale and tandem duplication events in Parasteatoda and Centruroides. (XLSX 52 kb)
Duplicated developmental genes in P. tepidariorum. (XLSX 50 kb)
Synteny of 39 P. tepidariorum scaffolds with greatest number of reciprocal hits. Circos plot of the subset of 39 scaffolds presenting the greatest numbers of hits on one another (as detected using SatsumaSynteny). One unit on the perimeter represents one Mbp. (PDF 819 kb)
Genomic and transcriptomic datasets used to build gene families. (DOCX 141 kb)
Phylogeny of 31 arthropod species used in comparison with P. tepidariorum. Species relationships are based on JC Regier, JW Shultz, A Zwick, A Hussey, B Ball, R Wetzer, JW Martin, and CW Cunningham [73], JE Bond, NL Garrison, CA Hamilton, RL Godwin, M Hedin, and I Agnarsson [144], PP Sharma and G Giribet [145]. The number of genes within P. tepidariorum-seeded gene families are shown parenthetically (n =), the number of speciation nodes observed in the gene families between P. tepidariorum and the other species (N =), and the median HKY distance between each speciation node and P. tepidariorum (HKY =) descendants are shown at the node. (DOCX 348 kb)
P values of Kolmogorov–Smirnov goodness-of-fit tests for five models of the HKY distance1 distribution of duplication nodes with non-P. tepidariorum descendants. Distributions unable to be rejected at an alpha level of 0.05 are in bold. Best fitting models are in italics. (DOCX 80 kb)
HKY distance distributions and Gaussian mixture models of duplication nodes from P. tepidariorum-seeded gene families for eight arachnid species. The HKY distances for duplication nodes were calculated as the mean HKY branch length from the duplication node to each of the descendent genes in the species of interest (A–H). In panel (I) the distribution is for all the duplication nodes with at least one P. tepidariorum descendant, using the mean HKY distance from the node to P. tepidariorum descendants. For each panel, the best match to one of five distributions (Uniform, exponential (G), or a Gaussian mixture model with 1, 2 (H), or 3 (A–F, I) distributions) is shown. The Gaussian mixture models were seeded with Gaussian mean and standard deviations estimated from the P. tepidariorum duplication nodes (Fig. 6a). (DOCX 430 kb)
Percent of duplication nodes assigned to three Gaussian distributions of HKY distances. The mean and standard deviation of the P. tepidariorum distributions (Fig. 6a) were used to estimate the other species’ Gaussian distributions. (DOCX 65 kb)
HKY distance distributions of all P. tepidariorum speciation nodes in P. tepidariorum-seeded gene families. The distributions are for mean HKY branch lengths from the P. tepidariorum speciation node to the descendant P. tepidariorum genes. (DOCX 244 kb)
Comparison of distributions for P. tepidariorum duplication and speciation nodes. Kolmogorov–Smirnov goodness-of-fit tests were used to compare the middle Gaussian distribution of HKY distances of duplication nodes (Fig. 6a, Additional file 13: Figure S6) to the log-normal distributions for speciation nodes. (DOCX 73 kb)
Shared paralog pair retention between P. tepidariorum and other arthropod species. Shaded species have complete genomes or deeply sequenced transcriptomes. (DOCX 92 kb)
Tandem duplications are abundant in young duplication events, but rare in older duplication events. HKY distance distributions and Gaussian mixture models of duplication nodes from P. tepidariorum-seeded gene families mirror those in Fig. 6a, but paralog pairs that are found in tandem (within five genes of each other on the same scaffold) are now shown in light grey, and dispersed paralogs in dark grey. (PDF 158 kb)
Analysis of the gene families containing a duplication pair from each of the Gaussian distributions (Fig. 6a), excluding tandem duplicates, show enrichment in several GO terms compared to gene families without duplication pairs. (XLSX 2339 kb)
Gene IDs of duplicated P. tepidariorum and C. sculpturatus genes compared to other arthropods. (XLSX 86 kb)
Frequency of tree topologies supporting different duplication scenarios. (XLSX 34 kb)
Orthology of P. tepidariorum genes giving full or partial support for WGD with respect to T. castaneum. (XLSX 19 kb)
P. tepidariorum proboscipedia-A expression. pb-A is first expressed in a thin stripe in the anterior of the embryo at stage 7 (A and B). The stripe broadens and as segmentation commences, it is found in the pedipalpal segment, and additionally, four weaker stripes start to appear in L1–L4 (C). The expression in the pedipalpal segment remains strongest, when by the end of stage 8.1, another stripe of weak pb-A expression is found in O1 (arrow) (D and E). During stage 9 and 10, pb-A expression is found in the mesoderm of the outgrowing pedipalp and walking legs, but also ectodermally in the distal tip of the outgrowing appendages of Pp-L4 (caret) (F–H). In the nervous system, pb-A is expressed most strongly in the Pp segment, starting at stage 9 (arrowhead), but it is also present in a smaller lateral domain of L1–O1 (arrow) (F–H). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–H). Anterior is to the left. Abbreviations: Pp pedipalpal segment, L walking leg segments, O opisthosomal segments. (JPG 6345 kb)
P. tepidariorum proboscipedia-B expression. Weak pb-B expression emerges at stage 8 in a domain spanning from Pp to L4 (A). The weak expression is located in the mesoderm, and at stage 8.2 also visible in O1 (arrow) (B). At stages 9 and 10, pb-B is expressed in the mesoderm of the outgrowing appendages of Pp–L4, as well as in dorsolateral parts of the neuroectoderm of segments Pp–O1 (arrows) (C and D). Each panel (except A) shows the same embryo, viewed laterally (left) and ventrally (center, right). Anterior is to the left. Abbreviations: see Additional file 27: Figure S11. (JPG 143 kb)
P. tepidariorum Hox3-A expression. Hox3-A expression appears first at stage 7 (not shown). At stage 8.1, the expression can be found in segmental stripes in the pedipalpal and the walking leg segments, and weak expression can also be seen in O1. The strongest expression is found in the first walking leg segment (A). At stage 8.2 and 9.1, Hox3-A is expressed in the mesoderm of the developing limb buds (B, C) and in the mesoderm of posterior segments. Note that Hox3-A expression is very weak and due to the high background in the head segments as well as in the opisthosomal segments, it is difficult to define anterior/posterior boundaries. Expression of Hox3-A vanishes after stage 9 (not shown). Panels show flat-mounted embryos, brightfield image above, nuclear stain of the same embryo below. Anterior is to the left. Abbreviations: see Additional file 27: Figure S11. (JPG 214 kb)
P. tepidariorum Hox3-B expression. Hox3-B is not expressed before stage 8, when expression starts in broad segmental stripes from Pp to L4, most likely in the mesoderm (A). Expression is strongest in L1 and L4 (A and B). In stage 9 and 10 embryos, Hox3-B is expressed in the mesoderm of the outgrowing limb buds, except for the pedipalps, which show a broad ring of expression (white arrow in C) that later refines into rings and additional expression at the tip of the pedipalp (D–F). Additionally, expression extends anterior-ventral to the limb buds in a triangular shape at stage 9 (arrowhead in C). This expression vanishes by stage 10. Instead, Hox3-B is now also expressed in the pedipalpal and walking leg segments in segmental groups of cells in the medial neuroectoderm (arrowheads in D–F). In stage 11 embryos, Hox3-B is additionally expressed in dots in the ventral neuroectoderm of every opisthosomal segment (carets in F). Furthermore, a dot of expression can be found in the first opisthosomal limb bud (arrow in F). Embryos in A and B are shown laterally, embryos C–E are shown laterally on the left and ventrally on the right. F shows ventral views of the head region (left) and the opisthosoma (right) of an embryo at a similar stage as the embryo in E. Anterior is to the left. Abbreviations: see Additional file 27: Figure S11. (JPG 4203 kb)
P. tepidariorum Deformed-A expression. Dfd-A is first expressed at stage 4, in almost all cells but the rim (arrowhead) of the germ disc (A). During stage 6, cells not expressing Dfd-A at the outer rim, the future anterior, have multiplied (arrowheads) (B). Later during stage 6, the expression clears from the posterior (former center of the germ disc, arrow) and the expression starts to form a broad stripe (bracket) (C). This uniform stripe of expression first subdivides into two stripes with lower expression between these stripes during stage 7. Then, the posterior domain splits into two more stripes and, in between the anterior and posterior domains, a new stripe gets inserted such that there are now four stripes of Dfd-A expression (asterisks) (D). These four stripes are later located in the four walking leg segments at stage 8 (E). In stage 9–11 embryos, Dfd-A is strongly expressed in the outgrowing walking legs with strongest expression in the tips of the legs (white carets) (F–H) and in the neuroectoderm (arrowheads in G and H). Each panel (except for A, B, C) shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left in all panels. Abbreviations: See Additional file 27: Figure S11. (JPG 6387 kb)
P. tepidariorum Deformed-B expression. Dfd-B expression is first detected in a weak, broad stripe in the developing germ band (bracket) (A). This stripe can be allocated to the first two walking leg segments at stage 8 (B). The anterior of the segments is stained stronger than the posterior. Additional, but much weaker expression can be seen in the L3 and L4 segments. At stage 9, Dfd-B is predominantly expressed in the ventral neuroectoderm (C). The anterior and posterior expression domain is marked by arrowheads in B and C, respectively. Each panel shows the same embryo viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 2272 kb)
P. tepidariorum Sex combs reduced-A expression. A stripe of Scr-A expression first appears at stage 6 (A). The stripe broadens during stage 7 (B), at the end of which it starts to split into two stripes, and a third new stripe appears posterior to the initial stripes (borders of expression marked by arrowheads) (C). At stage 8.1, there are four stripes located in the posterior parts of L2, L3, and L4 and the newest stripe appears in O1 (white arrow) (D and E). In limb bud stages, only the L3 and L4 limb buds carry Scr-A expression, mostly in their distal tips. Expression in L2 is restricted to the neuroectoderm (not shown), and expression in O1 is restricted to the posterior part of each hemisegment as well (F). At later stages, Scr-A expression in L3 and L4 refines to several rings in the distal part of the legs. Expression in L2 becomes undetectable. (G–I). At stage 10.1, Scr-A is visible in a neuroectodermal patch in L4 and a dot in O1 (arrow) (H). Each panel shows the same embryo viewed laterally (left) and ventrally (center and right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 266 kb)
P. tepidariorum Sex combs reduced-B expression. Scr-B is first expressed in a cap-like domain (arrowhead) in the center of the germ disc at stage 5. The position of the cumulus is marked by a ‘c’ (A). The expression widens during stage 6. Scr-B now forms an open ring, localized roughly halfway between anterior rim of the opening germ disc and the future posterior center of the germ disc. The cells posterior to the ring also express Scr-B, but at a much lower level (B). At stage 7, the ring splits up into two stripes (arrowheads) (C) and, later, two new stripes appear (carets) (D). The most anterior stripe of expression lies between L1 and L2 at stage 8, the second anterior stripe lies between L2 and L3. The two posterior stripes cover L3 and L4. The weak expression of Scr-B continues posterior to these stripes (E and F). Scr-B is predominantly expressed in the limb buds and the ventral neuroectoderm of stage 9.1 embryos (G). The anterior expression border is directly posterior to the L1 limb bud. Expression of Scr-B continues in the opisthosoma, but is much weaker, except for a domain in the SAZ (arrow) (G). This expression in the SAZ continues to the end of segmentation (H and I). No more expression is visible at the posterior end of the embryo at stage 11 (white caret) (J). Scr-B expression forms multiple rings in the legs (H–J), but it is much more strongly expressed in L3 compared to L2 and L4. At late stage 9, the expression of Scr-B in the neuroectoderm can also be found in every segment of the opisthosoma (arrowhead in H). Each panel shows the same embryo viewed laterally (left) and ventrally (center and right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 393 kb)
P. tepidariorum fushi tarazu expression. ftz is first expressed at stage 6 in a semicircle at the posterior end where the SAZ is forming (arrow) (A). The bracket marks an anterior broad stripe of ftz expression. This domain of expression gets weaker during stages 7 and 8, where mostly only the anterior border of the domain anterior to L2 (arrowhead in D) is visible. The expression is almost invisible when it is viewed ventrally. The expression in the SAZ persists, and does not clear from the posterior end (A–G). It emanates stripes at the anterior end of the SAZ (arrow in C–G). Stripes stay visible at the anterior border of the last formed segment (white arrow in D and G) until the next stripe of ftz expression forms. Only after segmentation finishes does the expression at the posterior disappear (arrow in H). Meanwhile, the anterior border of the Hox expression domain stretches from L2 to L4 and is predominantly found ventrally (black arrowheads in F and H). The legs on one side of a stage 11 embryo were removed to show the ventral Hox domain, which has concentrated to one spot per hemisegment (black arrowheads) within the L2–L4 domain (H). At stage 10, ftz expression appears in a ring near the distal tip of L3 (carets in G–H). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–H). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 7681 kb)
P. tepidariorum Antennapedia-A expression. Antp-A expression first develops in the SAZ at stage 7 (A). This expression transforms into a stripe and is followed by cells that show only weak expression of Antp-A (B), before new expression appears at the posterior end of the SAZ (C). The first two stripes can be allocated to the first two opisthosomal segments at stage 8 (D). New stripes keep appearing from the SAZ (arrows, E–H) until the end of segmentation (G). At stage 9, the anterior border of Antp-A expression reaches into the posterior half of L4 (arrowhead, E). Expression is now also seen in the ventral neuroectoderm, where it forms longitudinal rows in each hemisegment (white arrowheads, E–G), and rings in the L4 appendage (caret, F and G). Throughout development, Antp-A expression is always strongest in O1 and the anterior half of O2. Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–G). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 6657 kb)
P. tepidariorum Antennapedia-B expression. Antp-B first emerges in the first two opisthosomal segments at stage 8.2 (arrows) (A). The Antp-B domain expands anteriorly into the posterior part of L4 during stage 9 (white arrowheads) (B–D). Additionally, Antp-B forms rings of expression in the L4 appendage (carets in C and D). While initially the very posterior part of O2 is free of Antp-B expression (the O2/O3 border is demarcated by a black vertical line in D–F), during stage 10, expression can also be found at the O2/O3 border (arrows) (F). Furthermore, during stage 10, the expression pattern refines, in that the L4 and O1 Antp-B expression is mostly in the ventral neuroectoderm and not found dorsally, while the O2 expression is excluded from the most ventral region (E–G). Starting at stage 10, Antp-B is also expressed in one dot each in the opisthosomal limb buds on O4 and O5 (arrowheads) (E–G). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in E–G). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 5223 kb)
P. tepidariorum Ultrabithorax-A expression. Ubx-A is first expressed at stage 8.1 in the SAZ and in a weak stripe anterior to that (arrow) (A). This stripe gets stronger and the posterior domain enlarges (B and C) so that all new tissue added posteriorly expresses Ubx-A at equal levels (D–F). The anterior border is located in O2, while the very anterior part of O2 initially does not express Ubx-A (segmental boundaries in C–F indicated by black vertical lines) until the end of stage 9.2, when it is expressed dorsally and in the neuroectoderm also in the anterior part of O2 (white arrow in E). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 4678 kb)
P. tepidariorum Ultrabithorax-B expression. Ubx-B is first expressed at stage 8.2, in the SAZ, as well as in two weaker stripes in the O3 and O4 segments (arrows) (A). Expression is initially restricted to the posterior part of O3 (B), but eventually the anterior expression border extends ventrally into the posterior part of O2 (C–F), and is also found in the posterior part of the O2 limb bud (caret in E and F). Expression is strongest in O3, but otherwise is fairly uniform in all segments posterior to this one (D–F). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 4129 kb)
P. tepidariorum abdominalA-A expression. abdA-A expression starts in the SAZ during stage 9.1, posterior to O5 (A and B). By stage 10.1, the anterior border of abdA-A expression has extended into the posterior part of O3 (arrowhead, C). While the dorsal border of abdA-A expression remains there, in the ventral neuroectoderm, the expression expands anteriorly into the posterior half of O2 (arrowhead, D). These borders of expression persist during later development (E to G). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 5068 kb)
P. tepidariorum abdominalA-B expression. abdA-B expression first appears at stage 9.1 in the SAZ (A). Slightly later, shortly before the opisthosomal limb buds appear, abdA-B additionally emerges in the posterior part of O4 (B). From stage 9.2 onwards, abdA-B is expressed in the entire O4 segment and all posterior segments with strong expression in the opisthosomal limb buds (C–F). Each panel shows the same embryo, viewed laterally (left) and ventrally (right). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 4092 kb)
P. tepidariorum AbdominalB-A expression. AbdB-A is first expressed at stage 9.1 in O6 (arrowhead) and in a stripe in the anterior-most portion of the SAZ (A). The anterior expression border subsequently shifts anteriorly, first into the posterior part of O5 (arrowhead) (B and C), and the dorsal part of the AbdB-A domain then shifts into the posterior portion of O4 (arrowhead) (D and E). From the beginning of opisthosomal limb bud development, it is strongly expressed in the O5 limb buds. At stage 9.2, the ventral part of the AbdB-A domain expands into the posterior half of O2 (arrow). These anterior borders remain the same throughout the rest of embryonic development (E–G). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in D–G). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 245 kb)
P. tepidariorum AbdominalB-B expression. AbdB-B expression emerges with the appearance of the SAZ at stage 6 (A). It is continuously expressed in the SAZ until the end of segmentation (caret) (B–H). Additionally, from stage 9.1 on, weak AbdB-B expression is found posterior to the O3/O4 border (arrowhead) (F). Slightly later, additional expression appears in the O2 limb buds, in the prospective genital opening (arrow) (G and H). Dorsally, the AbdB-B expression domain extends from O5 to the posterior end (H–J), but in the ventral neuroectoderm, the AbdB-B expression border is located in the posterior half of O3 (arrowhead in I). The vertical line delineates the O2/O3 border, which the AbB-B expression does not reach in the neuroectoderm (J). Each panel shows the same embryo, viewed laterally (left) and ventrally (right, or center and right in G–J). Anterior is to the left. Abbreviations: See Additional file 27: Figure S11. (JPG 332 kb)
Hox gene expression in P. tepidariorum. Detailed description and comparison of the expression patterns of the Hox gene paralogs. (DOCX 143 kb)
Trinotate report of the best Blast hits, protein domain predictions and GO categories extracted from the Blast result, and the Pfam domain prediction for each of the aug3 predictions. (XLSX 7616 kb)