

Abstract
Acute neuromuscular paralysis (ANMP) is a clinical syndrome characterized by rapid onset muscle weakness progressing to maximum severity within several days to weeks (less than 4 wk). Bulbar and respiratory muscle weakness may or may not be present. It is a common neurological emergency which requires immediate and careful investigations to determine the etiology because accurate diagnosis has significant impact on therapy and prognosis. Respiratory failure caused by neuromuscular weakness is considered as more critical than lung disease because its development may be insidious or subtle until sudden decompensation leads to life threatening hypoxia. Also, the arterial blood gas finding of severe hypoxemia, hypercapnia, and acidosis may not be apparent until respiratory failure is profound. Hence, the requirement for respiratory assistance should also be intensively and promptly investigated in all patients with neuromuscular disease. The disorder is classified based on the site of defect in motor unit pathway, i.e., anterior horn cells, nerve root, peripheral nerve, neuromuscular junction or muscle. Identification of the cause is primarily based on a good medical history and detailed clinical examination supplemented with neurophysiologic investigations and sometimes few specific laboratory tests. Medical history and neurological examination should be focused on the onset, progression, pattern and severity of muscle weakness as well as cranial nerves testing and tests for autonomic dysfunction. Associated non neurological features like fever, rash or other skin lesions etc. should also be noted. Globally, Guillain-Barré syndrome is the most frequent cause of ANMP and accounts for the majority of cases of respiratory muscles weakness associated with neuromuscular disorders. Newly acquired neuromuscular weakness in intensive care unit patients consist of critical illness polyneuropathy, critical illness myopathy and drug induced neuromuscular weakness which may arise as a consequence of sepsis, multi-organ failure, and exposure to certain medications like intravenous corticosteroids and neuromuscular blocking agents.
Keywords: Neuromuscular weakness, Paralysis, Approach, Nerve, Muscle
Core tip: Acute neuromuscular paralysis is a clinical syndrome characterized by rapid onset muscle weakness progressing to maximum severity within several days to weeks. It is a neurological emergency which requires immediate and careful investigations to determine the etiology because accurate diagnosis has significant impact on therapy and prognosis. Disorder is classified based on the site of defect in motor unit pathway, i.e., anterior horn cells, nerve root, peripheral nerve, neuromuscular junction or muscle. Identification of the cause is primarily based on a good medical history and detailed clinical examination supplemented with neurophysiologic investigations and sometimes few specific laboratory tests. Medical history and neurological examination should be focused on the onset, progression, pattern and severity of muscle weakness as well as cranial nerves testing and tests for autonomic dysfunction.
INTRODUCTION
Acute neuromuscular paralysis (ANMP) is a common neurological emergency and can be defined as a clinical syndrome characterized by rapid onset muscle weakness progressing to maximum severity within several days to weeks (less than 4 wk)[1,2]. ANMP carries high mortality when it leads to bulbar palsy, respiratory muscle weakness or autonomic dysfunction. The disorder is caused by defect somewhere in the pathway of motor unit (MU), i.e., anterior horn cells, nerve root, peripheral nerve, neuromuscular junction or muscle (Figure 1). Immediate and careful evaluation to determine the etiology is crucial as the accurate diagnosis has significant implications on management and prognosis. Identification of the cause is primarily based on a good medical history and detailed clinical examination supplemented with neurophysiologic investigations and sometimes few specific laboratory tests.
Figure 1.
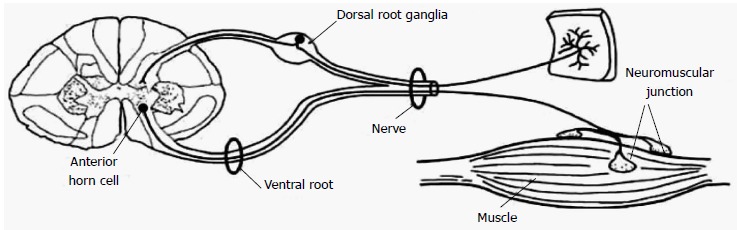
Schematic diagram showing the motor unit pathway.
The requirement for respiratory assistance should also be intensively and promptly investigated in patients with neuromuscular disease. Respiratory failure caused by neuromuscular weakness is considered as more critical than lung disease because its development may be insidious or subtle until sudden decompensation leads to life threatening hypoxia[2,3]. Also, the arterial blood gas finding of severe hypoxemia, hypercapnia, and acidosis may not be apparent until respiratory failure is profound[2,3].
Globally, Guillain-Barré syndrome (GBS) is the most frequent cause of ANMP and accounts for the majority of cases of respiratory muscles weakness associated with neuromuscular disorders[3-5]. Newly acquired neuromuscular weakness in intensive care unit (ICU) patients consist of critical illness polyneuropathy (CIP), critical illness myopathy and drug induced neuromuscular weakness which may arise as a consequence of sepsis, multi-organ failure, and exposure to certain medications like intravenous corticosteroids and neuromuscular blocking agents[1,2,6]. The disorders under the spectrum of ANMP are wide and based on the site of MU affection, ANMP can be classified as anterior horn cell disorder, polyradiculoneuropathy, peripheral neuropathy, disorders of myoneural junction and primary muscle diseases (Table 1).
Table 1.
Differential diagnosis of acute neuromuscular paralysis
| Anterior horn cell disorders |
| Poliomyelitis |
| West Nile virus |
| Peripheral neuropathy/polyradiculopathy |
| GBS |
| Porphyria |
| Diptheria |
| CMV polyradiculopathy |
| Lyme neuroborreliosis |
| Toxins (heavy metals, e.g., arsenic, mercury, hexacarbon, drug intoxication, organophosphate, Buckthorn) |
| Critical illness polyneuropathy |
| Tick paralysis |
| Vasculitic neuropathy |
| Neuromuscular junction disorder |
| MG |
| Lambert-Eaton syndrome |
| Neuroparalytic envenomation (e.g., tick and snake bites) |
| Botulism |
| Organophosphate and carbamate |
| Hypermagnesemia |
| Prolonged neuromuscular blockade |
| Overdose of anticholinesterases |
| Muscle disease |
| Periodic paralysis (hypokalemic: Hereditary and secondary, hyperkalemic) |
| Hypophosphatemia |
| Critical illness myopathy |
| Polymyositis, dermatomyositis, infectious myositis (e.g., dengue myositis) |
| Acute rhabdomyolysis |
Adapted from Maramattom et al[2]. GBS: Guillain-Barré syndrome; CMV: Cytomegalovirus; MG: Myasthenia gravis.
ANTERIOR HORN CELL DISORDER
Polio virus and West Nile virus (WNV) infections are two important causes of infection-associated acute muscular paralysis that primarily affect anterior horn cells. Poliovirus poliomyelitis is no longer prevalent nowadays. Afghanistan and Pakistan are two polio endemic countries[7-9]. WNV introduced to the United States in 1999 and has become endemic in North America and emerged as the commonest cause of epidemic meningoencephalitis. Presently, WNV is the leading cause of arboviral encephalitis in the United States[10].
Polio virus poliomyelitis
Poliomyelitis is a highly infectious disease caused by a virus belonging to the Picornaviridae family[9]. The clinical manifestations are varied, ranging from mild cases of respiratory symptoms, gastroenteritis, and malaise to severe forms of paralysis. These have been categorized into asymptomatic cases (90%-95%), mild illness or abortive poliomyelitis (4%-8%), aseptic meningitis (1%-5%), and paralytic poliomyelitis (0.1%-2%)[9]. Paralytic poliomyelitis is the most severe form presents as excruciating episodes of pain in back and limbs followed by motor weakness[9]. Weakness is rapid or gradual, asymmetric, predominantly proximal, and most commonly involves legs followed by the arms, abdominal, thoracic or even bulbar muscles. Respiratory failure may ensue in some patients either due to medullary involvement or phrenic or intercostal nerve paralysis[10]. Recovery may be complete in some patients but if the motor weakness persists beyond one year, lifelong disability occurs.
WNV associated paralysis
Acute, flaccid, and asymmetric motor weakness mimicking poliovirus may occur due to WNV[10]. Approximately 80% of WNV infections are asymptomatic, and 20% result in a self-limited disease referred to as West Nile fever. Less than 1% of patients develop neuroinvasive disease including meningitis, encephalitis, and acute flaccid paralysis or poliomyelitis[10]. As in poliovirus poliomyelitis, bulbar and respiratory muscles involvement may occur. Although anterior horn cells are primarily get affected, inflammatory changes may also involve muscles, peripheral nerves, spinal roots, spinal sympathetic neurons and ganglia. Rarely, WNV has been associated with demyelinating polyneuropathy similar to GBS.
The occurrence of meningoencephalitis, asymmetric pattern of weakness, normal sensory examination, lymphocytic pleocytosis in the cerebrospinal fluid (CSF) and reduced or absent compound muscle action potentials (CMAPs), preserved sensory nerve action potentials (SNAPs), and neurogenic electromyography (EMG) in a segmental pattern are core features that distinguish poliovirus or WNV paralysis from GBS[9]. Diagnosis is confirmed by detection of WNV-specific IgM antibody in CSF and serum nucleic acid amplification test is required in immunocompromised patients when antibody development is delayed or absent[9]. There is no specific treatment available for poliovirus and WNV poliomyelitis and management is primarily supportive care[10].
PERIPHERAL NEUROPATHY AND POLYRADICULONEUROPATHY
GBS
GBS is the most common and potentially life-threatening acute paralytic neuropathy worldwide with the reported annual incidence rate of 1-2 cases per 100000[11]. It is an acute-onset, rapidly progressive, immune-mediated symmetrical polyneuropathy with or without respiratory muscle involvement that often follows an antecedent infection. Two thirds of cases are usually preceded by systemic infection like upper respiratory tract infection or diarrhea[12]. Campylobacter jejuni is the most frequent antecedent infectious agent associated with subsequent development of the Guillain-Barré syndrome[13]. Epstein Barr virus, Cytomegalovirus (CMV), Mycoplasma, Human immunodeficiency virus are other common infectious agents that have been linked to GBS[13,14]. Studies have also documented the occurrence of GBS shortly after vaccinations or surgical procedures. Molecular mimicry between infectious antigen and surface components of peripheral nerve leads to activation of humoral and complement activation with membrane attack complex formation and nerve damage is the most accepted pathogenesis of disorder[15].
Any patient developing rapidly progressive, symmetrical limb weakness with/without sensory disturbances, hyporeflexia or areflexia and albuminocytological dissociation in CSF should first raise the diagnostic possibility of GBS. Neurological examination demonstrates proximal and often distal muscle involvement or sometimes limb weakness is global-both proximal and distal. Numbness, paresthesia and pain in the limbs are usual initial symptoms of GBS. The weakness progresses over a period of 12 h to 28 d before a plateau is reached and 80%-90% of patients with GBS become non-ambulatory during the illness[16]. Patients then have slow recovery phase that varies from weeks to months. Diagnostic criteria for the diagnosis of GBS as suggested by Asbury et al[14] and GBS Disability score are provided in Tables 2 and 3 respectively[14,17].
Table 2.
Diagnostic criteria for Guillain-Barré syndrome
| Features required for diagnosis |
| Progressive weakness in both arms and legs |
| Areflexia or hyporeflexia |
| Features that strongly support the diagnosis |
| Progressive motor weakness up to 4 wk |
| Relative symmetry of symptoms |
| Mild sensory involvement |
| Cranial nerve involvement, especially bilateral facial |
| Weakness |
| Autonomic dysfunction |
| Pain |
| Albuminocytological dissociation in CSF |
| Electrodiagnostic features of demyelination |
| Features that should raise doubt about the diagnosis |
| Respiratory failure with limited weakness of limbs at onset |
| Severe sensory signs at onset |
| Bladder or bowel dysfunction at onset and persistence of dysfunction in the disease course |
| Fever at onset |
| Sharp sensory level |
| Slow progression with limited weakness without |
| Respiratory involvement |
| Persistent asymmetry of motor weakness |
| Mono/polymorphonuclear leukocytosis in CSF |
Adapted from Asbury et al[14]. CSF: Cerebrospinal fluid.
Table 3.
Guillain-Barré syndrome Disability score[17]
| 0 Healthy state |
| 1 Minor symptoms and capable of running |
| 2 Able to walk 10 m or more without assistance but unable to run |
| 3 Able to walk 10 m across an open space with help |
| 4 Bedridden or chair bound |
| 5 Requiring assisted ventilation for at least part of the day |
| 6 Dead |
Respiratory insufficiency occurs in 25% of patients, and major complications, including pneumonia, sepsis, pulmonary embolism, and gastrointestinal bleeding, autonomic dysfunctions develop in 60% of mechanically ventilated patients[18]. Among the cranial nerves, the facial nerves are most commonly affected followed by bulbar and ocular motor nerves. Despite the appropriate treatment, mortality occurs in 4%-15% of cases and about 20% of severely-affected patients remain non-ambulatory after 6 mo of symptoms onset[18].
Based on the electrophysiological and pathological studies, GBS is classified into axonal and demyelinating subtypes[16,18]. Acute inflammatory demyelinating polyneuropathy is the most common GBS subtype, which is characterized pathologically by demyelination, lymphocytic infiltration, and macrophage-mediated clearance of myelin. The two axonal variant of GBS are acute motor axonal neuropathy (AMAN), characterized by pure motor neurological deficit, and acute motor sensory axonal neuropathy in which sensory fibers are also involved. However, detailed studies have suggested that mild sensory changes may occur in some patients with AMAN. The Miller Fisher syndrome is the least common type of GBS and appears to be more common among peoples who live in eastern Asia. It is characterized by a triad of ophthalmoplegia, ataxia, and areflexia. Facial and lower cranial nerve involvement, limb weakness, respiratory failure, and mild sensory involvement may occur in various combinations[16,18].
CSF examination in GBS typically reveals increased protein with normal CSF leukocyte count and termed as albumino-cytological dissociation. The protein concentrations are often normal in the first week, but increased in more than 90% of the patients at the end of the second week[16,18]. Increased CSF leukocyte count should raise the possibility of illness like leptomeningeal malignancy, Lyme’s disease, WNV infection, HIV-related GBS, or poliomyelitis[18,19]. Electrophysiological studies have an important role in disease confirmation, subtype classification, and prognostication. Nerve conduction study of at least 4 motor nerves, at least 3 sensory nerves, F waves, and H reflexes provide sufficient electrodiagnostic information for the diagnosis of GBS[16,18]. Nerve conduction studies often reveal evidence of patchy demyelination, manifested as conduction block, slowed motor conduction velocities, prolonged distal latencies, and temporal dispersion of CMAPs (Figure 2). Similar to CSF analysis, electrodiagnostic testing may be entirely normal in the early phase of GBS. Inconsistent or absent F wave, prolonged F wave and distal motor latencies, reduced nerve conduction velocities, absent H response and abnormal upper extremity SNAP combined with a normal sural SNAP are the characteristic electrophysiological findings in early GBS[16,18].
Figure 2.
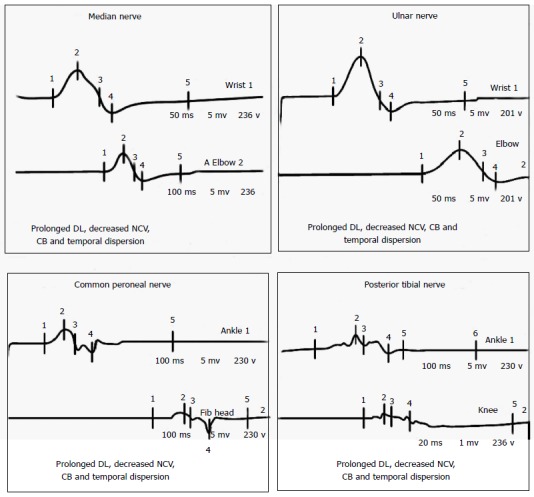
Motor nerve conduction study of a 24-year male patient presented with acute onset flaccid quadriparesis without sensory involvement. Nerve conduction study performed on day 2 shows typical findings of acquired demyelinating polyneuropathy. DL: Distal latency; NCV: Nerve conduction velocity; CB: Conduction block.
Immunological treatment along with meticulous supportive care to prevent or manage complications is required[19-21]. Frequent monitoring of respiratory function by measurement of vital capacity, cardiac and hemodynamic monitoring, prophylaxis for deep vein thrombosis, management of bladder and bowel dysfunction, early initiation of physiotherapy, and pain management should be done. Intravenous immunoglobulin (IVIg) and plasma exchange (PE) are effective immunotherapies if given during the initial phase of disease. PE or IVIg is indicated in severely-affected patients with inability to walk unaided or GBS disability score ≥ 3. Immunotherapy should be started as soon as possible in these patients before irreversible nerve damage has taken place. Although equally effective, IVIg is preferred over PE because of its ease of administration and fewer side effects. It is unclear whether IVIg is effective in mildly-affected patients (GBS disability score ≤ 2) or in Miller Fisher syndrome. The recommended dose of IVIg is 0.4 g/kg for 5 d and the usual regimen of PE is 5 times during 2 wk, with a total exchange of about 5 plasma volumes. In mildly-affected patients only 2 plasma volume may suffice and the dosage of IVIg conventionally administered (2 g/kg) may be suboptimal in some patients[18,19]. In clinical trials, no difference was found between IVIg and PE with respect to the improvement in disability grade after 4 wk, the duration of mechanical ventilation, mortality, or residual disability. The combination of PE and IVIg is not significantly better than PE or IVIg alone and oral or intravenous steroids are not beneficial[22]. About 10% of patients treated with IVIg or PE may deteriorate after initial improvement or stabilization and can be benefited by repeated treatment (2 g IVIg/kg in 2-5 d)[16-19].
Acute intermittent porphyria
Porphyrias are rare disorders of heme metabolism, characterized by a defect in an enzyme required for the synthesis of heme[23]. Acute intermittent porphyria (AIP) is an autosomal dominant disorder, results from a partial defect of porphobilinogen deaminase caused by a mutation in the hydroxymethylbilane synthase gene[23]. Neurological manifestations are characterized by acute polyneuropathy (predominantly motor), confusion, delirium, visual field defects, and seizures. Certain triggers like corticosteroids, other drugs, alcohol or fasting can precipitate an attack. The porphyric crisis typically begins with moderate to severe abdominal pain. Peripheral neuropathy is caused by axonal degeneration and predominantly affects motor nerves with minimal or no sensory involvement. Initially, weakness involves the proximal muscles of upper limbs[23]. Ankle jerk is frequently preserved. The polyneuropathy can affect cranial nerves and respiratory muscles requiring mechanical ventilation. Neuropsychiatric manifestations are common and seizures may occur in up to 20% of cases. Autonomic symptoms including tachycardia, cardiac arrhythmias, hypertension, constipation, and urinary retention are frequent and may lead to sudden death. The primary tool for diagnosis is measurement of porphyrin levels in urine, stool, and blood during an acutely symptomatic state. In an acute attack of AIP, there is elevated urinary excretion of aminolevulinic acid (ALA), porphobilinogen (PBG), uroporphyrin, and coproporphyrin; erythrocyte PBG deaminase may be normal or decreased[23]. The urine turns dark when standing due to formation of porphobilin. The study of CSF is normal or may reveal slightly raised protein levels. Treatment is based on glucose supplementation, prohibition of drugs that may worsen an attack and hematin (4 mg/kg daily for 4 d) to inhibit synthesis of ALA synthetase[23]. Neuropathy begins to improve within days and may continue to improve over several days.
Diphtheria
Diphtheria is a contagious disease caused by toxin-producing strains of the bacterium Corynebacterium diphtheriae[24]. It is a biphasic illness with initial symptoms of fever, throat congestion, neck swelling and ipsilateral palatal weakness followed by diphtheric polyneuropathy. The latency in development of diphtheritic polyneuropathy varies from 18 to 46 d after the initial infection. It is an acute demyelinating polyneuropathy, occurs in about 20% of patients with diphtheria[24]. The classic features of include accommodation disturbances, convergence or pupillary light reflex disturbance, anisocoria, ptosis, mydriasis, malfunction of extraocular muscles and dysfunction of the other cranial nerves followed by quadriparesis[24]. Various combinations of these clinical manifestations may be seen. Respiratory muscle and diaphragmatic paresis leading to respiratory failure is a common life-threatening neurological complication. Improvement in cranial nerves occurs with evolving motor disturbance in the trunk and extremities. Autonomic dysfunction is common in diphtheritic polyneuropathy, with the incidence ranging from 36% to 100% in severe diphtheritic polyneuropathy[24]. Diphtheria antitoxin is ineffective if administered after one or two days of diphtheritic symptoms. Death from diphtheria occurs by autonomic dysfunction, cardiac arrhythmias, myocarditis, aspiration pneumonia or respiratory paralysis.
Lyme’s disease and CMV related polyradiculoneuropathy
Lyme’s disease is focally endemic in temperate climates of the northern hemisphere. It is the most common tick-borne disease in United States. It is a multistage and multi-system disease caused by Borrelia spirochetes, which are transmitted by ixodes ticks. It is focally endemic in temperate climates of the northern hemisphere. Early manifestations of the disease include fever, headache, fatigue, and a characteristic skin rash called erythema migrans. Weeks to months later, neurological or cardiac symptoms develop. Neurological manifestations are characterized by aseptic meningitis or fluctuating meningoencephalitis with cranial or peripheral neuropathies. Treatment is oral doxycycline or amoxicillin. Late or severe disease requires ceftriaxone or penicillin G. Single-dose doxycycline (200 mg orally) can be used as prophylaxis in selected patients. Preventive measures should be emphasized to patients to help reduce risk.
CMV polyradiculopathy is clinically characterized by lower extremity weakness, urinary retention, and sacral dysesthesias. It is an important cause of polyradiculitis in immunocompromised individuals including malignancies, organ transplant recipients and persons with acquired immunodefiency syndrome. CSF examination reveals pleocytosis. CMV polyradiculopathy is rapidly fatal without treatment. Detection of CMV in CSF is mandatory to confirm the diagnosis. Treatment with foscarnet or ganciclovir may improve or stabilize the condition.
Acute toxic polyneuropathies
Various environmental and industrial toxins including heavy metals, pesticides, organophosphates, and organic solvents can affect peripheral nerves. Although these toxins usually cause subacute or chronic neuropathy, acute polyneuropathy resembling GBS can also occur. Common toxins that can lead to acute neuropathy are described below.
Arsenic: Suicidal, homicidal or accidental ingestion of arsenic may lead to rapidly-evolving polyneuropathy 1-3 wk after acute poisoning. The neuropathy is of axonal type. Severe gastrointestinal symptoms including abdominal pain, diarrhoea, vomiting, and shock are other manifestations of acute arsenic poisoning. Urinary excretion of arsenic > 0.1 mg/L is abnormal and the levels can reach up to 1 mg/L after acute exposure. Once polyneuropathy has occurred it does not respond to chelating therapy with British anti-Lewisite agent.
Thallium: Patients with acute thallium poisoning present with abdominal pain, vomiting and diarrhea with later development of limb pain and paresthesia. Rapidly progressive muscle weakness develops soon, which is more prominent in distal muscles. Sensory impairment for pain is more markedly affected than other sensory modalities. Other neurological manifestations like cranial neuropathies, nystagmus, and optic neuritis may occur. Alopecia is another important clinical finding of thallium poisoning.
Organophosphate poisoning: Organophosphate compounds are widely used as pesticides and insecticides in agriculture and household. Organophosphorus poisoning may result from occupational, accidental or intentional exposure. Tri-ortho-cresyl phosphate is a common organophosphate compound leading to neurological disorders. Three typical patterns of neurological manifestations usually occur following organophosphates poisoning. Type 1 paralysis or acute cholinergic crisis occurs because of excessive muscarinic receptor stimulation by acetylcholine. Type 2 paralysis or intermediate syndrome usually appears 24-96 h after the apparent recovery from acute cholinergic phase. Dysfunction of neuromuscular junction caused by downregulation of presynaptic and postsynaptic nicotinic receptors is the proposed hypothesis in the pathogenesis of intermediate syndrome. The clinical features consist of proximal muscles weakness, neck flexors weakness, and respiratory paralysis. As the clinical manifestations occurred after the acute cholinergic phase but before organophosphate-induced delayed polyneuropathy, it is called “intermediate syndrome”. Organophosphate-induced delayed neuropathy is a distal symmetric sensory motor (predominantly motor) axonal neuropathy, which occurs 2-5 wk after exposure. Cramping muscle pain in the lower limbs and paraesthesia occur, followed by progressive muscle weakness and diminished deep tendon reflexes.
Buckthorn poisoning: Buckthorn shrub is found mainly in Texas and Mexico. Ingestion of the green or ripe fruit of the buckthorn or tullidora shrub can cause a rapidly progressive, symmetric, progressive and severe axonal type neuropathy. Facial and pharyngeal weakness may also occur and neurological picture resembles GBS or other polyradiculoneuropathies.
Vasculitic neuropathy: Although, vasculitic neuropathies usually present in a subacute or chronic manner, sometimes aggressive vasculitic neuropathy may manifest acutely and resembles GBS[25]. Pseudo-conduction block in vasculitic neuropathy may be mistaken for conduction block typically seen in GBS. Presence of fever, history of multifocal involvement that very rapidly became confluent and pain accompanying focal weakness are important clinical features suggestive of vasculitic neuropathy. Nerve conduction studies in vasculitic neuropathy usually show only conduction block but no other features of demyelination. Also, conduction block disappears subsequently because axonal degeneration follows as a consequence of nerve infarct[25].
CIP
CIP is an acute reversible neuropathy that develops during the treatment of critically-ill patients and has an important impact on the outcome of patients in the ICU[26-28]. Difficulty in weaning from the mechanical ventilator in the absence of cardiopulmonary compromise and generalized muscle weakness in critically-ill patient should always lead to suspicion of CIP. It has been reported to occur in 70% of patients with sepsis and multiorgan failure[6,26]. Other causes of acute onset flaccid paralysis and areflexia in critically-ill patients needs to be ruled out before diagnosis of CIP is made. The severity of weakness ranges from moderate paresis to complete quadriplegia. The muscle weakness is predominantly distal and involves the lower limbs. The cranial nerves are usually spared, although facial weakness has been occasionally reported. Hyporeflexia or areflexia is common, although deep tendon reflexes may be normal in about one-third of the patients. The course of CIP is monophasic and self-limiting and shows significant recovery if the patient survives the underlying critical illness[26-28]. Sepsis, multi-organ dysfunction syndrome, multi-organ failure, female sex, use of corticosteroids, severe asthma, ionic abnormalities, malnutrition and immobility are frequently reported risk factors of CIP. Electrophysiological studies shows diminished compound motor and sensory nerve action potential amplitudes with normal conduction velocities suggesting axonal neuropathy. Needle EMG shows fibrillation potentials and positive sharp waves suggesting denervation. Abnormal phrenic nerve conduction (bilateral reduced or absent diaphragmatic CMAP) is reported in about 50%-80% of patients. There is no specific treatment for CIP and management is primarily supportive[6,26,27].
DISORDERS OF NEUROMUSCULAR JUNCTION
Myasthenia gravis
Myasthenia gravis (MG) is the most common neuromuscular junction disorder featured by fluctuating motor weakness that has a predilection for the ocular and bulbar musculature. Incidence of MG has been reported to be 0.25-2 patients per 100000 populations[29]. It is an immune mediated disorder due to circulating antibodies directed against the postsynaptic acetylcholine receptors. Most common clinical manifestations include fatigue, diplopia, ptosis, dysphagia, difficulty in mastication, dysarthria, proximal and neck muscle weakness[29]. Involvement of respiratory muscles may lead to acute respiratory insufficiency. Although the subacute and chronic presentation is more common, a subset of patients with MG can present with ANMP. Diagnosis is essentially based on a positive edrophonium test, decremental response on repetitive nerve stimulation, and presence of serum acetylcholine receptor antibodies. About 85% of patients with generalized MG are seropositive for acetylcholine receptor antibodies. Patients with MG typically require admission to the ICU for myasthenic crisis or cholinergic crisis. The term myasthenic crisis refers to respiratory weakness in patients with acquired, autoimmune form of MG. The life-time risk of myasthenic crisis in patients with MG is about 20%-30% and it usually occurs during the course of first symptomatic presentation in the young and later in the course of the illness in elderly[29]. Two-thirds to 90% of patients with myasthenic crisis require intubation and mechanical ventilation. Most patients need immunosuppression, in addition to symptomatic therapy with acetylcholinesterase inhibitors. The most commonly used symptomatic drug in MG is pyridostigmine and also the faster acting neostigmine. Prednisolone and azathioprine are the first choice among immunosuppressants. Several second choice drugs like cyclosporine and mycophenolate mofetil are methotrexate may also be used. Thymectomy should be performed in MG with thymoma and in generalized, early-onset MG. For MG crisis and other acute exacerbations, IVIg and PE are equally effective and safe treatments. Whenever difficult to differentiate between myasthenic and cholinergic crisis, acetylcholinesterase inhibitors should be stopped and the patient should be observed in the ICU.
Botulism
Botulism is a food-borne illness caused by the exotoxin of Clostridium botulinum, which acts by blocking the presynaptic release of acetylcholine. The clinical manifestations usually begins 12-36 h after consumption of the tained food with bulbar symptoms, nasal intonation, blurred vision, ophthalmoplegia, fixed dilated pupils, and autonomic dysfunction including dry mouth, constipation, and urinary retention. The severity of muscle weakness is variable and may present with progressive descending flaccid paralysis and sometimes respiratory involvement. Deep tendon reflexes and gag reflex may be preserved except in cases of severe generalised weakness. Sensory symptoms are absent. The diagnosis should be suspected based on history of ingestion of improperly sterilized home-canned foods followed by the development of the clinical manifestations described above. It is confirmed by detection of toxin in serum, feces or contaminated food scraps and is supported by electrophysiological studies, which show small amplitude motor responses that increase in amplitude at high rates of repetitive nerve stimulation. Treatment involves administration of trivalent antitoxin to neutralize circulating neurotoxin in the serum. Mechanical ventilation may be required in severely-affected patients.
Snake bite and other neuroparalytic envenomation
Snake bite is common in the rural parts of developing countries and carries high mortality if not adequately managed. The venom of elapid snakes, cobra and krait are predominantly neurotoxic, causing a selective neuromuscular block. Post-synaptic neurotoxins in snake venom such as bungarotoxin and cobrotoxin bind to acetylcholine receptors at motor end plates, while pre-synaptic neurotoxins such as bungarotoxin, crotoxin, and taipoxin interferes the release of acetylcholine at the neuromuscular junction. The nerve conduction study plays an important role in supplementing the diagnosis of snake bite and may also help to differentiate it from other causes of neuromuscular paralysis. Cobra and krait venom affect mainly the ocular, bulbar, and respiratory muscles leading to respiratory failure. Early morning neuroparalytic syndrome or pseudomyasthenic syndrome commonly seen among farmers and slum dwellers is a presentation of the krait bite as their bites are generally painless. Timely administration of anti-snake venom serum and institution of supportive treatment is associated with good outcome.
The neurotoxin produced by the Rocky Mountain wood tick, Dermacentor andersoni causes rapidly progressive ascending paralysis that can lead to respiratory failure and death. Weakness usually starts after about 5-6 d after the insect has embedded itself into the skin. The toxin acts by inhibiting the release of acetylcholine from the presynaptic nerve terminal.
Drug-induced neuromuscular junction disorders
Several groups of drug including aminoglycosides, quinolones, polymyxin antibiotics, calcium-channel blockers, beta-blockers, quinidine, procainamide, and neuromuscular blocking agents have been reported to produce or potentiate neuromuscular weakness[30]. Patients treated with high doses of nondepolarizing neuromuscular blocking agents such as vecuronium and pancuronium may have persistent weakness and difficult weaning from the ventilator even after drug has been stopped. At high doses, acetylcholinesterase inhibitors given to myasthenic patients can produce neuromuscular blockade and cause respiratory weakness. This overdose reaction is termed as cholinergic crisis and is characterized by nausea, diarrhea, miosis, bradycardia, muscle fasciculations, and hypersalivation[30].
MUSCLE DISORDERS
Periodic paralysis
Hypokalemic periodic paralysis is the classical and most common form of periodic paralysis. It is a calcium channel disorder manifests with acute muscle weakness that closely mimics GBS. Attacks usually begin in adolescence and are precipitated by exercise followed by rest, high carbohydrate and sodium content meals or sudden changes in temperature. The weakness evolves rapidly over minutes to several hours. Limbs are affected more than trunk and weakness is predominantly proximal. Deep tendon reflexes may be normal, decreased or absent. Ankle reflex is usually preserved even at the peak of weakness. Facial, ocular, bulbar, and respiratory muscles are rarely involved. Serum potassium levels are low. Hypokalemic periodic paralysis can be primary/hereditary (transmitted in an autosomal dominant pattern) or secondary caused by conditions such as thyrotoxicosis, barium poisoning, aldosteronism, and renal tubular acidosis. Treatment consists of large doses of oral potassium (0.25 mEq potassium chloride/kg) or potassium chloride intravenous solution in refractory cases and prevention with diet rich in potassium and low in carbohydrates and sodium. Acetazolamide can be used to prevent attacks. Hyperkalemic periodic paralysis is an autosomal dominant, inherited sodium channelopathy. It begins during childhood or the second decade of life and presents with crises of varying severity after exercise, cold, and fasting that usually last 1-2 h. The serum potassium level is high or borderline.
Polymyositis and dermatomyositis
Both polymyositis and dermatomyositis are inflammatory muscle disease which manifest in a subacute or chronic manner, although acute presentation can occur in rare cases[31]. In contrast to MG, extraocular muscles are never affected. Facial, bulbar, and respiratory muscles involvement is rare[31]. The clinical diagnosis of polymyositis and dermatomyositis is confirmed by elevated serum muscle enzyme concentrations, EMG, and muscle biopsy. In dermatomyositis, the inflammation is predominantly perivascular or in the interfascicular septae and around rather than within the fascicles; whereas in polymyositis, multifocal lymphocytic infiltrates surround and invade healthy muscle fibers. Prednisolone is the first-line drug and the addition of another immunosuppressive drug may be necessary in subjects who do not show improvement even after 3 mo of adequate dose of corticosteroids. In the first double-blind trial conducted for dermatomyositis, IVIg was reported to be effective in improving muscle strength and resolving the underlying immunopathology[32]. No controlled studies have been undertaken in polymyositis, but IVIg seems to be effective in about 70% of patients. Plasmapheresis was not found to be helpful in a double-blind, placebo-controlled study[33].
Several viruses (coxsackieviruses, infl uenza, parvoviruses, paramyxoviruses, CMV, Epstein-Barr virus, dengue) and bacteria (Borrelia burgdorferi, streptococci) have also been reported to be associated with acute myositis and muscle paralysis. Rhabdomyolysis trauma, sepsis, alcohol abuse, certain viral infections like influenza, dengue, etc. and various medications can lead to acute rhabdomyolysis. Rapid-onset muscle pain, swelling, tenderness, predominant proximal or generalized weakness, acute renal failure, myoglobinuria, and markedly raised serum ceatinine kinase are the core features. Sometimes weakness is severe enough to cause respiratory failure. Electromyography shows myopathic changes and spontaneous fibrillations. A muscle biopsy is confirmatory and shows massive muscle fiber necrosis and often numerous regenerating fibers with minimal inflammatory changes[33,34].
CONCLUSION
ANMP is a common neurological emergency and should be promptly investigated and treated. Sometimes, neurological examination in the emergency department or in ICU can be difficult. Combined clinical and electrophysiological assessment helps to locate the site of MU affection, i.e., anterior horn cell, radical, nerve, muscle, and neuromuscular junction disorders. Algorithmic approach to a patient with acute neuromuscular weakness is shown in the Figure 3. Complications of critical illness, including critical illness neuropathy, critical illness myopathy and prolonged neuromuscular blockade, are now considered as the principal cause of new onset weakness in the seriously ill patients. These disorders should to be differentiated from other neurological conditions that may develop after admission to the ICU. A proper protocol based clinical and investigational approach is essential in every emergency department to manage such cases.
Figure 3.
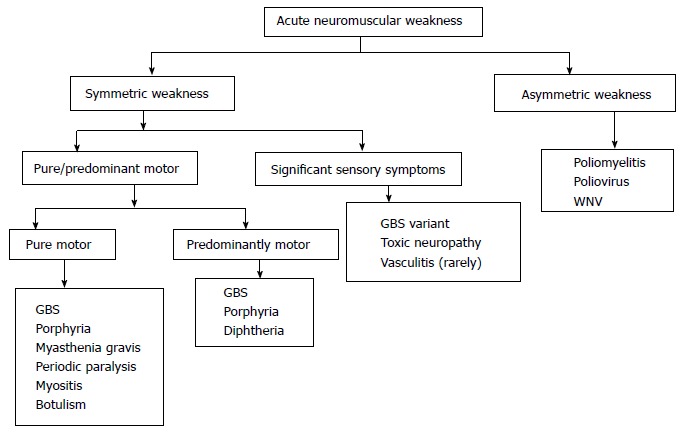
Althorithm: Practical approach to the patient with acute neuromuscular paralysis. GBS: Guillain-Barré syndrome; WNV: West Nile virus.
ACKNOWLEDGMENTS
I sincerely thank Dr. Sapna Chakrnarayan and Dr. Pramod Kumar for their assistance in English editing, preparing figures, tables and audio clip.
Footnotes
Conflict-of-interest statement: There is no conflict-of-interest.
Manuscript source: Invited manuscript
Specialty type: Medicine, research and experimental
Country of origin: India
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: February 12, 2017
First decision: March 7, 2017
Article in press: May 15, 2017
P- Reviewer: Kai K, Sergi CM S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Young GB, Hammond RR. A stronger approach to weakness in the intensive care unit. Crit Care. 2004;8:416–418. doi: 10.1186/cc2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maramattom BV, Wijdicks EF. Acute neuromuscular weakness in the intensive care unit. Crit Care Med. 2006;34:2835–2841. doi: 10.1097/01.CCM.0000239436.63452.81. [DOI] [PubMed] [Google Scholar]
- 3.Hutchinson D, Whyte K. Neuromuscular disease and respiratory failure. Pract Neurol. 2008;8:229–237. doi: 10.1136/pn.2008.152611. [DOI] [PubMed] [Google Scholar]
- 4.Dimachkie MM, Barohn RJ. Guillain-Barré syndrome and variants. Neurol Clin. 2013;31:491–510. doi: 10.1016/j.ncl.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ropper AH. The Guillain-Barré syndrome. N Engl J Med. 1992;326:1130–1136. doi: 10.1056/NEJM199204233261706. [DOI] [PubMed] [Google Scholar]
- 6.Latronico N, Bolton CF. Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis. Lancet Neurol. 2011;10:931–941. doi: 10.1016/S1474-4422(11)70178-8. [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention (CDC) Progress toward interruption of wild poliovirus transmission--worldwide, January 2011-March 2012. MMWR Morb Mortal Wkly Rep. 2012;61:353–357. [PubMed] [Google Scholar]
- 8.Atkinson W, Hamborsky J, McIntyre L, Wolfe S. Epidemiology and Prevention of Vaccine-Preventable Diseases (Pink Book) 11th ed. Washington DC: Public Health Foundation; 2009. pp. 231–244. [Google Scholar]
- 9.Mehndiratta MM, Mehndiratta P, Pande R. Poliomyelitis: historical facts, epidemiology, and current challenges in eradication. Neurohospitalist. 2014;4:223–229. doi: 10.1177/1941874414533352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carson PJ, Borchardt SM, Custer B, Prince HE, Dunn-Williams J, Winkelman V, Tobler L, Biggerstaff BJ, Lanciotti R, Petersen LR, et al. Neuroinvasive disease and West Nile virus infection, North Dakota, USA, 1999-2008. Emerg Infect Dis. 2012;18:684–686. doi: 10.3201/eid1804.111313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiò A, Cocito D, Leone M, Giordana MT, Mora G, Mutani R. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003;60:1146–1150. doi: 10.1212/01.wnl.0000055091.96905.d0. [DOI] [PubMed] [Google Scholar]
- 12.Hughes RA, Rees JH. Clinical and epidemiologic features of Guillain-Barré syndrome. J Infect Dis. 1997;176 Suppl 2:S92–S98. doi: 10.1086/513793. [DOI] [PubMed] [Google Scholar]
- 13.Alter M. The epidemiology of Guillain-Barré syndrome. Ann Neurol. 1990;27 Suppl:S7–S12. doi: 10.1002/ana.410270704. [DOI] [PubMed] [Google Scholar]
- 14.Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. 1990;27 Suppl:S21–S24. doi: 10.1002/ana.410270707. [DOI] [PubMed] [Google Scholar]
- 15.Green DM, Ropper AH. Mild Guillain-Barré syndrome. Arch Neurol. 2001;58:1098–1101. doi: 10.1001/archneur.58.7.1098. [DOI] [PubMed] [Google Scholar]
- 16.Burns TM. Guillain-Barré syndrome. Semin Neurol. 2008;28:152–167. doi: 10.1055/s-2008-1062261. [DOI] [PubMed] [Google Scholar]
- 17.Hughes RA, Swan AV, Raphaël JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 2007;130:2245–2257. doi: 10.1093/brain/awm004. [DOI] [PubMed] [Google Scholar]
- 18.Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barré syndrome. Lancet. 2016;388:717–727. doi: 10.1016/S0140-6736(16)00339-1. [DOI] [PubMed] [Google Scholar]
- 19.Rajabally YA. Treatment of Guillain-Barré syndrome: a review. Inflamm Allergy Drug Targets. 2012;11:330–334. doi: 10.2174/187152812800959059. [DOI] [PubMed] [Google Scholar]
- 20.Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349:225–230. [PubMed] [Google Scholar]
- 21.Hughes RA, Raphaël JC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2006;1:CD002063. doi: 10.1002/14651858.CD002063.pub3. [DOI] [PubMed] [Google Scholar]
- 22.Hughes RA, Newsom-Davis JM, Perkin GD, Pierce JM. Controlled trial prednisolone in acute polyneuropathy. Lancet. 1978;2:750–753. doi: 10.1016/s0140-6736(78)92644-2. [DOI] [PubMed] [Google Scholar]
- 23.Schaumburg HH, Berger AR, Thomas PK. Disorders of peripheral nerves, 2nd ed. Philadelphia: Davis; 1992. pp. 786–789. [Google Scholar]
- 24.Piradov MA, Pirogov VN, Popova LM, Avdunina IA. Diphtheritic polyneuropathy: clinical analysis of severe forms. Arch Neurol. 2001;58:1438–1442. doi: 10.1001/archneur.58.9.1438. [DOI] [PubMed] [Google Scholar]
- 25.Suggs SP, Thomas TD, Joy JL, Lopez-Mendez A, Oh SJ. Vasculitic neuropathy mimicking Guillain-Barré syndrome. Arthritis Rheum. 1992;35:975–978. doi: 10.1002/art.1780350820. [DOI] [PubMed] [Google Scholar]
- 26.Zifko UA, Zipko HT, Bolton CF. Clinical and electrophysiological findings in critical illness polyneuropathy. J Neurol Sci. 1998;159:186–193. doi: 10.1016/s0022-510x(98)00164-6. [DOI] [PubMed] [Google Scholar]
- 27.Bolton CF. Neuromuscular manifestations of critical illness. Muscle Nerve. 2005;32:140–163. doi: 10.1002/mus.20304. [DOI] [PubMed] [Google Scholar]
- 28.Hermans G, Van den Berghe G. Clinical review: intensive care unit acquired weakness. Crit Care. 2015;19:274. doi: 10.1186/s13054-015-0993-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juel VC. Myasthenia gravis: management of myasthenic crisis and perioperative care. Semin Neurol. 2004;24:75–81. doi: 10.1055/s-2004-829595. [DOI] [PubMed] [Google Scholar]
- 30.Spitzer AR, Giancarlo T, Maher L, Awerbuch G, Bowles A. Neuromuscular causes of prolonged ventilator dependency. Muscle Nerve. 1992;15:682–686. doi: 10.1002/mus.880150609. [DOI] [PubMed] [Google Scholar]
- 31.Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991;325:1487–1498. doi: 10.1056/NEJM199111213252107. [DOI] [PubMed] [Google Scholar]
- 32.Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, Dinsmore ST, McCrosky S. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000. doi: 10.1056/NEJM199312303292704. [DOI] [PubMed] [Google Scholar]
- 33.Miller FW, Leitman SF, Cronin ME, Hicks JE, Leff RL, Wesley R, Fraser DD, Dalakas M, Plotz PH. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N Engl J Med. 1992;326:1380–1384. doi: 10.1056/NEJM199205213262102. [DOI] [PubMed] [Google Scholar]
- 34.Reed AM, Ytterberg SR. Genetic and environmental risk factors for idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:891–916. doi: 10.1016/s0889-857x(02)00029-7. [DOI] [PubMed] [Google Scholar]