

Abstract
DNA microarrays represent an important new method for determining the complete expression profile of a cell. In “spotted” microarrays, slides carrying spots of target DNA are hybridized to fluorescently labeled cDNA from experimental and control cells and the arrays are imaged at two or more wavelengths. In this paper, we perform statistical analysis on images of microarrays and show that quantitating the amount of fluorescent DNA bound to microarrays is subject to considerable uncertainty because of large and small-scale intensity fluctuations within spots, nonadditive background, and fabrication artifacts. Pixel-by-pixel analysis of individual spots can be used to estimate these sources of error and establish the precision and accuracy with which gene expression ratios are determined. Simple weighting schemes based on these estimates are effective in improving significantly the quality of microarray data as it accumulates in a multiexperiment database. We propose that error estimates from image-based metrics should be one component in an explicitly probabilistic scheme for the analysis of DNA microarray data.
Large-scale expression profiling has emerged as a leading technology in the systematic analysis of cellular physiology (1). Expression profiling involves the hybridization of fluorescently labeled cDNA, prepared from cellular mRNA, to microarrays carrying up to 105 unique sequences. Several types of microarrays have been developed (2), but microarrays printed by pin transfer are among the most popular (3). Typically, a set of target DNA samples representing different genes is prepared by PCR and transferred to a coated slide to form a 2-D array of spots with a center-to-center distance (pitch) of about 200 μm. In the budding yeast Saccharomyces cerevisiae, for example, an array carrying about 6,200 genes provides a pan-genomic profile in an area of 3 cm2 or less (4, 5). cDNA samples from experimental and control cells are labeled with different color fluors and hybridized simultaneously to microarrays, and the relative levels of mRNA for each gene are then determined by comparing red and green signal intensities. An elegant feature of this procedure is its ability to measure mRNA levels for many genes at once with relatively simple technology.
Computation is required to extract meaningful information from the large amounts of data generated by expression profiling (6, 7). The development of bioinformatics tools and their application to the analysis of cellular pathways are topics of great interest. Several databases of transcriptional profiles are accessible on-line and proposals are pending for the development of large public repositories (8). However, relatively little attention has been paid to the computation required to obtain accurate intensity information from microarrays (but see refs. 9 and 10). The issue is important, however, because microarray signals are weak and biologically interesting results are usually obtained through the analysis of outliers. In this paper, we show that pixel-by-pixel information present in microarray images can be used in the formulation of metrics that assess the accuracy with which an array has been sampled. Because measurement errors can be high in microarrays, a statistical analysis of errors combined with well established filtering algorithms is effective in improving significantly the reliability of databases containing information from multiple expression experiments.
Methods
Scanning Technology.
Microarray slides were imaged with a modified fluorescence microscope designed for scanning large areas at high resolution (arrayWoRx, Applied Precision, Issaquah, WA). Fluorescence illumination was obtained from a metal halide arc lamp focused onto a fiber optic bundle, the output of which was directed at the microarray slide and emission recorded through a microscope objective (Nikon) onto a cooled CCD (charge-coupled device) camera (Apogee Instruments, Tucson, AZ). Interference filters (Chroma Technology, Brattleboro, VT) were used to select the excitation and emission wavelengths corresponding to the Cy3 and Cy5 fluorescent probes (Amersham Pharmacia). Each image covered a 2.4 × 2.4 mm area of the slide at 5-μm resolution. To scan the entire microarray, a series of images (“panels”) were acquired by moving the slide under the microscope objective in 2.4-mm increments.
Determining Spot Positions.
A mask containing a map of the microarray geometry was manually aligned to the image and then refined by determining the center-of-mass for each spot. The fluorescence intensity in a circular region 80% the diameter of a typical spot was integrated to determine mean fluorescence.
Process Control Flags.
Of a total of 14 process control flags in the arrayWoRx software, the following were used in this study: spot not at expected location, red/green negative signal after background subtraction, ratio mismatch, correlation error, and infinite ratio/divide by zero (further details available on request).
Reliability Measures for Individual Spots.
The probability that the distribution of Zi covers the population distribution was determined, and one minus this value used as the probability of difference. To eliminate excessive sensitivity to outliers, the upper and lower population half-distributions were truncated at 2σ and the mean and standard deviations recalculated. In addition, only 90% of the ratio distribution of Zi (±1.65σ) was used to determine overlap. Although a more rigorous method is desired, this overlap technique gives a basic measure of the similarity between two distributions without being excessively sensitive to outliers.
Results
As a representative source of expression data, we analyzed microarrays containing 6,200 spotted cDNAs from known and potential S. cerevisiae ORFs. We are interested in artifacts present in images of DNA microarrays that appear to be intrinsic to expression profiling methods and therefore chose, from a large collection of S. cerevisiae array data obtained at the Fred Hutchinson Cancer Microarray Center (courtesy of J. Delrow), images that had the highest overall quality (the “model array”) and, from the Department of Chemistry at Harvard University (courtesy of J. Tong and J. Hardwick), more typically noisy images. The precise nature of the experiments is not important for our analysis, and the arrays consisted of comparisons between two different strains of wild type S. cerevisiae or of a wild type and a strain deleted for a single gene selected blind. Following methods established by Brown and colleagues (3, 4), mRNA from strain A was copied to cDNA and labeled with Cy3 (“green”), mRNA from strain B was labeled with Cy5 (“red”), and the cDNAs were hybridized to a spotted DNA microarray. Images of hybridized microarrays were acquired with a modified fluorescence microscope that scans a slide-size area at several wavelengths by stitching together images acquired with a CCD camera (this instrument has been commercialized as the Applied Precision arrayWoRx Scanner). Typically, the fluorescence signal from a 100-μm-diameter spot was captured on about 200 camera pixels, yielding a large number of independent measurements of red and green intensities (Fig. 1a). Real microarrays deviate from ideal grids with round spots and we therefore recorded information about various imperfections in each spot as a series of binary flags (“process control flags”) that denote poor spot position, channel misregistration, low signal-to-noise ratio, etc. (see Methods for details).
Figure 1.

Comparing local and best-fit methods for determining array background. (a) Gene expression graph of Cy5 vs. Cy3 intensity for 6,200 yeast genes with local background used to offset signal intensities. Not shown are 98 spots with negative intensities. (Inset) Local background was determined by averaging red and green intensities over 16 pixels, at each of the four corners (marked B) of a rectangular region surrounding each spot. (b) Distribution of Cy5 intensities for a region of the slide away from the hybridization area (dotted blue line), a spot with negative intensity (a “black hole,” red line), local background surrounding the black hole (solid blue line), and a nearby low-intensity spot (green line). Each distribution is derived from over 150 sampled pixels. (c) Gene expression graph as in b, but with best-fit background. Only one (obviously scratched) spot has negative intensity and is not shown.
To assess the differential expression for each gene, we need to determine accurately the amount of real and background fluorescence at each spot on the array. The mean intensity for the ith spot (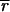
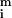 in the red and
in the red and 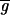
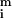 in the green) consists of the fluorescent cDNA signal hybridized to the spot (
in the green) consists of the fluorescent cDNA signal hybridized to the spot (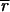 i and
i and  i), background arising from nonspecific binding by probe DNA (
i), background arising from nonspecific binding by probe DNA (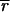
 and
and 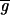
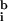 ), and intensity variation arising from pattern noise, electronic noise, and photon counting error (r
), and intensity variation arising from pattern noise, electronic noise, and photon counting error (r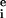 and g
and g ):
):
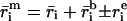 |
1 |
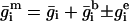 |
2 |
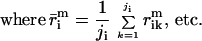 |
3 |
where j is the number of pixels in each spot included in the measurement. The extent of induction or repression of the ith gene, the expression ratio Zi, is then
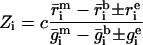 |
4 |
The distributions about 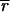
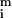 and
and 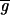
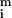 (on a pixel-by-pixel basis) are approximately normal, as judged by using the method of Bowman and Shenton [P values for normality were typically greater than 0.9 (11)]; as were the distributions for Zi so that
(on a pixel-by-pixel basis) are approximately normal, as judged by using the method of Bowman and Shenton [P values for normality were typically greater than 0.9 (11)]; as were the distributions for Zi so that
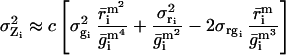 |
5 |
where σri and σgi are the standard deviations about 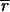
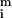 and
and 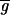
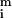 , σrgi is the red–green covariance, and c is a constant that corrects for differences in the gains of the red and green channels.
, σrgi is the red–green covariance, and c is a constant that corrects for differences in the gains of the red and green channels.
Determining 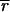 i and
i and 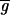 i is nontrivial because 5–10% of the spots in a typical array are close to background in intensity. Following published methods (9), we determined
i is nontrivial because 5–10% of the spots in a typical array are close to background in intensity. Following published methods (9), we determined 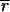
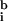 and
and 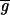
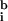 by measuring local background at the four corners of a rectangular region of interest surrounding the ith spot (Fig. 1a). With our model array we found that the expression ratio for all genes was near to one (after normalization), but 98 genes had negative intensities (Fig. 1a and data not shown). When background subtraction was applied to our set of typical arrays, 50–500 spots per array had negative intensities. Similar problems appear to plague published studies. Negative expression is nonsensical and suggestive of a flaw in using local background to estimate nonspecific hybridization.
by measuring local background at the four corners of a rectangular region of interest surrounding the ith spot (Fig. 1a). With our model array we found that the expression ratio for all genes was near to one (after normalization), but 98 genes had negative intensities (Fig. 1a and data not shown). When background subtraction was applied to our set of typical arrays, 50–500 spots per array had negative intensities. Similar problems appear to plague published studies. Negative expression is nonsensical and suggestive of a flaw in using local background to estimate nonspecific hybridization.
Why is local background such a poor measure of nonspecific fluorescence for microarray spots? When absolute intensities were compared on a pixel-by-pixel basis for a spot that looked like a “black hole,” a region surrounding the black hole (the local background), a weakly fluorescent spot, and a point on the slide outside the hybridization area, significant overlaps were observed in the intensity distributions (Fig. 1b). The center of the black hole was clearly less intense than the local background and the weakly fluorescent spot was only slightly brighter. The problem of negative intensities does not arise because the spot is being incorrectly located during image segmentation, but rather because more fluorescent probe is actually binding to the area surrounding the spot than to the spot itself. We suspect that nonspecific and specific hybridization signals are nonadditive as a consequence of differences in the chemistry of nonspecific binding of probe to nonhomologous spotted DNA and to DNA-free substrate (in our experiments, polylysine-treated glass).
We therefore explored ways to calculate the background by taking advantage of the ratiometric design of expression profiling experiments. Successful calculation is expected to yield background values for each channel greater than zero and less than the intensity of the weakest spot. Three precisely determined hybridization standards would in principal allow Eq. 4 to be solved for the entire data set. In cases where the majority of genes are unaffected by experimental conditions it is also valid to solve Eq. 4 from the expression data itself. In this case, the average expression ratio for the whole array is approximately one, 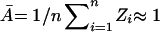 (where n is the number of spots in the array). Some advantages of this approach are that hybridization standards are not required and the contribution of noise is reduced by averaging across thousands of spots. However, the entire array is assumed to have constant background, which may not be correct. Moreover, if the overall levels of transcription change (as might be expected in a miniarray in which only selected genes are being analyzed), then the assumption that Ā = 1 is not valid and it is necessary to use control spots.
(where n is the number of spots in the array). Some advantages of this approach are that hybridization standards are not required and the contribution of noise is reduced by averaging across thousands of spots. However, the entire array is assumed to have constant background, which may not be correct. Moreover, if the overall levels of transcription change (as might be expected in a miniarray in which only selected genes are being analyzed), then the assumption that Ā = 1 is not valid and it is necessary to use control spots.
Because the microarrays analyzed in this study did not contain hybridization standards, a best-fit method was used to determine c and background levels from the experimental spots themselves. We observed that the best-fit values for background for the model array (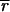
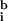 ,
, 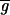
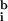 = 55, 39) were close to the background measured away from the hybridized area of the slide (i.e., outside the coverslip;
= 55, 39) were close to the background measured away from the hybridized area of the slide (i.e., outside the coverslip; 
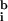 ,
, 
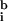 = 53, 30) and typically lower than the local background (
= 53, 30) and typically lower than the local background (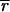
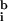 ,
, 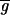
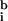 ≈ 67, 60; a bias value of 50 counts has been subtracted from all values). In addition, when ratios were calculated by using a best-fit background (Fig. 1c), essentially all negative expression values were eliminated and a significant number of transcripts were induced or repressed (Table 1; at 99.99% confidence, see below). We conclude that a computational approach finds optimized values for background from the spot intensities themselves, that these values are typically lower than those obtained by using local background, and that the computed background eliminates the problem of negative intensities. We believe, but have not yet proven, that this computation could be made more reliable through the use of multiple, nonhomologous hybridization controls.
≈ 67, 60; a bias value of 50 counts has been subtracted from all values). In addition, when ratios were calculated by using a best-fit background (Fig. 1c), essentially all negative expression values were eliminated and a significant number of transcripts were induced or repressed (Table 1; at 99.99% confidence, see below). We conclude that a computational approach finds optimized values for background from the spot intensities themselves, that these values are typically lower than those obtained by using local background, and that the computed background eliminates the problem of negative intensities. We believe, but have not yet proven, that this computation could be made more reliable through the use of multiple, nonhomologous hybridization controls.
Table 1.
Statistical analysis of changes in gene expression ratios
| Significant changes in gene expression*†
| |||||
|---|---|---|---|---|---|
| Background method | Change in expression | Average change | Number of spots differentially expressed
|
||
| Total | 1 < Zi < 2 | 0.5 < Zi < 1 | |||
| Local | Induced | 4.3 | 4 | 0 | |
| Repressed | 0.29 | 3 | 0 | ||
| Best-fit | Induced | 3.2 | 14 | 2 | |
| Repressed | 0.36 | 18 | 7 | ||
| Insignificant changes in gene expression† | |||||
| Background | Zi > 2 | Zi < 0.5 | |||
| determination | |||||
| Local | 20‡ | 90 | |||
| Best-fit | 8 | 3 | |||
Data from the model array in Figure 1.
Determined at 99.99% confidence by assessing overlap between the spot ratio distribution and the population distribution, as described in the text.
Excludes 57 divide by 0 spots found using local background subtraction and one (scratched spot) with best-fit methods.
A Statistical Metric for Microarray Data Quality–Spot Ratio Variability.
Careful inspection of typical DNA microarrays reveals that in addition to black holes, a significant fraction (5–20%) of all spots have nonuniform red–green ratios (Fig. 2a). Data obtained by others and posted to the web are also characterized by uneven spot morphology. In some spots, the red and green probes almost completely separate from each other or form bright clumps, presumably during hybridization. To explore systematically these intensity variations across a 6,200-spot array we plotted the standard deviation of the pixel-by-pixel intensities for each spot (averaged across both channels) against the spot's average signal intensity (Fig. 2b). The standard deviations in the pixel-by-pixel intensity distributions were high in absolute magnitude and the trend-line rose linearly as the signal intensity increased (red line). In contrast, measurement noise, including photon counting noise, rises only as the square root of the signal (green line). The unexpectedly high fluctuation in signal intensities across spots is not imposed by our analytic methods, nor does it arise from limitations in the instrumentation. Instead, it is a feature of the granularity of microarray spots, generated, we assume, during array fabrication and processing. For all but the dimmest spots, the inherent granularity of spots is the primary contributor to variations in red and green fluorescence and not photon counting uncertainty.
Figure 2.

Analysis of spot morphologies. (a) A gallery of spots from cDNA based microarrays and their corresponding red–green ratio is graphed three-dimensionally. Clockwise from the top left: a high quality spot, a spot exhibiting dye separation, a scratched spot, and a clumped spot. (b) Relationship between signal intensity ([
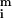 +
+ 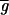
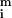 ) and standard deviation in the pixel-by-pixel intensities ([σri + σgi]/2) for all 6,200 spots in the model array. The red dotted line shows the trend-line; the green-dotted line shows expected instrument noise based on photon counting statistics.
) and standard deviation in the pixel-by-pixel intensities ([σri + σgi]/2) for all 6,200 spots in the model array. The red dotted line shows the trend-line; the green-dotted line shows expected instrument noise based on photon counting statistics.
Although all spots are characterized by intensity fluctuations (granularity), even well fabricated ones, some spots are much more irregular than others. As a simple measure of the irregularity of a spot, we propose the normalized standard deviation of the ratio measurement, the spot ratio variability (SRV):
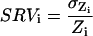 |
6 |
Intuitively, it seems likely that gene expression ratios calculated from spots with highly irregular morphologies would be less reliable than those obtained from uniform spots. Highly irregular spots are difficult to segment away from the surrounding background of the slide and extreme irregularity presumably reflects some underlying problem with array fabrication or hybridization. This finding is illustrated by two spots chosen to have Zi ≈ 2, but an SRV of 0.15 (spot A) or 0.45 (Spot B; Fig. 3a). Whereas spot A is very uniform in color, spot B contains an irregular clump of probe (Fig. 3b). An examination of spots across the model array confirms that artifacts in spot morphology readily apparent on inspection of microarray images give rise to high SRV values and often produce poorly determined expression ratios.
Figure 3.

Comparison of two spots with similar induction ratios but different SRVs. (a) Gene expression graph as in Fig. 1d based on best-fit-background, with spots differing from the average induction ratio by more than 99.99% probability (as determined by SRV values) denoted by red squares for induction and green dots for repression. Two spots have been chosen for further analysis; Spot A with Z = 1.7 and Spot B with Z = 1.94. (b) Pixel by pixel Cy5/Cy3 ratios for Spot A (green) and Spot B (orange) and normal approximations to these values compared with the population distribution of all Zi for the entire model array. The population distribution is approximated by two log-normal half-distributions (gray and blue lines), one used for Zi above the mean and the other for Zi those below the mean. Based on the extent of overlap between individual spot ratio distributions and the population distribution, Spot A has a P = 0.87 and Spot B has P = 0.01.
Using SRV to Assign Significance Estimates of Expression Ratios.
One application of SRV values is to identify genes whose expression ratios Zi are significantly different from the norm when the reliability of the measurement is taken into account. The basic notion is that when SRV is small, we can be confident in the significance of a relatively small difference between Zi and Ā, but when SRV is large, the difference between Zi and Ā must be greater. To ascertain whether Zi differs significantly from Ā, we determined the extent of overlap between the ratio distribution of the ith spot and the population distribution for all Zi (representing roughly 6,200 ratios in the entire array; Fig. 3b). As described above, the ratio distribution for the ith spot can be approximated by Zi, σZi, but the population distribution is inherently skewed because Zi cannot go below zero, its most probable value is ca. one, and it can be arbitrarily large. We therefore split the population distribution into a lower half-distribution below the peak value and an upper half above the peak, and approximated both half-distributions as either normal or log-normal (log-normal for the model array). To make probability estimates robust to outliers, the distributions were truncated to eliminate the tails (see Methods). A confidence limit of 99.99% was chosen to threshold the probability of overlap because with 6,200 measurements it is expected to yield only one false positive (obviously it can be lowered if less rigorous discrimination is desired). By using these simple methods, 14 genes in the model array were found to be significantly induced an average of 2.3-fold (±0.4) and 18 genes were repressed an average of 2.0-fold (±0.4) (Table 1). It is expected that the future application of more sophisticated robust statistics will be even better at identifying ratios that differ significantly from the population as a whole.
Measuring Scan Quality.
In many cases we observed that red–green intensity varied several-fold across a spot, but that the SRV value was quite low, indicating that the two signals were rising and falling together. To examine this quantitatively, pixel-by-pixel intensities of spots A and B were plotted relative to each other (Fig. 4a). In both the relatively uniform spot A (low SRV) and the less uniform spot B (high SRV), a high degree of covariance was observed relative to variance, resulting in ellipsoidal distributions. When covariance was plotted relative to the product of the variance in the red and green channels (Fig. 4b, blue dots) for all 6,200 spots, the data fell on a straight line, consistent with the idea that red and green signals exhibit highly correlated intensity fluctuations (Fig. 4b). As expected, spots that lie below the variance–covariance trend-line are those in which the normal granularity is perturbed and appear irregular (Fig. 4c). Obtaining data in which intensity fluctuations are correlated is not trivial however; image misalignment between red and green channels on the scale of 1–2 pixels is very common with many instruments and this drastically diminishes the average correlation, increases SRV, and reduces information content. We believe that the average correlation coefficient 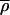 is a good indicator of overall scan quality, and illustrate the effect of a scan problem by introducing a single pixel shift computationally into an arrayWoRx image (Fig. 4b, green dots).
is a good indicator of overall scan quality, and illustrate the effect of a scan problem by introducing a single pixel shift computationally into an arrayWoRx image (Fig. 4b, green dots).
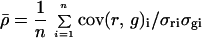 |
7 |
When SRV values for an entire array are compared with a recently published error model that parameterizes sources of error in microarray analysis (Fig. 4c; ref. 12), we note that the model tracks the general trend for SRV, and correctly predicts increasing error at low signal intensities, but does not capture the behavior of the large number of spots that lie away from the trend-line. Thus, it is likely to be less effective in predicting the precision with which individual Zi are determined than spot-by-spot analysis.
Figure 4.

Intensity variance and covariance for microarray spots. (a) Plot of Cy3 vs. Cy5 signal on a pixel by pixel basis for the Spot A and Spot B of Fig. 3. Arrows denote the direction of covariance, in which intensities in the red and green rise and fall together, and of ratio variance (SRV), in which the channels rise and fall independently. (b) Relationship of covariance and intensity variance for 6,200 spots (blue dots and red trend-line). (Insets) Images of spots lying well away from the trend-line (indicated by red circles). To illustrate the effect of a misalignment of one or more pixels, a common problem with laser scanners, a single pixel shift was introduced into the original scan data (green dots, pink trend-line). The average correlation coefficient 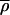 decreased from 0.85 to 0.41 (1.00 is the maximum possible) and the average SRV increased almost 3-fold. (c) Comparison of SRV values (blue dots) to a recently published parameterized error model (red line; ref. 12).
decreased from 0.85 to 0.41 (1.00 is the maximum possible) and the average SRV increased almost 3-fold. (c) Comparison of SRV values (blue dots) to a recently published parameterized error model (red line; ref. 12).
Using SRV to Improve Data Quality.
Because of their high cost, gene array experiments samples are rarely assayed with enough repetitions to support an effective statistical analysis of the data. We therefore asked whether quality metrics such as SRV derived from a single microarray experiment might provide useful estimates of reliability in lieu of information from truly independent measurements of the same sample. Ten repeat mRNA profiles from a comparison of wild-type yeast and a deletion strain were chosen blind. The lowest quality array, as judged visually, was discarded. For each of the 6,200 spots in the remaining nine arrays, the average of the expression ratio for the ith spot across nine microarrays 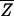 i, the standard deviation in this value
i, the standard deviation in this value 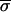 Zi, and the spot ratio variability for the nine arrays
Zi, and the spot ratio variability for the nine arrays 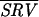 i were determined. Encouragingly,
i were determined. Encouragingly, 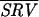 i, an image metric derived from the pixel-by-pixel analysis of images of single spots, showed reasonable correlation with
i, an image metric derived from the pixel-by-pixel analysis of images of single spots, showed reasonable correlation with 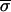 Zi, a conventional measure of variability derived from repeating the experiment (Fig. 5a). The relationship did not hold, however, if local background subtraction was used to determine fluorescent intensities, emphasizing the importance of correct image processing (Fig. 5b).
Zi, a conventional measure of variability derived from repeating the experiment (Fig. 5a). The relationship did not hold, however, if local background subtraction was used to determine fluorescent intensities, emphasizing the importance of correct image processing (Fig. 5b).
Figure 5.

Relationship between SRV and true experiment-to-experiment variability. (a) Nine microarrays were probed with the same cDNA preparation in parallel and data collected from 9 × 6,200 spots. The standard deviation in expression ratios for each spot as measured on nine arrays using a best-fit background and plotted relative to the average SRV for the same spot. The trend-line is indicated. (b) As in a, but with local background subtraction. (c) Generation of unbiased minimum-variance estimates for Zi by applying SRV and process control flags to inversely weight ratio data as it enters an operational database (see text for details).
Next, we attempted to use SRV information to improve the quality of a database containing information from all nine microarray experiments. Data quality was compared for a database in which the nine determinations of expression ratio for each gene in the array were simply averaged, and a database in which each measurement of expression ratio was weighted by 1/SRV before averaging. Using a simple measure of data quality for all spots in the database
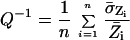 |
8 |
where n is the number of spots in each array, we observed that data quality increased 14% when data were weighted by 1/SRV before being averaged. When 1/SRV weighting was supplemented by process control flags to eliminate spots that were significantly shifted from their ideal positions, that had zero intensity in the green channel, or were characterized by other obvious flaws (see Methods), Q improved 38% relative to an unfiltered database. This increase in overall quality raised from 3 to 14 the number of genes that differed significantly in expression ratio from the norm at 99.99% confidence. At the same time, process control flags suggested that three genes that were initially considered significant should be excluded. We conclude from these findings, that image metrics (SRV) and process control flags derived from the analysis of individual arrays can be used to improve significantly the quality of a database containing microarray data. This is possible because SRV values are a fairly good predictor of actual experimental variability.
Discussion
In this paper we explore methods for extracting and manipulating data from images of cDNA-based microarrays. Despite widespread interest in microarray technology and expression profiling, the analysis of measurement uncertainties has attracted relatively little attention (9, 13). The issue is important however, because it is the small fraction of genes whose expression differs most from the average that are often of interest in an expression profile. To evaluate these outliers it is necessary to determine whether they reflect biological reality or simply error. We must therefore use probabilities that account for error and noise as measures of gene expression rather than single values. Because error and noise in expression profiling have multiple origins (including sampling error, biological fluctuation, etc.; see ref. 13), a different probability distribution must be considered for each expression ratio. In this paper, we focus on probability distributions derived from statistical analysis of microarray images and make the simple assumption that a gene must differ in expression ratio (Zi) from the mean ratio (Ā) by more than the measurement error for the difference to be statistically significant. Because fabrication and signal processing errors appear to be among the most quantitatively significant errors in expression profiling experiments, our focus on these sources of error is reasonable. However, it has recently been shown that different genes in yeast have different degrees of biological variability in their expression levels (for as-yet unknown reasons; ref. 12) and that this must be taken into account while formulating conditional probabilities associated with each gene's expression (Fig. 5c).
Image analysis of DNA microarrays suggests that two issues are of greatest importance in obtaining good data: determining the background and reducing the impact of poor quality spots on the data set. We find that the widely used method of subtracting local background from spot intensity (9) is not accurate and causes about 1–5% of spots in a typical array to have nonsensical negative intensities. As an alternative, we propose a best-fit method of determining background that uses the ratiometric nature of gene arrays to compute a background. This method has the advantage of producing a self-consistent and noise-free estimate of background. In the work presented here, we have assumed that transcription across all genes is unaltered (Ā ≈ 1); but in future experiments we intend to include a series of hybridization and negative control spots and to use them in the best-fit determination of background (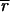
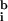 and
and 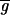
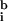 ) and ratio normalization (c).
) and ratio normalization (c).
To determine what distinguishes good data from bad, we have explored the impact of intensity fluctuations across a spot. A statistical analysis of spot intensities is possible with typical images of microarrays because many (ca. 100–200) independent intensity measurements are available for each spot. We find that spots in hybridized microarrays are characterized by intensity fluctuations among pixels substantially higher than would be expected on the basis of sampling statistics and instrument limitations. In most spots, this fluctuation is highly correlated between red and green channels and appears to arise from an intrinsic granularity generated during array fabrication and hybridization. In some spots however, red and green signals fluctuate independently of each other (Fig. 2a), causing the apparent gene expression ratio to vary from one place in the spot to the next. As a simple measure of ratio inhomogeneity that succinctly summarizes the reliability of the expression ratio for a spot, we calculate the normalized standard deviation of the ratio distribution, a value we refer to as spot ratio variability (SRV). Pixel-by-pixel ratio distributions across spots are nearly normal so that the mean and normalized variance for the red–green ratio—Zi and SRV—constitute reasonable and easily manipulated estimates for the distribution of Zi. However, we have observed that not all anomalies in DNA arrays are captured by the SRV value. A series of flags that denote other obvious problems such as severe mispositioning (and thus, likely overlap with a neighboring spot), extremely low signal levels, and abnormally high local background are also important.
Applications of Image Metrics in Microarray Analysis.
We envision three uses for image-derived quality metrics in the analysis of spotted DNA microarrays. The first is to provide information for statistical quality control during array fabrication and processing. By monitoring average SRV and the average correlation coefficient (Fig. 4b) we can determine which methods give the best quality data. The second is to determine whether the expression ratio of a gene is significantly different from that of the population as a whole when measurement error is taken into account. To estimate the probability of difference, we have used a simple method of comparing the overlap between the ratio distribution for a single spot and the overall distribution of Zi for all spots and argued that it is more rigorous than the current standard of using a 2-fold change as a threshold for significance (10). However, more sophisticated methods will undoubtedly yield better results.
The third application of image metrics is to weight data when populating a database with results from multiple experiments (Fig. 5c). As we have seen, the precision with which a microarray is measured varies from spot to spot. As data from multiple microarrays are combined, we must ensure that good data are not contaminated by bad. The approach we illustrate with nine transcriptional profiles is to record nine Zi values for each gene and to then calculate a weighted average in which each measurement is inversely scaled according to its precision (i.e., 1/SRV with additional information from process control flags). This is known to produce an unbiased minimum-variance estimate for the data and in our case results in a significant improvement in overall database quality. In a fully developed scheme, biological error models would also be incorporated (Fig. 5c; refs. 12 and 13).
In conclusion, we describe simple methods for using information in images of microarrays to calculate the precision of individual measurements of gene expression levels. As hoped, measurement precision appears to be a good indicator of overall data quality, presumably because a major source of error in microarray experiments lies not in setting up hybridization reactions, but in the fabrication and quantitation of spots. Future extensions of the basic statistical models in this paper include a fully developed Bayesian analytic scheme based on image analysis to assign conditional probabilities to each ratiometric measurement.
Acknowledgments
We are particularly indebted to J. Tong, J. Hardwick, and J. Delrow for the microarray data used in this paper and A. J. Sinskey, C. Shamu, K. Duggar, and R. Britton for help with the manuscript. This work was supported by Applied Precision, Inc., the Harvard Institute of Chemistry and Cell Biology, National Institutes of Health Grant GM51464, and Biomeasure, Inc.
Abbreviation
- SRV
spot ratio variability
References
- 1.Young R A. Cell. 2000;102:9–15. doi: 10.1016/s0092-8674(00)00005-2. [DOI] [PubMed] [Google Scholar]
- 2.Duggan D J, Bittner M, Chen Y, Meltzer P, Trent J M. Nat Genet. 1999;21:10–14. doi: 10.1038/4434. [DOI] [PubMed] [Google Scholar]
- 3.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 4.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 5.Holstege F C, Jennings E G, Wyrick J J, Lee T I, Hengartner C J, Green M R, Golub T R, Lander E S, Young R A. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 6.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ermolaeva O, Rastogi M, Pruitt K D, Schuler G D, Bittner M L, Chen Y, Simon R, Meltzer P, Trent J M, Boguski M S. Nat Genet. 1998;20:19–23. doi: 10.1038/1670. [DOI] [PubMed] [Google Scholar]
- 8.Brazma A, Robinson A, Cameron G, Ashburner M. Nature (London) 2000;403:699–700. doi: 10.1038/35001676. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Dougherty E R, Bittner M L. J Biomed Optics. 1997;4:364–374. doi: 10.1117/12.281504. [DOI] [PubMed] [Google Scholar]
- 10.Newton, M. A., Kendziorski, C. M., Richmond, C. S., Blattner, F. R. & Tsui, K. W. (2000) J. Comput. Biol., in press. [DOI] [PubMed]
- 11.Bowman K O, Shenton L R. Biometrika. 1975;62:243–250. [Google Scholar]
- 12.Hughes T R, Roberts C J, Dai H, Jones A R, Meyer M R, Slade D, Burchard J, Dow S, Ward T R, Kidd M J, et al. Nat Genet. 2000;25:333–337. doi: 10.1038/77116. [DOI] [PubMed] [Google Scholar]
- 13.Schuchhardt J, Beule D, Malik A, Wolski E, Eickhoff H, Lehrach H, Herzel H. Nucleic Acids Res. 2000;28:E47i–E47v. doi: 10.1093/nar/28.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]