

Abstract
We introduce a general technique for making statistical inference from clustering tools applied to gene expression microarray data. The approach utilizes an analysis of variance model to achieve normalization and estimate differential expression of genes across multiple conditions. Statistical inference is based on the application of a randomization technique, bootstrapping. Bootstrapping has previously been used to obtain confidence intervals for estimates of differential expression for individual genes. Here we apply bootstrapping to assess the stability of results from a cluster analysis. We illustrate the technique with a publicly available data set and draw conclusions about the reliability of clustering results in light of variation in the data. The bootstrapping procedure relies on experimental replication. We discuss the implications of replication and good design in microarray experiments.
DNA microarrays (1) are a revolutionary high-throughput tool for the study of gene expression. The ability to simultaneously study thousands of genes under a multitude of conditions presents a huge challenge to comprehend and interpret the resulting mass of data. Early, pioneering research with cDNA microarrays that demonstrated the promise of the technology also influenced the direction of research to answer this challenge (2, 3). Specifically, an assortment of clustering techniques have been developed and applied to identify groups of genes with similar patterns of expression (3–6). A great deal of effort has gone into identifying the best clustering techniques for microarray data. However, another question that is at least as important has received less attention: How does one make statistical inference based on the results of clustering? The input into any clustering technique is a set of estimates of relative gene expression from a microarray experiment. In current practice, these estimates are taken to be precisely known quantities, ignoring the fact that every estimate has a margin of error. Consider two genes that cluster together. Are the patterns of expression for these genes sufficiently similar beyond any reasonable doubts raised by the uncertainty of the estimates, or could these genes have clustered together by chance? We propose a bootstrap method to assess the reliability of clustering results in a statistically quantifiable manner. The bootstrap is widely accepted as a method to assess the reliability of reconstructed phylogenetic trees (7), which is the primary inspiration for this work. We first describe our bootstrap methodology in generality, and then illustrate the implementation on the clustering technique used by Chu et al. (2).
By “clustering” genes, we mean organizing genes into groups, which may be predefined or data-driven, or organizing genes into a structure to represent some measure of distance between them. In a cluster analysis we start with raw data y, which we use to estimate the relative expression r of the genes among the mRNA samples. We use r̂ to estimate a clustering C. (The ^ notation denotes estimated quantities.) Schematically, we might write the process as
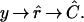 |
The specifics of the transformation from r̂ to Ĉ and the structure of C depend on the clustering method. The simplest kind of clustering (case 1) assigns genes to prespecified groups. This is the case for our example below, where genes are clustered by calculating the correlation of an observed profile with a collection of fixed target profiles. Thus, the group “centers” are known and there is no ambiguity about group labels. A second situation (case 2) occurs when we divide genes into groups but the group identities (and perhaps even the number of groups) are not defined in advance. This is similar to the first case, but the group labels are not well defined. The third kind of clustering we wish to consider is hierarchical clustering (case 3) (3). In this case, genes are organized into a bifurcating tree structure. The bootstrapping procedure proposed here applies equally well to any of these cases, and possibly others. Differences between the cases arise in how one evaluates and summarizes the bootstrapping results. We explore this issue further in Discussion.
Our methodology exists within the following paradigm: If we knew the precise differences in gene expression among the samples, we would have the “true” clustering C. In other words, if we knew r we could simply calculate C. Instead, we have estimates of relative gene expression r̂ with which we produce Ĉ as an estimate of C. Just as one wants error bars on any univariate parameter estimate, one would like to know how much confidence to put on the clustering resulting from microarray data. How much is Ĉ like C? We stress that our interest is not in evaluating the merits of any particular clustering algorithm. Rather, we present a method for assessing how much confidence one should have in clustering results in light of the error in estimation.
Experimental design determines how well one can estimate a quantity of interest and whether it is possible to assess the error in the estimate (8). Insufficient replication may lead to a situation where one can estimate the quantities of interest, but lacks the information to assess the accuracy of those estimates. Increased replication achieves more accurate estimation and also may improve one's ability to examine assumptions of the analysis. Limitations imposed by experimental design will arise in our example, and we will revisit this issue in Discussion.
Statistical Framework
The basis of this methodology is a statistical model for microarray data. We use analysis of variance (ANOVA) models, developed in ref. 9, to both estimate relative gene expression and to account for other sources of variation in microarray data. The exact form of the ANOVA model depends on the particular data set. In other words, one should evaluate each data set individually to determine which sources of variation are present, and construct the model accordingly. A typical ANOVA model is
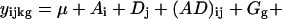 |
1 |
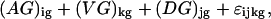 |
where yijkg is the measured intensity from array i, dye j, variety k, and gene g on an appropriate scale (typically the log scale). We use the generic term “variety” to refer to the mRNA samples under study. For example, the varieties may be treatment and control samples, cancer and normal cells, or time points of a biological process as in the example we will discuss later. The terms A, D, and AD account for all effects that are not gene-specific. The gene effects Gg capture the average levels of expression for genes and the array-by-gene interactions (AG)ig capture differences due to varying sizes of spots on arrays. The dye-by-gene interactions (DG)jg represent gene-specific dye effects. Although we did not originally anticipate such effects, they have appeared repeatedly. None of these effects are of biological interest, but amount to a normalization of the data for ancillary sources of variation. The effects of interest are the interactions between genes and varieties, (VG)kg. These terms capture differences from overall averages that are attributable to the specific combination of variety k and gene g. Differences among these variety-by-gene interactions comprise our estimates of relative gene expression r̂. In other words, to estimate the relative expression of gene g in varieties 1 and 2, one should estimate (VG)1g − (VG)2g. We assume the error terms ɛijkg are independent with mean 0 and variance σ2 but do not make any other distributional assumption.
Bootstrap Clustering
In a typical application of clustering, an investigator will estimate the relative expression of genes by using a ratio method (3), filter out uninformative genes, and cluster the remaining genes with a chosen algorithm. Our approach differs in two important respects. First, we estimate relative expression by using ANOVA instead of ratios. ANOVA models allow us to estimate relative expression while simultaneously accounting for other sources of variation. In addition, residuals from the fitted ANOVA model provide an empirical estimate of the error distribution—the “noise” in the data. This estimated error distribution serves as the basis of the technique we introduce here, which is to add a bootstrapping step (10) to evaluate clustering results.
Bootstrapping cluster analysis begins with creating a number of simulated datasets based on the statistical model. If 1 is the appropriate model, then bootstrap-simulated data sets y* are created
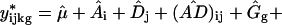 |
2 |
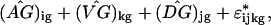 |
where a ^ over a term means the estimate from the original model fit. The ɛ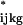 are drawn with replacement from the studentized residuals of the original model fit. Studentized residuals (11) are the fitted residual rescaled to have the same variance as the corresponding theoretical distribution. One repeats the clustering procedure on each simulated data set:
are drawn with replacement from the studentized residuals of the original model fit. Studentized residuals (11) are the fitted residual rescaled to have the same variance as the corresponding theoretical distribution. One repeats the clustering procedure on each simulated data set:
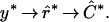 |
The result is a clustering of the original data Ĉ accompanied by a collection of bootstrap clusterings {Ĉ*}, which can be regarded as a sample of clusterings that are close to Ĉ in space of all possible clustering. What constitutes “close” is determined by the accuracy of the estimates of relative expression r̂ and the nature of the clustering method. The accuracy of r̂ as an estimate of r is a function of the the level of noise in the data and the experimental design (8, 16). When accuracy is high, the bootstrap estimates of relative expression will be more like the original estimates and the bootstrap clusterings will be more like the original clustering.
Example
We illustrate bootstrap clustering with the data from the experiment by Chu et al. (2). In this experiment, spotted cDNA microarrays containing 97% of the known genes of Saccharomyces cerevisiae (yeast) were used to study gene expression during meiosis and spore formation. Yeast cells were transferred to a nitrogen-deficient medium to induce sporulation and mRNA samples were taken at seven time points: 0, 30 min, and 2, 5, 7, 9, and 12 h. The “varieties” in this experiment are the time points. For each time point, the scientists prepared a “red”-labeled cDNA pool. In addition, they prepared a “green”-labeled cDNA pool from the time-0 sample. Seven microarrays were used in the study, one for each of the seven time points. Each array was probed with the green-labeled sample mixed with one of the seven red-labeled samples. In effect, time 0 serves as a reference for all of the samples. This experimental setup has some peculiar consequences for analysis that we will discuss later.
The data set contains four measurements for each spot: green signal, green background, red signal, and red background. As their estimate of relative expression of a gene at time k compared with time 0, Chu et al. use the background-corrected ratio (red signal − red background)/(green signal − green background) from the array containing red-labeled cDNA from time k and green-labeled cDNA from time 0. ANOVA modeling of the non-background-corrected data showed systematic trends in the residuals, pointing to model inadequacy. We investigated several ANOVA models for the log background-corrected data, and found the data supported the model
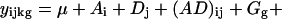 |
3 |
 |
for i = 1, . . . , 7 arrays; j = 1,2 dyes; k = 0, . . . , 6 varieties (time points); and g = 1, . . . , 6,118 genes. Table 1 gives the analysis of variance. We note that it is possible to fit the larger model (1), which includes dye-by-gene effects, to this data. However, with this experimental design, 0 residual degrees of freedom remain so it is not possible to evaluate the adequacy of the model. On the other hand, with Model 2 and this experimental design, all of the nonzero residuals come from the time 0 vs. time 0 self-comparison array. This is a highly undesirable situation, because one is forced to assume that this array is representative of all of the others. The residual plot showed no systematic trend or evidence against our assumption of constant error variance, although there was insufficient data to consider testing for gene-specific variance because there are only two residuals per gene.
Table 1.
Analysis of variance for sporulation data
| Source | SS | df | MS |
|---|---|---|---|
| Array | 6,616.33 | 6 | 1,102.72 |
| Dye | 187.57 | 1 | 187.57 |
| Array × Dye | 92.33 | 6 | 15.39 |
| Gene | 48,329.71 | 6,117 | 7.90 |
| VG,AG | 22,907.16 | 73,404 | 0.31 |
| Residual | 89.18 | 6,117 | 0.0146 |
| Adjusted Total | 78,222.28 | 85,651 |
SS, sum of squares; df, degrees of freedom; MS, mean square.
Chu et al. are interested in genes induced during sporulation. Their filter excludes genes that do not show a minimum increase relative to time 0. Their clustering procedure matches genes to seven temporal patterns or “profiles” of induced transcription of special interest. Each profile is defined by a “prototypical” expression pattern calculated by averaging a hand-picked set of 3–8 genes per profile. The clustering method matches genes to these profiles based on the correlation between the 7-vector of log ratios and the profile prototype. A gene is matched to a profile if its correlation with that profile is larger than the with the other profiles and also above a threshold. Of about 1,000 genes that pass their filter, about 450 are assigned to one of the seven profiles.
We modify the Chu et al. clustering to incorporate bootstrapping and assess the reliability of the results. First, we estimate the difference in gene expression for gene g at time k compared with time 0 with ( )kg − (
)kg − ( )0g. In addition, we construct 99% bootstrap confidence intervals for these estimates (9, 10). We chose bootstrap confidence intervals to avoid making distributional assumptions about the error. Fig. 1 shows estimated profiles for two genes with 99% bootstrap confidence intervals based on 10,000 bootstrap simulations.
)0g. In addition, we construct 99% bootstrap confidence intervals for these estimates (9, 10). We chose bootstrap confidence intervals to avoid making distributional assumptions about the error. Fig. 1 shows estimated profiles for two genes with 99% bootstrap confidence intervals based on 10,000 bootstrap simulations.
Figure 1.

Temporal profiles for select genes. The solid line gives the profile estimated by using Model 3. Error bars around the profiles are 99% bootstrap confidence intervals. The dotted line gives the temporal profile estimated with log ratios, rescaled to have the same standard deviation as the solid line.
Next, we created model profiles based on the same representative genes identified by Chu et al. (Fig. 2). (Two genes, MRD1 and NAB4, for profile 3 and two genes, KNR4 and EXO1, for profile 4 could not be found in the publicly available data file. We constructed profiles 3 and 4 with the remaining genes.) As our filter, we exclude any gene that does not satisfy the following criteria: for at least one time point k not zero ( kg − (
kg − (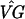 )0g > 0 and the 99% confidence interval for (VG)kg − (VG)0g does not contain 0. Thus we mimic the filter used by Chu et al., but with a statistically based criterion. For each gene g passing the filter, we calculate the correlation coefficient rgp for that gene and the p = 1, . . . , 7 profiles. Gene g is assigned to profile p if rgp > 0.9 and rgp is the largest of {rg1, . . . , rg7}. From columns a and b in Table 2, we see that the number of genes clustering to each profile is somewhat larger here than for Chu et al., except for profile 2 (Early I). This can be attributed to the fact that our filter is not as stringent as that in ref. 2 and passes almost twice as many genes, close to 2,000.
)0g > 0 and the 99% confidence interval for (VG)kg − (VG)0g does not contain 0. Thus we mimic the filter used by Chu et al., but with a statistically based criterion. For each gene g passing the filter, we calculate the correlation coefficient rgp for that gene and the p = 1, . . . , 7 profiles. Gene g is assigned to profile p if rgp > 0.9 and rgp is the largest of {rg1, . . . , rg7}. From columns a and b in Table 2, we see that the number of genes clustering to each profile is somewhat larger here than for Chu et al., except for profile 2 (Early I). This can be attributed to the fact that our filter is not as stringent as that in ref. 2 and passes almost twice as many genes, close to 2,000.
Figure 2.

The seven model profiles used for clustering. The profiles are rescaled to a have standard deviation of 1, which does not affect the clustering results because clustering is based on correlations.
Table 2.
Number of genes matching to each profile
| Profile | Clustering method
|
|||
|---|---|---|---|---|
| a | b | c | d | |
| 1 | 52 | 65 | 3 | 8 |
| 2 | 61 | 51 | 7 | 11 |
| 3 | 45 | 74 | 3 | 11 |
| 4 | 95 | 151 | 12 | 27 |
| 5 | 158 | 241 | 86 | 120 |
| 6 | 61 | 145 | 17 | 36 |
| 7 | 5 | 15 | 2 | 6 |
(a) Chu et al. clustering, (b) modified clustering with no reliability measure, (c) modified clustering requiring 95% stability, and (d) modified clustering requiring 80% stability. Column d is included because a 95% stability requirement is somewhat arbitrary.
The next step is bootstrapping to assess the reliability of the clusters. We create 499 bootstrap data sets y*ijkg and, for each simulated data set, we construct a bootstrap temporal pattern based on the estimates (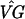 )*kg − (
)*kg − (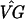 )*0g for each gene. We repeat the filtering and clustering steps with these bootstrap estimates. The same filter is used in each bootstrap iteration (i.e., we do not repeat the first level of bootstrapping). Similarly, the profile “prototypes” are not recalculated. The result is 500 clusterings, 1 based on the actual data and 499 bootstrap-simulated clusterings. The match of a gene to a profile is declared “95% stable” if it occurs in the analysis of the actual data and in at least 95% of the bootstrap clusterings.
)*0g for each gene. We repeat the filtering and clustering steps with these bootstrap estimates. The same filter is used in each bootstrap iteration (i.e., we do not repeat the first level of bootstrapping). Similarly, the profile “prototypes” are not recalculated. The result is 500 clusterings, 1 based on the actual data and 499 bootstrap-simulated clusterings. The match of a gene to a profile is declared “95% stable” if it occurs in the analysis of the actual data and in at least 95% of the bootstrap clusterings.
Column c of Table 2 shows the numbers of 95% stable matches per profile. Fig. 3 plots the profiles of the 95%-stable genes. When we reduce the criterion to 80% stability, the number of stable genes in each cluster remains many fewer than the number of nominal matches. For the most part, the 95%-stable genes are a subset of the Chu et al. genes. The exceptions are seven stable genes that matched to profiles 5, 6, or 7 but did not match to any profile by Chu et al. One of these, YPL280W, is in Fig. 1. Like YPL280W, the other six genes have fairly flat profiles, so the difference is likely due to the less stringent filter.
Figure 3.

Ninety-five-percent-stable genes for the seven model profiles based on 500 bootstrap clusterings. The plotted profiles have been rescaled to have a standard deviation of 1. See Fig. 5, an expanded version of this figure, which is published as supplemental data on the PNAS web site.
Bootstrapping draws attention to some attributes of this particular clustering procedure. When profiles are themselves highly correlated, one can expect that genes with high correlation to one profile will also have high correlation to the other. Eight of 21 pairs of profiles have correlation at least 0.75 (see Table 3 and Figs. 5 and 6, which are published as supplemental data on the PNAS web site, www.pnas.org). The largest correlation is 0.95 for profiles 4 and 5. Fifty-eight percent of genes that are nominal matches to profile 4 match to profile 5 in more than 5% of bootstrap iterations. Twenty-seven percent of genes that are nominal matches to profile 5 match to profile 4 in more than 5% of bootstrap clusterings. Thus, many genes fail to be 95% stable matches to profile 4 simply because of the presence of profile 5, and vice versa. Given the level of noise in the data, these two profiles are too similar to be readily distinguished.
Discussion
The goal of bootstrap clustering is to make statistical inference about a discrete structure, the clustering C, which we estimate with Ĉ. The estimated clustering Ĉ has some unknown sampling distribution around the true clustering C. Bootstrapping uses the sampling distribution of Ĉ* around Ĉ to infer the sampling distribution of Ĉ around C. Efron, Halloran, and Holmes (13) discuss bootstrap inference for such discrete objects. In discussing our methodology we avoid the terms “confidence” and “significance” because they are technically incorrect in this setting. However, the arguments in ref. 13 for the appropriateness of bootstrapping to make inferences from reconstructed phylogenetic trees (7) apply here. The “stability” of a gene—the percent of bootstrap clusterings in which it matches to the same cluster—is a reasonable first approximation to the confidence of the match.
We chose the Chu et al. example to demonstrate bootstrap clustering because of its simplicity. The bootstrapping procedure does not change with other clustering methods, but additional complexity may arise in the step of summarizing and evaluating the results. With a case-2 clustering method, group labels are not well defined across bootstrap clusterings Ĉ*. Several approaches are possible. We suggest looking at pairs of genes that cluster together in Ĉ and counting the frequency with which such pairs cluster together in the Ĉ*. One hopes that stable clusters of genes emerge where each pair of members clusters together reliably.
Case 3, hierarchical clustering, is exactly the situation addressed by Felsenstein (7) in his original work on bootstrapping phylogenies. Following the terminology in phylogenetics, we refer to a group of genes descending from a common node as a clade. To summarize bootstrapping results, we count the number of occurrences of each clade in the observed tree T̂ in the bootstrap sample of trees {T̂*}. The resulting frequency is placed on the dendrogram T̂ at the node that marks the clade. These frequencies provide a way to gauge the approximate significance of each component of the tree (13).
Our approach for bootstrapping procedure differs from Felsenstein's (7) in the way bootstrap data sets y* are generated. With phylogenetic data, one resamples columns from a data set under the assumption that the columns are independent, identically distributed (i.i.d.) realizations of a random process. With microarray data, there is no such i.i.d. structure. Instead, we use a structural model to estimate systematic sources of variation and separate those from the noise, which we assume is i.i.d. given the correct structural model.
In the example, we did not see any evidence in the residuals against our assumption of constant error variance. In principle, heteroscedasticity does not present a limitation to the methodology. If each gene has its own error distribution, one could incorporate this in the resampling scheme by using only the residuals for gene g to produce y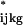 . However, this will only be possible when the experimental design provides enough replication. Intermediate solutions are available for bootstrapping in the presence of intensity-dependent heteroscedasticity (12).
. However, this will only be possible when the experimental design provides enough replication. Intermediate solutions are available for bootstrapping in the presence of intensity-dependent heteroscedasticity (12).
The computational burden of bootstrapping is an obvious concern. In the example, computation was less than one minute per bootstrap data set on a 400-MHz personal computer with interpreted code (MATLAB). We chose 500 for the bootstrap sample size to put the computational time on the scale of “overnight.” Clearly, computing time could be reduced with more computing power and efficient, compiled code. The primary limit to the practicality of bootstrapping will be the computational intensity of the clustering algorithm.
Bittner et al. (14) use a different approach for evaluating cluster results. They add normally distributed noise with mean 0 directly to the log-ratios and recluster the results. This method implicitly assumes that ratios are unbiased estimates of relative expression. The technique in ref. 14 further assumes normally distributed error, whereas we generally find error distributions to be heavy-tailed. Furthermore, Bittner et al. use an estimate of variance that includes variation due to differential expression. The advantage of our model-based approach is that it separates systematic sources of variation from noise and uses an empirical estimate of error, which is free of distributional assumptions.
It would be interesting to reanalyze the clustering in ref. 14 the same way as the Chu et al. data, but this is not possible for two reasons. First, the publicly available data provides only ratios, so the information needed for ANOVA modeling is lost. More fundamentally, the experiment in ref. 14 does not have even the minimal replication of the Chu et al. experiment. A model such as 3 would be saturated and no estimate of the error distribution would be possible. This situation highlights the importance of replication in microarray experiments, which has been noted in several publications (9, 15, 16). Replication is a fundamental principle of good experimental design and serves two purposes. First, replication increases the precision of estimated quantities. Second, and perhaps most important, it provides information about the uncertainty of estimates (8). Only with an appropriately designed experiment that includes replication can statistically valid conclusions be drawn (17).
In the yeast sporulation experiment that is reanalyzed here, a kind of replication is achieved by making a self-comparison of the time-0 sample. Although this is adequate for providing error degrees of freedom, it is not an ideal situation. All of the nonzero residuals from the ANOVA analysis come from the self-comparison array—all other data points are fit exactly because they are not replicated. If the self-comparison array is not typical of the experiment as a whole, one can be misled in imputing the same level of variation to the other arrays.
Although perhaps counterintuitive, it is possible to replicate all samples without using additional arrays. For example, samples could be arranged in a loop as shown in Fig. 4, so that samples from each time point appear on two arrays. Fitting Model 1 with this design, residuals are obtained from every array. In addition to the built-in replication, varieties (V) are balanced with respect to dye (D). This balance has certain advantages for the data analysis (16), although there is additional cost associated with the number of labeling reactions required. With the loop design, the variance of gene-specific differences in time points depends on the relative position of the corresponding samples in the loop. Because adjacent time points are estimated most precisely, it is most efficient to estimate profiles by using those comparisons rather than by using time 0 as a fixed reference point. With the loop design, estimates of (VG)k+1,g − (VG)kg for adjacent time points have variance 85.7% as large as estimates of (VG)kg − (VG)0g with the design used by Chu et al. This increased precision, balance among design factors, and the fact that residuals are obtained from every array make this design one alternative worthy of consideration.
Figure 4.

An alternative experimental design for the sporulation study, represented as a directed graph. The boxes represent RNA samples and the arrows represent microarrays. The tail of an arrow is, say, the “red” dye and the head of an arrow is the “green” dye. Thus, the arrow from the time-0 sample to the half-hour sample means to hybridize an array with red-labeled time-0 mRNA and green-labeled mRNA from the half-hour sample. Such a design has advantages over the plan used by Chu et al. in balance, precision of estimates, and distribution of residuals.
In scientific experimentation, results depend on experimental designs that yield precise estimates of quantities of interest, as well as estimates of the precision achieved. Furthermore, the design should produce data that allows the assumptions of analysis to be verified. Microarray experiments are no exception. It is certainly an interesting exercise to run a clustering algorithm on gene expression data. However, without an assessment of the reliability of the clusters one cannot make valid inferences about similarly behaving genes. Whatever clustering algorithm is chosen, it is imperative to assess whether the results are statistically reliable relative to the level of noise in the data. Bootstrapping is a useful tool to accomplish this.
Supplementary Material
Acknowledgments
This work is supported in part by a Burroughs–Wellcome postdoctoral fellowship from the Program in Mathematics and Molecular Biology (to M.K.K.), and National Institutes of Health Grant CA88327 and National Heart, Lung, and Blood Institute Program in Genomics Applications Grant 66620 (to G.A.C.).
References
- 1.Brown P O, Botstein D. Nat Genet Suppl. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 2.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 3.Eisen M, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hastie, T., Tibshirani, R., Eisen, M. B., Alizadeh, A., Levy, R., Staudt, L., Chan, W. C., Botstein, D. & Brown, P. (August 4, 2000) Genome Biol., http://genomebiology.com/2000/1/2/research/0003/. [DOI] [PMC free article] [PubMed]
- 6.Lazzeroni L, Owen A. Stanford Biostatistics Series: Technical Report 211. Stanford, CA: Stanford Univ.; 2000. [Google Scholar]
- 7.Felsenstein J. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 8.Fisher R A. The Design of Experiments. 6th Ed. Edinburgh: Oliver and Boyd; 1951. [Google Scholar]
- 9.Kerr M K, Martin M, Churchill G A. J Comp Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- 10.Efron B, Tibshirani R J. An Introduction to the Bootstrap. London: Chapman & Hall; 1994. [Google Scholar]
- 11.Draper N R, Smith H. Applied Regression Analysis. 3rd Ed. New York: Wiley; 1998. [Google Scholar]
- 12.Davison A C, Hinkley D V. Bootstrap Methods and their Application. Cambridge, U.K.: Cambridge Univ. Press; 1997. [Google Scholar]
- 13.Efron B, Halloran E, Holmes S. Proc Natl Acad Sci USA. 1996;93:13429–13434. doi: 10.1073/pnas.93.23.13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, et al. Nature (London) 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 15.Lee M-L T, Kuo F C, Whitmore G A, Sklar J. Proc Natl Acad Sci USA. 2000;97:9834–9839. doi: 10.1073/pnas.97.18.9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerr M K, Churchill G A. Biostatistics. 2000;2:183–201. doi: 10.1093/biostatistics/2.2.183. [DOI] [PubMed] [Google Scholar]
- 17.Kerr M K, Churchill G A. Genet Res. 2001;7:819–837. doi: 10.1017/s0016672301005055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.