

Abstract
Targeted delivery of anti-inflammatory osteoarthritis treatments have the potential to significantly decrease undesirable systemic side effects and reduce required therapeutic dosage. Here we present a targeted, non-invasive drug delivery system to decrease inflammation in an osteoarthritis model. Hollow thermoresponsive poly(N-isopropylacrylamide) (pNIPAM) nanoparticles have been synthesized via degradation of a N,N′-bis(acryloyl)cystamine (BAC) cross-linked core out of a non-degradable pNIPAM shell. Sulfated 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPSA) was copolymerized in the shell to increase passive loading of an anti-inflammatory mitogen-activated protein kinase-activated protein kinase 2 (MK2)-inhibiting cell-penetrating peptide (KAFAK). The drug-loaded hollow nanoparticles were effective at delivering a therapeutically active dose of KAFAK to bovine cartilage explants, suppressing pro-inflammatory interleukin-6 (IL-6) expression after interleukin-1 beta (IL-1β) stimulation. This thermosensitive hollow nanoparticle system provides an excellent platform for the delivery of peptide therapeutics into highly proteolytic environments such as osteoarthritis.
Keywords: N-isopropylacrylamide, Thermosensitive polymer, Anti-inflammatory peptides, Inflammation, Osteoarthritis
Graphical abstract
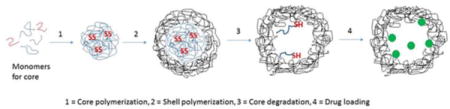
INTRODUCTION
Osteoarthritis (OA) is the leading cause of chronic disability in the United States, affecting over 20 million patients[1, 2] and costing over $100 billion annually[3]. Osteoarthritis is characterized by a persistent cycle of cartilage inflammation and degradation resulting in pain during movement. Pathological persistent activation of immune cells such as macrophages causes sustained release of pro-inflammatory cytokines such as interleukin-1 (IL-1)[4, 5], interleukin-6 (IL-6)[6–8], and tumor necrosis factor-alpha (TNF-α)[8] which can aggravate or accelerate damage to the surrounding tissues. The elevated levels of inflammatory cytokines result in upregulation of proteolytic enzymes that degrade key components of the extracellular matrix such as aggrecan and collagen[9, 10], facilitating penetration of inflammatory factors deeper into the cartilage.
Initial treatment of OA often includes orally administered anti-inflammatory compounds such as non-steroidal anti-inflammatory drugs (NSAIDs) or newer compounds that inhibit specific pro-inflammatory cytokines such as Enbrel or Humira (both targeting TNF-α[11]) and Kineret (targeting IL-1[12]). Further treatment strategies include intra-articular glucocorticoid injections which suppress cell-mediated immunity and reduce inflammation[13]. A significant challenge with these treatments is their short therapeutic lifetimes due to proteolysis and rapid clearance by immune cells[14]. Additionally, long-term glucocorticoid use is associated with adverse side effects such as musculoskeletal disorders[15] and liver toxicity[16], while long-term use of NSAIDs are associated with increased gastrointestinal[17, 18], cardiovascular[19], and kidney[20] complications. Thus, there is need for a delivery system that is able to be directly administered to the site of inflammation and provide a controlled release of an anti-inflammatory therapeutic, providing long-term relief while minimizing the side effects common in systemic and non-targeted administration. Drug-loaded nanoparticles provide a biocompatible delivery system that is able to protect the encapsulated therapeutic from enzymatic degradation while delivering it directly to the target tissue.
Brugnano et al[21], previously reported a 23-mer cell-penetrating anti-inflammatory peptide (KAFAKLAARLYRKALARQLGVAA, abbreviated KAFAK) that reduces the expression of pro-inflammatory cytokines by inhibiting mitogen-activated protein kinase-activated protein kinase 2 (MK2), which modulates the expression of many pro-inflammatory cytokines such as TNF-α, IL-1, and IL-6[22–25]. However, poor bioavailability of peptides in the presence of serum and gastric acid peptidases limits their efficacy and commercial application[26–29]. To combat this, our lab has developed a biocompatible nanoparticle system that is able to load these therapeutic peptides and release them in a controlled manner, allowing for increased therapeutic efficacy while providing local delivery to the injured area[30–32].
Thermosensitive poly(N-isopropylacrylamide) (pNIPAM) has been extensively studied in a variety of biomedical applications[33–35], as it exhibits a lower critical solution temperature (LCST) at ~31–33°C[36, 37]. pNIPAM-based nanoparticles are water soluble at temperatures below the LCST, allowing for loading of water soluble therapeutics via passive diffusion[31, 37]. The particles then undergo hydrophobic collapse at physiological temperatures, entrapping the loaded drug and allowing for controlled release while protecting the drug from the proteolytic environment [31, 38]. Additionally, Bartlett et al[31, 32] demonstrated that the loading efficiency of cationic therapeutic peptides could be increased by co-polymerizing with the sulfated monomer 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS). Although these particles had a loading efficiency up to ~40%, they only released ~5% of the loaded peptide in the first 48 hours, with a maximum release of ~12% after 10 days, suggesting that much of the loaded peptide was permanently associated with the inner core of the particle. Previously, we have shown that reducible pNIPAM nanoparticles crosslinked with N,N′-bis(acryloyl)cystamine (BAC) can release up to ~35% of their loaded peptide when exposed to a reducing environment, such as those found within lysosomal compartments following cellular uptake[39]. However, these particles have a slightly decreased loading efficiency of ~35% while still releasing less than half of their peptide load. The development of hollow pNIPAM nanoparticles should provide increased loading capacities and improved release, with the potential to tune release rates through control of shell thickness and crosslink density [40, 41].
In this paper, we demonstrate that hollow PEGylated (hNGPEGMBA) and non-PEGylated (hNGMBA) pNIPAM nanoparticles have increased loading and release of KAFAK compared to solid pNIPAM nanoparticle counterparts (NGPEGMBA and NGMBA). Additionally, we show that these particles maintain biocompatibility, are readily taken up by macrophages, and yield improved therapeutic efficacy of KAFAK when administered directly to arthritic cartilage in an ex vivo osteoarthritis model.
MATERIALS AND METHODS
Cell Culture
RAW 264.7 macrophages were donated by Dr. Philip Low at Purdue University. The cells were grown as a monolayer in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS), penicillin (50 units/mL), and streptomycin (50 μg/mL) at 37 °C and 5% CO2[42]. Cells were used between passages 3 and 9.
Materials
N-Isopropylacrylamide (NIPAm, ≥98%) was acquired from Polysciences Inc. (Warrington, PA). Dialysis membrane tubing was purchased from Spectrum Laboratories (Dominguez, CA). (1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (≥98%, HBTU), N-hydroxybenzotriazole (>97%, HOBT), N,N’-methylenebisacrylamide (99%, MBA), sodium dodecyl sulfate (SDS; 10% w/v in water), 2- acrylamido-2-methyl-1-propanesulfonic acid (99%, AMPSA), dithiothreitol (98%, DTT), fluorescein o-acrylate (98%), N,N-Diisopropylethylamine (99%, DIPEA), potassium persulfate (99%, K2S2O8), N,N’-bis(acryloyl)cystamine (98%, BAC), trifluoroacetic acid and dimethyl sulfoxide (DMSO) were acquired from Sigma-Aldrich (St. Louis, MO). NIPAm, MBA, and AMPSA were stored under nitrogen at 4°C. Acrylate-PEG2000 was purchased from Laysan Bio (Arab, AL). Triisopropyl silane was purchased from TCI America (Boston, MA), and ethane dithiol was purchased from Alfa Aesar (Ward Hill, MA). All water used in synthesis, dialysis, and testing was treated by a Millipore Milli-Q system (Billerica, MA; 18.2 MΩ•cm resistivity).
Peptide Synthesis and Purification
Therapeutic peptides (KAFAK) were synthesized on knorr resin by 9-fluorenylmethyloxycarbonyl (FMOC) solid phase methodology[39]. Knorr amine resin was swollen with dichloromethane (DCM) followed by dimethylformamide (DMF). A solution of 25% piperidine in DMF was added to the resin, and nitrogen was bubbled for 15 min. The resin was washed with DMF (3x) and isopropyl alcohol (i-PrOH, 3x). For amino acid coupling, a solution of Fmoc-amino acid (3 equiv), HBTU (3 equiv), HOBT (3 equiv), and DIPEA (5 equiv) in DMF was added. Nitrogen was bubbled for 2 h, and resin was washed with DMF (3x), DCM (3x) and i-PrOH (3x). Fmoc deprotection was performed by adding a solution of 25% piperidine in DMF to the resin and bubbling nitrogen for 20 min; the solvent was removed and the resin was washed with DMF (3x) and i-PrOH (3x). The above sequence was repeated for coupling of each amino acid.
The completed peptide was cleaved from the resin with a cocktail of trifluoroacetic acid, triisopropyl silane, ethane dithiol, and Milli-Q water, and then concentrated under vacuum. The concentrated product was immediately precipitated in ice cold ether, centrifuged, solubilized in Milli-Q water, and lyophilized. Peptides were purified on an FPLC AKTA Explorer (GE Healthcare, Pittsburgh, PA) with a 22/250 C18 prep-scale column (Grace Davidson, Deerfield, IL) and an acetonitrile gradient with 0.1% trifluoroacetic acid. Peptide purity was determined by HPLC to be >98%. Peptide molecular weight was confirmed by matrix-assisted laser desorption ionization time-of-flight (MALDI TOF) mass spectrometry with a 4800 Plus MALDI TOF/TOF Analyzer (Applied Biosystems, Foster City, CA).
Nanoparticle Synthesis
Hollow NIPAm nanoparticles with 5% AMPS were prepared using a precipitation polymerization reaction[43–46]. For core synthesis, 49 mL of Milli-Q water was heated to 70°C in a 3-neck round bottom flask and refluxed under nitrogen for 30 minutes. 178 mg of NIPAm, 6 mol% BAC, and 20 mg of SDS were dissolved in 10 mL of degassed Milli-Q water and added to the flask. Finally, 35 mg of potassium persulfate was dissolved in 1 mL of Milli-Q water and added to the flask to initiate the reaction. The polymerization was carried out for 5 h under nitrogen. The resultant particles were filtered through a Whatman filter to remove any aggregates, and then cleaned several times by centrifugation and resuspension in Milli-Q water.
To create the pNIPAM shell, 4 mg of SDS was added to 5 mL of the disulfide crosslinked cores in a 3-neck round bottom flask and heated to 70°C under nitrogen. A separate solution of 50 mg NIPAm, 5 mol% AMPSA, 2 mol% MBA and, for PEGylated nanoparticles, 3 mol % Acrylate-PEG2000 were prepared in 30 mL of degassed Milli-Q water and allowed to incubate to room temperature under nitrogen for 45 minutes before being added to the heated core solution. To initiate polymerization, 6 mg of potassium persulfate was dissolved in 1 mL Milli-Q water and added to the flask. After 5 hours, the core-shell nanoparticles were collected and dialyzed against Milli-Q water for 6 days using a 15 kDa MWCO dialysis membrane (Spectra-Por).
Following dialysis, the core was degraded by re-suspending the core-shell particles in a 1 mM DTT solution and allowing the solution to react under stirring for 7 days. The hollow particles were then cleaned by centrifugation and resuspension in Milli-Q water, frozen, and lyophilized. For fluorescent nanoparticle synthesis, 1 mol% fluorescein o-acrylate was pre-dissolved in 3% DMSO and then added to the shell polymer mixture and allowed to equilibrate for 30 minutes before the reaction being initiated with the addition of potassium persulfate.
Nanoparticle Characterization
Hydrodynamic radius of the particles was determined using dynamic light scattering (DLS) on a Nano-ZS90 Zetasizer (Malvern, Westborough, MA). Nanoparticles were dissolved at 0.1 mg/mL in MilliQ water in a disposable polystyrene cuvette, and were subjected to a minimum of 12 runs after equilibrating at the desired temperature for 5 minutes. Zeta (ζ) potentials and electrophoretic mobility were obtained on a Nano-ZS90 Zetasizer at the same sample concentration in PBS using folded capillary cells. All Transmission electron microscopy (TEM) work was performed at the electron microscopy core laboratory within the UC Davis School of Medicine on a FEI CM120 (Hillsboro, OR) transmission electron microscope. Discharged TEM grids were places on a 5 μL droplet of MilliQ water containing suspended nanoparticles for 10 minutes before staining with uranyl acetate. Samples were then dried and imaged at room temperature. As one of the mechanisms for drug loading and release is electrostatic attraction, PBS was selected for zeta potential measurements in order to gain information on the particles surface charge in the high ionic strength solutions found in our drug release studies. Confirmation of core degradation was performed using particles with cores labeled with 0.1 mol% fluorescein o-acrylate. Core degradation with and without 1mM DTT was monitored over 7 days using a Spectramax M5 plate reader (Molecular Devices, Sunnyvale, CA) to measure the absorbance of the nanoparticle core.
Drug Loading and Release
To load the nanoparticles, 1 mg of purified KAFAK and 1 mg of lyophilized hollow nanoparticles were incubated in 1 mL of Milli-Q water and stored at 4°C for 24 hours. The loaded particles were then collected via centrifugation at 18,000g and 25°C for 1 hour and the supernatant was collected for analysis. In order to aid in later resuspension, the pellet was broken up by brief suspension in ~200 μL of Milli-Q before being frozen and lyophilized. Due to the complexities of detecting our peptide in cell culture media, peptide release was measured in 1mL of PBS and the particles were incubated on a plate shaker at 37°C and 200 RPM. At each time point, releasing nanoparticles were centrifuged at 18,000g and the supernatant run on a fluoraldehyde o-phthalaldehyde (OPA) assay to quantify the amount of peptide released. For time points beyond 4 hours, the pellet was then resuspended in 1x PBS and placed back on the plate shaker. Briefly, for the fluoraldehyde OPA assay, 1:10 mixtures of sample and fluoraldehyde OPA were created in opaque 96-well plates (VWR), and the fluorescence was measured at an excitation of 360 nm and emission of 455 nm on a SpectraMax M5 plate reader.
Values were compared against a standard to determine the amount of KAFAK present in each solution. Drug loading efficiency was then determined by measuring the amount of peptide loaded per milligram of nanoparticle.
Nanoparticle Cytotoxicity
In order to determine the toxicity of KAFAK loaded nanoparticles, a CellTiter Aqueous One assay was performed, according to manufacturer instructions, to measure the number of viable cells before and after nanoparticle treatment. Briefly, RAW 264.7 cells were seeded at a density of 20,000 cells/cm2 on a tissue culture treated 96-well plate, and incubated at 37°C for 24 hours to allow to adhere. The media was then aspirated and treatments of 4 mg/mL nanoparticles in media were added and allowed to incubate for 24 hours. Following incubation, the media was removed and cells were washed with PBS. Finally, 100 μL of culture media and 20 μL of CellTiter (Promega, Madison, WI) reagent was added to each well and allowed to incubate for 3 hours at 37°C. Following incubation, the absorbance was read on a Spectramax M5 plate reader.
In Vitro Cell Imaging
For confocal microscopy studies, RAW 264.7 cells were seeded at 10,000 cells/well in an 8-well culture slide (Lab-Tek) and maintained at 37 °C. After adhering, fluorescein-labeled nanoparticles at a final concentration of 1.5 mg/mL in culture media were added to the wells and allowed to incubate for 4 h or 24 h at 37 °C. Culture media used in nanoparticle experiments contained FBS (as indicated in the cell culture section) unless otherwise noted. After incubation, cells were washed with media to remove unbound nanoparticles and then stained with lysotracker DND-99 (Life Technologies, Grand Island, NY) according to manufacturer instructions, in order to confirm endosomal/lysosomal uptake of the nanoparticles. Following staining, cells were imaged with Olympus FV1000 confocal microscopy (Center Valley, PA). Semi-quantitative fluorescence measurements were taken using ImageJ to determine total fluorescent area and intensity of the green channel normalized to the red channel within each field of view (n=3). Results of the PEGylated and non-PEGylated hollow and solid nanoparticles were then compared to determine relative nanoparticle uptake rates.
Ex vivo Osteoarthritis Model
Cartilage plugs were obtained from 3 month old bovine knee joints obtained from an abattoir within 24 h of slaughter (Dutch Valley Veal, South Holland, IL). The plugs were removed from the load bearing region of the femoral condyle using a 3 mm diameter cork borer. They were then washed three times in serum free medium and equilibrated for 3 days in 5% FBS supplemented media. OA like conditions were simulated by removal of native aggrecan using a previously described protocol by Poole et al[47]. Briefly, plugs were treated with 0.5% (wt/vol) trypsin in HBSS for 3h at 37°C to remove aggrecan. After trypsin treatment plugs were washed three times in HBSS and incubated with 20% FBS for 20 minutes to inactivate residual trypsin activity. Inflammation was initiated in the plugs by treating with 20 ng/ml IL-1β as mentioned previously by Pratta et al[48]. Nanoparticle treatments were added after day 2 of culture. Fresh IL-1β and nanoparticles were added every 2 days for an 8 day culture period. Media aliquots were collected and stored in low bind tubes at −80°C until further analysis.
IL-6 production in the explanted cartilage was measured with a bovine IL-6 ELISA development kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s protocol. Briefly, capture antibody was coated overnight onto Nunc MaxiSorp 96-well plates. The plate was washed and blocked for 1h with 5% sucrose and 4% bovine serum albumin (Sera Life Sciences, Milford, MA) in PBS. After blocking, plates were washed with PBS containing 0.05% Tween-20, samples and standards were then added to the plate and incubated with gentle shaking for 1h. After washing, the plates were incubated with a detection antibody for 1 hour, washed, and incubated with Streptavidin-HRP for 30 min. Finally, the samples were developed by the addition of 3,3′,5,5′-tetramethylbenzidine (TMB) solution and incubating for 20 minutes before adding a 0.16M sulfuric acid stop solution. The plate was read on an absorbance plate reader at 450 nm with a correction at 550 nm. IL-6 production was normalized to individual plug weight as well as to the negative control where OA-like conditions were induced in the plugs with trypsin.
Nanoparticle uptake in ex vivo inflammatory model
Bovine cartilage plugs were either trypsin treated to induce OA-like conditions or left in cell culture media to maintain a healthy phenotype. Fluorescently labeled nanoparticles were re-suspended in PBS at 0.5 mg/ml and incubated with the cartilage plug, to allow the nanoparticles to diffuse through the cartilage. After the treatment, plugs were washed 3 times with PBS and incubated for 30 minutes at 37°C to remove any unbound NPs. A 30 μm cryo-mid-sagittal cut was made through the plug to examine diffusion into the plugs from the articular surface. Diffusion of NPs was monitored using 488 nm laser excitation on an Olympus FV1000 confocal microscope. ImageJ was then used to measure the fluorescent intensity of three separate areas along the articular surface of each sample. Values are presented as average ± SEM.
Statistical Analysis
Student’s t-tests were used to determine statistical significance between treatment groups using a significance level of p < 0.05. Data is expressed as mean values ± standard deviation unless otherwise noted.
RESULTS AND DISCUSSION
Nanoparticle Characterization
Due to the importance of complete core removal in the formation of hollow pNIPAM nanoparticles, fluorescently-labeled (FITC) nanoparticle cores were fabricated to track degradation and removal following exposure to DTT. Figure 1A shows an absorbance sweep with maximum absorbance at 490nm due to the fluorescently-labeled core. Therefore, we used particle absorbance at 490nm to monitor the rate of core degradation over 7 days (Figure 1B). The data shows decreased particle absorbance following exposure to DTT, while particles incubated without DTT show no decrease in absorbance for 7 days. This indicates that the crosslinked cores were stable within the particle until being exposed to DTT, at which point the degraded core oligomers were able to diffuse out through the outer shell of the nanoparticle. Additionally, this demonstrates that the observed decrease in absorbance is due to the dissolution and removal of the fluorescent core, and not a general decrease in absorbance over time. As seen in Figure 1, minimal absorbance is observed ~5 days after exposure to 1 mM DTT, suggesting complete core dissolution. During this process, it is possible for the polymer chains within the shell to undergo rearrangement into the hollow core, resulting in nanoparticles with a low-density, rather than hollow, core; either of which would allow for increased loading of therapeutic peptides.
Figure 1.
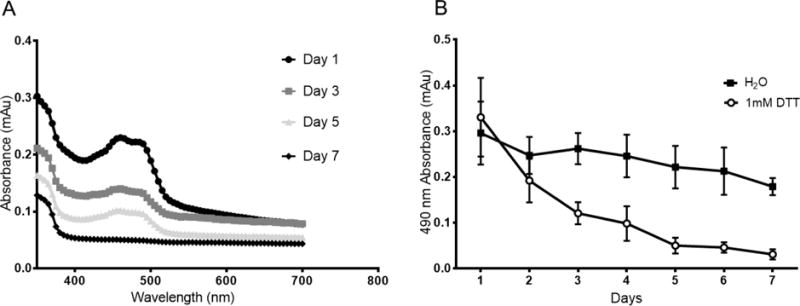
Degradation of BAC crosslinked core with and without exposure to 1 mM DTT. A) Absorbance of fluorescent FITC-labeled core as a function of wavelength showing maximum absorbance at 490 nm. B) 490 nm absorbance of fluorescently labeled core, with and without DTT, over 7 days of dialysis against water
Following core removal, the particles were analyzed with TEM to verify their size and shape as well as gain insight into their density following core removal. As seen in figure 2, the nanoparticles exhibited a spherical morphology with sizes similar to those reported by DLS at temperatures above the LCST. Due to heating of the sample during imaging, it is likely that TEM images represent collapsed particles with hollow nanoparticles demonstrating a decreased core density as seen by areas of increased brightness in the center of the nanoparticles. This is clearly seen in Figure 2(1B), but is less prominent in Figure 2(2B), likely due to their smaller size resulting in a more dense overall structure. The TEM images, combined with our fluorescent data, suggest that following removal of the core, the particles are left with a hollow or low density structure, which should allow for increased loading of therapeutic peptides. Additionally, a halo effect can be observed around PEGylated nanoparticles suggesting that these particles were able to form a PEG layer on their outer surface. Following synthesis, we investigated whether the hollow nanoparticles retained the temperature sensitivity necessary to effectively load cationic therapeutics such as KAFAK. The temperature sensitivity and effect of PEG incorporation into the hollow particles is shown in Figure 3. As expected of pNIPAM based nanoparticles, we observed a decrease in hydrodynamic diameter at temperatures above the LCST, with the solid and hollow particles decreasing 44% and 45%, respectively, and the PEGylated solid and hollow particles decreasing 41% and 83%, respectively. The hollow nanoparticles demonstrated a more pronounced LCST transition, likely due to their hollow cores which would provide less resistance to collapse than a solid polymer core, allowing for more rapid size change as the temperature increased. The hollow PEGylated nanoparticles also had a greater magnitude of collapse then their solid counterpart. This is likely due to the PEG chains increasing the particles hydrophilicity, which would increase particle swelling below the LCST. This increased size would then result in a dramatic decrease in hydrodynamic radius following hydrophobic collapse.
Figure 2.
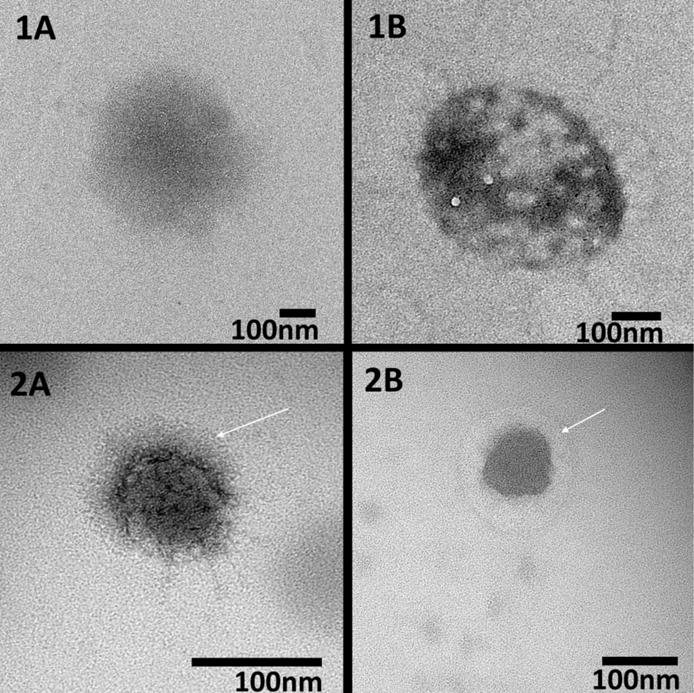
TEM images of non-PEGylated (1A, 1B) and PEGylated (2A, 2B) solid (1A, 2A) and hollow (1B, 2B) nanoparticles demonstrating decreased density in the hollow particles as well as halos around PEGylated particles (white arrows). Particles were stained with uranyl acetate prior to imaging. Scale bar = 100 nm
Figure 3.
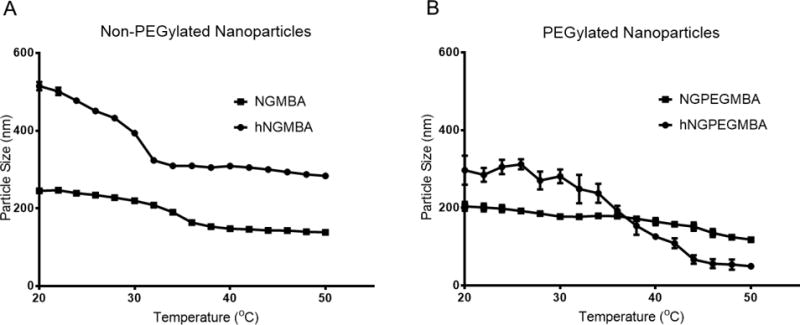
Dynamic light scattering hydrodynamic diameter temperature sweep from 20°C to 50°C of non-PEGylated (A) and PEGylated (B) solid and hollow nanoparticles
Table 1 and Figure 3 indicate that the hollow non-PEGylated nanoparticles were larger than solid particles, with DLS indicating a ~30% larger hydrodynamic radius then solid particles at 25°C and a 7% larger radius at 37°C. This can be attributed to the fabrication process where the shell is added to a preexisting core, resulting in overall larger particles. Interestingly, the addition of PEG to the nanoparticle backbone served to decrease the hydrodynamic radius of both the solid and hollow particles, with the hollow particles decreasing in size by 60% and 35% at 25°C and 37°C respectively. This may be due to reduced polymer incorporation into the particles during synthesis due to the large excluded volume of the PEG chains. What is interesting is the smaller increase in size between the solid and hollow PEGylated particles (30% increase for non-PEGylated nanoparticles and 20% increase for PEGylated at 25°C). This is likely due to the presence of the hydrophilic PEG chains, which would decrease monomer incorporation during polymerization and drive the LCST to higher temperatures, resulting in a smaller size increase for the hollow PEGylated particles at the temperatures studied here [49].
Table 1.
Size, Drug Loading, and ζ Potential. Size presented represents the z-average diameter of measured nanoparticles.
| Nanoparticle | Size (nm) ± stdev 25°C | PDI ± stdev | Size (nm) ± stdev 37°C | PDI ± stdev | Drug Loading (mg/mg) ± stdev | Zeta Potential (mV) ± stdev |
|---|---|---|---|---|---|---|
| NGMBA | 361 ± 1.6 | 0.141 ± 0.001 | 303 ± 7.4 | 0.119 ± 0.031 | 0.251 ± 0.013 | −5.38 ± 1.2 |
| hNGMBA | 469 ± 1.0 | 0.542 ± 0.007 | 324 ± 15.9 | 0.100 ± 0.065 | 0.470 ± 0.018 | −6.18 ± 1.9 |
| NGPEGMBA | 238.4 ± 18.1 | 0.492 ± 0.04 | 178 ± 22.3 | 0.358 ± 0.039 | 0.297 ± 0.093 | −6.96 ± 3.1 |
| hNGPEGMBA | 293 ± 1.4 | 0.193 ± 0.004 | 239 ± 5.9 | 0.182 ± 0.022 | 0.498 ± 0.087 | −8.48 ± 3.5 |
To maximize the loading of cationic peptide, particle shells were co-polymerized with 5% AMPS, which has been shown to be the highest percentage that allowed for stable nanoparticle formation[31, 50]. We investigated particle ζ potentials to verify retention of anionic charge following core dissolution and the addition of PEG (Table 1). As shown in Table 1, PEG addition did not significantly impact ζ potential, suggesting that PEGylated particles should exhibit the increased peptide loading capacity observed in other sulfated nanoparticles[31, 50, 51]. Hollow particles had similar ζ potentials to their solid counterparts due to a constant 5 mol% AMPS co-monomer.
Hollow nanoparticles exhibited greater loading of KAFAK compared to their solid counterparts (Table 1). This can primarily be attributed to the low density core which would decrease the resistance to diffusion, allowing for increased KAFAK loading via a decreased diffusion barrier compared to solid particles. Additionally the hollow, or low density, cores support a greater swelling, suggesting a decreased porosity and lower diffusion barrier allowing more peptide to be loaded by the particle. These characteristics allow for greater electrostatic loading of KAFAK into the particles low density core and increased peptide loading. This electrostatic attraction then causes some of the loaded peptide to become strongly associated with the particle, resulting in the incomplete release observed in figure 4. Work is being done on a degradable poly(NIPAM) shell which will allow for the dissolution of the shell and release of the remaining entrapped peptide.
Figure 4.
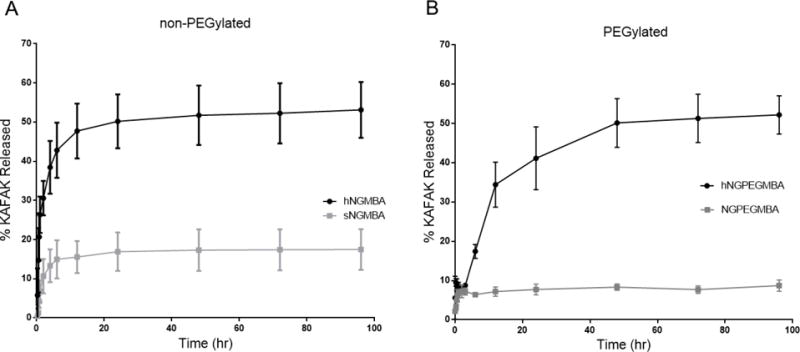
Drug release profiles of KAFAK-loaded non-PEGylated (A) and PEGylated (B) solid and hollow nanoparticles in PBS (pH 7.4) over a 96 h period at 37 °C
The release of KAFAK from the particles over 4 days is shown in Figure 4, with all particles exhibiting a high initial burst release caused by the quick release of peptide electrostatically associated with the outer shell. This effect may have been increased by the particles brief resuspension in water prior to lyophilization, which may have allowed some of the peptide to diffuse out of the particle and be read as part of the initial burst during the first time point. In addition to loading more KAFAK, the hollow nanoparticles also released a significantly larger percentage of their payload, over a longer time, compared to the solid nanoparticles. The solid nanoparticles released 16% of their loaded KAFAK, (0.04 mg KAFAK per mg of particle), with over 80% of this release occurring within the first 2 hours. Conversely, the hollow nanoparticles released 53% of their loaded peptide (0.25 mg KAFAK per mg particle), with 89% of this release occurring within 12 hours. The PEGylated particles showed a similar trend with solid PEGylated particles releasing 8.3% of their loaded peptide (0.02 mg KAFAK per mg particle) and the Hollow PEGylated particles releasing 51% of their loaded peptide, (0.25 mg KAFAK per mg particle). However, as seen in figure 4, the addition of large PEG chains to the hollow nanoparticles slowed the rate of release likely by generating a large excluded volume that inhibited diffusion of KAFAK out of the nanoparticle, allowing for 79% of their release to occur within 24 hours, as opposed to 12 hours for the non-PEGylated particles to achieve a similar level of release. This represents over a 50% decrease in the release rate, and should allow for improved therapeutic efficacy by prolonging KAFAK treatment time. The higher overall KAFAK release from the hollow particles is due to both their higher loading efficiency and the thinner outer shell. Additionally, the lack of a dense anionically-charged core would allow for easier diffusion of the loaded drug out of the nanoparticle and a lower potential for permanent entrapment of KAFAK. These factors combine to provide a large diffusion gradient as well as a lower diffusion barrier, resulting in an increased release rate and a higher overall peptide release which occurs over a longer period of time.
Nanoparticle cytotoxicity was tested at a concentration of 4 mg/mL to determine if the particles are non-cytotoxic at a concentration that far exceeds those required to achieve a therapeutic dose, found to be 2 mg/mL in our ex vivo studies. At these high concentrations, the loaded particles did not cause any reduction in viable cell count when compared to untreated control, as seen in Figure 5. This indicates that the particles are minimally cytotoxic and are not expected to adversely impact cellular viability when used in vivo.
Figure 5.
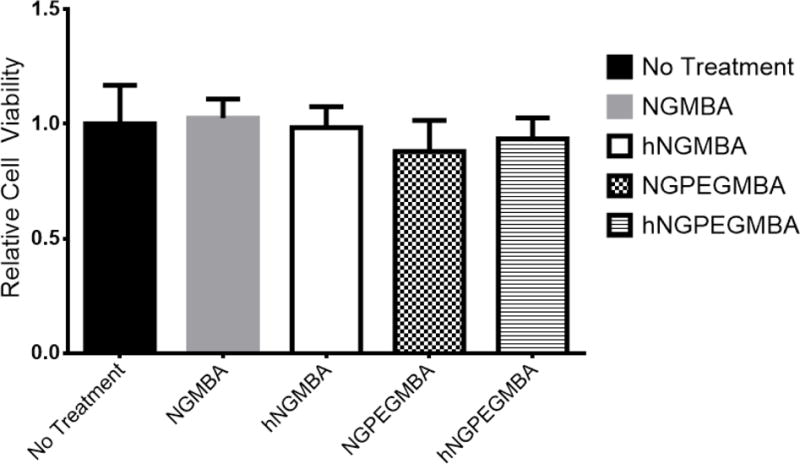
Normalized cell viability of RAW 264.7 cells treated with non-PEGylated and pegylated solid and hollow nanoparticles over a 48 h period at 37 °C
Nanoparticle Uptake
In order to remain viable drug delivery vehicles, the nanoparticles need to be able deliver KAFAK directly to the immune cells largely responsible for the local inflammatory state. To this end, we used confocal microscopy to determine the uptake of fluorescently labeled nanoparticles by RAW 264.7 macrophages (figures 6 and 7). The data indicates that PEGylated nanoparticles were taken up by the macrophages within 24 hours (figure 7), while fewer non-PEGylated particles were taken up within the same time period. While changes in intracellular pH within the endo/lysosomes can alter the fluorescent intensity of our FITC-tagged nanoparticles, the colocalization of our nanoparticles with lyostracker indicates that the PEGylated and non-PEGylated particles are taken up, albeit at different rates. This increased uptake of PEGylated nanoparticles matches what we have observed previously[39]. Taken together with KAFAK release data, this suggests that the particles will achieve up to 79% of their overall KAFAK release in the local environment, which can treat multiple cell types. By 24 hours, the nanoparticles are taken up into endolysosomal vesicles by inflamed macrophages (Figure 7A and 7B), indicating that the remaining KAFAK would be released intracellularly. This allows for maximal effect of the remaining KAFAK as it is released directly to the inflamed cells.
Figure 6.
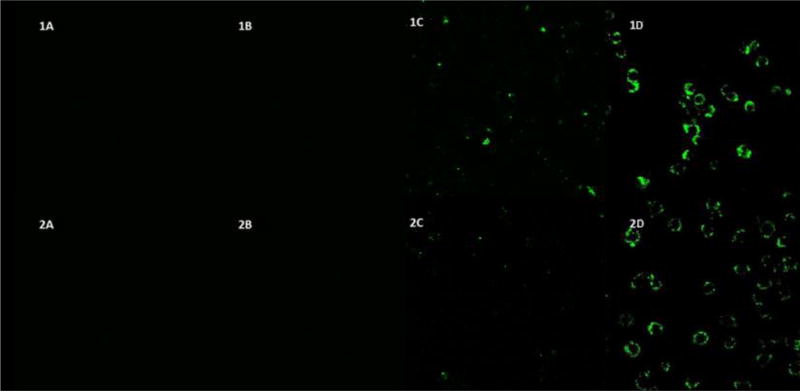
Confocal fluorescence microscopy of RAW 264.7 cells incubated @ 37°C with hollow (Row 1) and solid (Row 2) nanoparticles. 4 hour incubation of fluorescent NGMBA (1A) hNGMBA (2A) NGPEGMBA (1B) and hNGPEGMBA (2B); 24 hour incubation of fluorescent NGMBA (1C) hNGMBA (2C) NGPEGMBA (1D) and hNGPEGMBA (2D) Cells were visualized at λex = 490nm, λem = 520nm.
Figure 7.
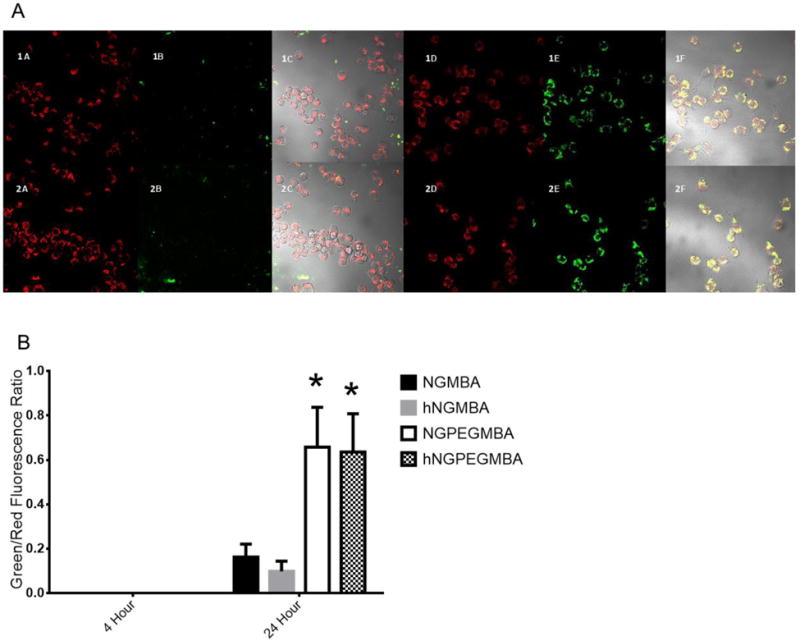
A) Lysotracker DND-99 fluorescence indicating co-localization of fluorescent hollow (top row) and solid (bottom row) nanoparticles in endolysosomal compartments. (1A), (2A), (1D) and (2D) are lysotracker labeled endolysosomes. (1B) NGMBA, (2B) hNGMBA, (1E) hNGPEGMBA and (2E) NGPEGMBA show fluorescent particle uptake in RAW 264.7 cells. (1C), (2C), (1F) and (2F) are overlay images. B) Semi-quantitative measurements of colocalization of nanoparticles and lysotracker DND-99, showing uptake of cells into endolysosomal compartments
Nanoparticle diffusion into damaged cartilage
Due to their prolonged release rate and increased macrophage uptake, hollow PEGylated nanoparticles were the most promising platform for inhibiting inflammation in an ex vivo osteoarthritis model. In order to best model the application of the nanoparticles via intra-articular injection, we investigated the ability of KAFAK-loaded hNGPEGMBA particles to infiltrate into cartilage that was aggrecan-depleted to simulate an osteoarthritic environment (Figure 8). The nanoparticles were able to penetrate beyond the articular zone of the osteoarthritic cartilage, allowing them to access the inflamed environment deeper in the cartilage. This allows the KAFAK to be released at the site of inflammation, which would serve to attenuate the progression of osteoarthritic damage. In contrast, the nanoparticles demonstrated minimal diffusion into the healthy cartilage, indicating that aggrecan is successfully providing a barrier to infiltration.
Figure 8.
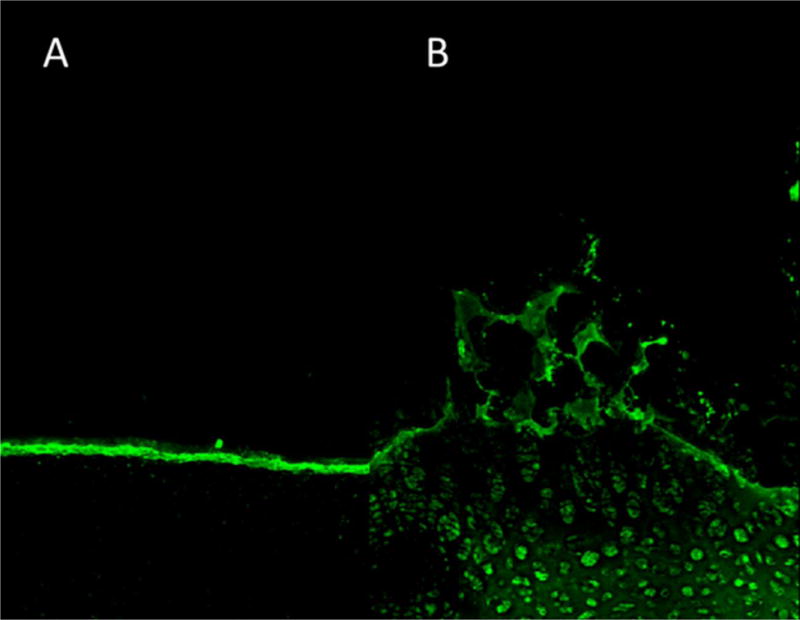
Sagittal cross-sectional area of load bearing region of bovine knee explants representing articular surface (cartilage–synovial fluid interface) taken with 488-nm excitation of FITC-labeled hNGPEGMBA (bar scale is 100μm). 7A represents normal healthy cartilage. 7B represents aggrecan-depleted cartilage explants.
Inflammatory inhibition of Ex Vivo cartilage explant
To verify that the particles were able to successfully attenuate osteoarthritic inflammation, we measured pro-inflammatory cytokine levels for eight days in explanted cartilage before and after treatment with KAFAK-loaded nanoparticles. Figure 9 shows that we were able to stimulate an inflammatory environment within the explants that was maintained for 8 days (Figure 9b). Interestingly, treatment with free KAFAK yielded no decrease in IL-6 production, likely due to proteolytic enzymes present in the environment as well as interactions with local proteoglycans which would prevent uptake by inflamed cells. Both of these factors prevent KAFAK from inhibiting inflammation deeper in the cartilage. Conversely, the KAFAK-loaded nanoparticles showed significant reductions in IL-6 from day 4 through day 8. Interestingly, while the level of inhibition caused by the solid particles remained constant, IL-6 in the samples treated with the hollow particles continued to decrease from day 4 to day 8. This can be attributed to the prolonged release of KAFAK from the hollow nanoparticles, which maintains therapeutic doses of KAFAK, resulting in the observed increasing inhibition over time and yielding continually decreasing IL-6 levels.
Figure 9.
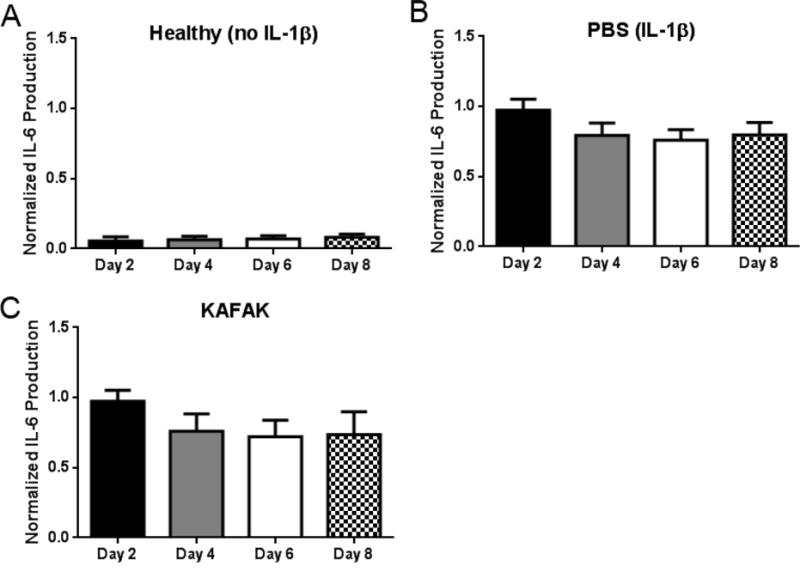
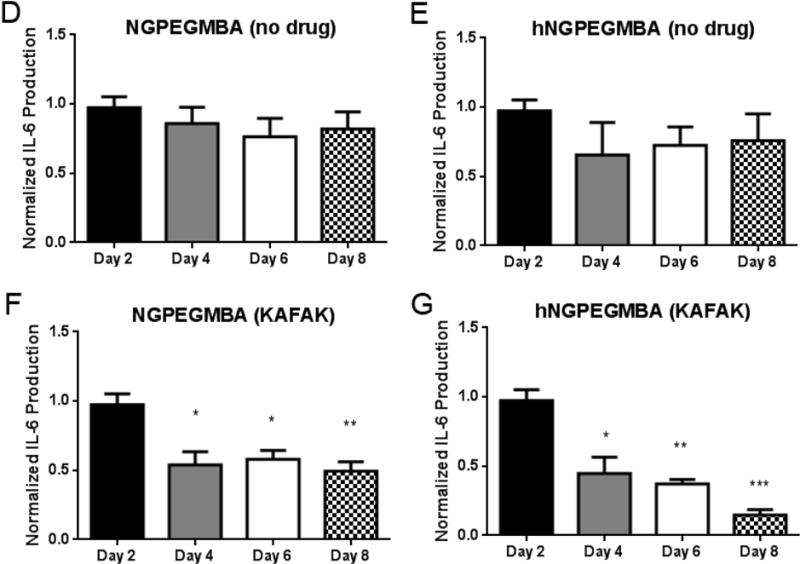
Therapeutic time study of IL-6 production in cartilage plugs when dosed nanoparticles loaded with KAFAK. A) Healthy cartilage plugs (no IL-1β stimulation). Plots A–E represent aggrecan-depleted plugs dosed with IL-1β. Plots are normalized to individual plug weight and IL-6 production from the control and represent average ± SEM (n=4). * denotes ρ < 0.05 and ** ρ < 0.01 with respect to IL-1β only treatment.
CONCLUSION
Overall, the data demonstrates that hollow, sulfated pNIPAM nanoparticles are an effective platform for the loading and delivery of anti-inflammatory cell penetrating peptides, with higher loading capacity, and prolonged release profiles compared to their solid counterparts. In in vitro studies, the nanoparticles were successfully taken up into the endolysosomal compartments of RAW 264.7 cells, thereby allowing for intracellular delivery of any KAFAK loaded into the particle. Additionally, uptake of the particles did not adversely affect cell viability, indicating that the particles are biocompatible and making them promising candidates for the in vivo delivery of cationic therapeutics such as KAFAK.
In ex vivo studies, the hollow PEGylated nanoparticles demonstrated the ability to penetrate into aggrecan-depleted cartilage explants. Additionally, nanoparticles demonstrated the ability to protect their cargo from proteolytic degradation, as observed in their ability to reduce inflammation in an ex vivo osteoarthritis model. In this model, hollow nanoparticles showed progressive inhibition of inflammation, with IL-6 levels decreasing continually for 8 days. This study demonstrates the promise of hollow PEGylated pNIPAM nanoparticles as a platform for the delivery of cationic therapeutics.
Scheme 1.
Fmoc synthesis of KAFAK
Acknowledgments
We wish to thank Dr. Shaili Sharma for her valuable assistance.
FUNDING
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number AR065398 and the National Institute of Diabetes and Digestive and Kidney Disease for JL under award number T32DK101001. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Manen MD, Nace J, Mont MA. Management of primary knee osteoarthritis and indications for total knee arthroplasty for general practitioners. J Am Osteopath Assoc. 2012;112:709–715. [PubMed] [Google Scholar]
- 2.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!) Osteoarthritis Cartilage. 2013;21:16–21. doi: 10.1016/j.joca.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 3.Jeffries MA, Donica M, Baker LW, Stevenson ME, Annan AC, Humphrey MB, James JA, Sawalha AH. Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol. 2014;66:2804–2815. doi: 10.1002/art.38762. [DOI] [PubMed] [Google Scholar]
- 4.Fukui N, Zhu Y, Maloney WJ, Clohisy J, Sandell LJ. Stimulation of BMP-2 expression by pro-inflammatory cytokines IL-1 and TNF-α in normal and osteoarthritic chondrocytes. The Journal of Bone & Joint Surgery. 2003;85:59–66. doi: 10.2106/00004623-200300003-00011. [DOI] [PubMed] [Google Scholar]
- 5.Bondeson J, Wainwright SD, Lauder S, Amos N, Hughes CE. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis research & therapy. 2006;8:R187. doi: 10.1186/ar2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attur M, Patel I, Patel R, Abramson S, Amin A. Autocrine production of IL-1 beta by human osteoarthritis-affected cartilage and differential regulation of endogenous nitric oxide, IL-6, prostaglandin E2, and IL-8. Proceedings of the Association of American Physicians. 1997;110:65–72. [PubMed] [Google Scholar]
- 7.Firestein G, Berger A, Tracey D, Chosay J, Chapman D, Paine M, Yu C, Zvaifler N. IL-1 receptor antagonist protein production and gene expression in rheumatoid arthritis and osteoarthritis synovium. The Journal of Immunology. 1992;149:1054–1062. [PubMed] [Google Scholar]
- 8.Stannus O, Jones G, Cicuttini F, Parameswaran V, Quinn S, Burgess J, Ding C. Circulating levels of IL-6 and TNF-α are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthritis and Cartilage. 2010;18:1441–1447. doi: 10.1016/j.joca.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 9.Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529–543. doi: 10.2741/1817. [DOI] [PubMed] [Google Scholar]
- 10.Mort JS, Dodge GR, Roughley PJ, Liu J, Finch SJ, Dipasquale G, Poole AR. Direct evidence for active metalloproteinases mediating matrix degradation in interleukin 1-stimulated human articular cartilage. Matrix. 1993;13:95–102. doi: 10.1016/s0934-8832(11)80068-5. [DOI] [PubMed] [Google Scholar]
- 11.Wick M, Ernestam S, Lindblad S, Bratt J, Klareskog L, Van Vollenhoven R. Adalimumab (Humira®) restores clinical response in patients with secondary loss of efficacy from infliximab (Remicade®) or etanercept (Enbrel®): results from the STURE registry at Karolinska University Hospital. Scandinavian journal of rheumatology. 2005;34:353–358. doi: 10.1080/03009740510026887. [DOI] [PubMed] [Google Scholar]
- 12.Burger D, Dayer JM, Palmer G, Gabay C. Is IL-1 a good therapeutic target in the treatment of arthritis? Best Practice & Research Clinical Rheumatology. 2006;20:879–896. doi: 10.1016/j.berh.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Gamble R, Wyeth-Ayerst J, Johnson EL, Searle WA, Beecham S. Recommendations for the medical management of osteoarthritis of the hip and knee. Arthritis & Rheumatism. 2000;43:1905–1915. doi: 10.1002/1529-0131(200009)43:9<1905::AID-ANR1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 14.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 15.McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Current opinion in rheumatology. 2008;20:131–137. doi: 10.1097/BOR.0b013e3282f51031. [DOI] [PubMed] [Google Scholar]
- 16.Marinó M, Morabito E, Brunetto MR, Bartalena L, Pinchera A, Marocci C. Acute and severe liver damage associated with intravenous glucocorticoid pulse therapy in patients with Graves’ ophthalmopathy. Thyroid. 2004;14:403–406. doi: 10.1089/105072504774193276. [DOI] [PubMed] [Google Scholar]
- 17.Rostom A, Dube C, Wells G, Tugwell P, Welch V, Jolicoeur E, McGowan J. Prevention of NSAID-induced gastroduodenal ulcers. Cochrane Database Syst Rev. 2002:CD002296. doi: 10.1002/14651858.CD002296. [DOI] [PubMed] [Google Scholar]
- 18.Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, FitzGerald GA, Goss P, Halls H, Hawk E, Hawkey C, Hennekens C, Hochberg M, Holland LE, Kearney PM, Laine L, Lanas A, Lance P, Laupacis A, Oates J, Patrono C, Schnitzer TJ, Solomon S, Tugwell P, Wilson K, Wittes J, Baigent C. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–779. doi: 10.1016/S0140-6736(13)60900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gislason GH, Rasmussen JN, Abildstrom SZ, Schramm TK, Hansen ML, Fosbol EL, Sorensen R, Folke F, Buch P, Gadsboll N, Rasmussen S, Poulsen HE, Kober L, Madsen M, Torp-Pedersen C. Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure. Arch Intern Med. 2009;169:141–149. doi: 10.1001/archinternmed.2008.525. [DOI] [PubMed] [Google Scholar]
- 20.Lee A, Cooper MG, Craig JC, Knight JF, Keneally JP. Effects of nonsteroidal anti-inflammatory drugs on postoperative renal function in adults with normal renal function. Cochrane Database Syst Rev. 2007:CD002765. doi: 10.1002/14651858.CD002765.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brugnano J, McMasters J, Panitch A. Characterization of endocytic uptake of MK2-inhibitor peptides. Journal of Peptide Science. 2013;19:629–638. doi: 10.1002/psc.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engel K, Ahlers A, Brach MA, Herrmann F, Gaestel M. MAPKAP kinase 2 is activated by heat shock and TNF-alpha: in vivo phosphorylation of small heat shock protein results from stimulation of the MAP kinase cascade. J Cell Biochem. 1995;57:321–330. doi: 10.1002/jcb.240570216. [DOI] [PubMed] [Google Scholar]
- 24.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nature Reviews Immunology. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 25.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? The lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 26.Bhaskar Kompella U, VHL Lee. (C) Means to Enhance Penetration: (4) Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Advanced Drug Delivery Reviews. 1992;8:115–162. doi: 10.1016/s0169-409x(00)00137-x. [DOI] [PubMed] [Google Scholar]
- 27.Bundgaard H. (C) Means to enhance penetration: (1) Prodrugs as a means to improve the delivery of peptide drugs. Advanced Drug Delivery Reviews. 1992;8:1–38. [Google Scholar]
- 28.Scott Swenson E, Curatolo WJ. (C) Means to enhance penetration: (2) Intestinal permeability enhancement for proteins, peptides and other polar drugs: mechanisms and potential toxicity. Advanced Drug Delivery Reviews. 1992;8:39–92. [Google Scholar]
- 29.Shen WC, Wan J, Ekrami H. (C) Means to enhance penetration: (3) Enhancement of polypeptide and protein absorption by macromolecular carriers via endocytosis and transcytosis. Advanced Drug Delivery Reviews. 1992;8:93–113. [Google Scholar]
- 30.Anderson R, Franch A, Castell M, Perez-Cano FJ, Brauer R, Pohlers D, Gajda M, Siskos AP, Katsila T, Tamvakopoulos C, Rauchhaus U, Panzner S, Kinne RW. Liposomal encapsulation enhances and prolongs the anti-inflammatory effects of water-soluble dexamethasone phosphate in experimental adjuvant arthritis. Arthritis Res Ther. 2010;12:R147. doi: 10.1186/ar3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartlett RL, 2nd, Medow MR, Panitch A, Seal B. Hemocompatible poly(NIPAm-MBA-AMPS) colloidal nanoparticles as carriers of anti-inflammatory cell penetrating peptides. Biomacromolecules. 2012;13:1204–1211. doi: 10.1021/bm300173x. [DOI] [PubMed] [Google Scholar]
- 32.Bartlett RL, 2nd, Panitch A. Thermosensitive nanoparticles with pH-triggered degradation and release of anti-inflammatory cell-penetrating peptides. Biomacromolecules. 2012;13:2578–2584. doi: 10.1021/bm300826v. [DOI] [PubMed] [Google Scholar]
- 33.Galperin TJL, Ratner Degradable ABD. Thermo-Sensitive Poly(N-isopropyl acrylamide)-Based Scaffolds with Controlled Porosity for Tissue Engineering Applications. Biomacromolecules. 2010;11:2583–2592. doi: 10.1021/bm100521x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overstreet RH, Jarbo DJK, McLemore RY, Vernon BL. In situ forming, resorbable graft copolymer hydrogels providing controlled drug release. J Biomed Mat Res A. 2013;101A:1437–1446. doi: 10.1002/jbm.a.34443. [DOI] [PubMed] [Google Scholar]
- 35.Overstreet RYM, Doan DJBD, Farag A, Vernon BL. Temperature-Responsive Graft Copolymer Hydrogels for Controlled Swelling and Drug Delivery. Soft Materials. 2013;11:294–304. [Google Scholar]
- 36.Mishra S, De A, Mozumdar S. Synthesis of thermoresponsive polymers for drug delivery. Methods Mol Biol. 2014;1141:77–101. doi: 10.1007/978-1-4939-0363-4_4. [DOI] [PubMed] [Google Scholar]
- 37.Schmaljohann D. Thermo- and pH-responsive polymers in drug delivery. Adv Drug Del Rev. 2006;58:1655–1670. doi: 10.1016/j.addr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Gan DJ, Lyon LA. Tunable swelling kinetics in core-shell hydrogel nanoparticles. J Am Chem Soc. 2001;123:7511–7517. doi: 10.1021/ja010609f. [DOI] [PubMed] [Google Scholar]
- 39.Poh S, Lin JB, Panitch A. Release of Anti-inflammatory Peptides from Thermosensitive Nanoparticles with Degradable Cross-Links Suppresses Pro-inflammatory Cytokine Production. Biomacromolecules. 2015;16:1191–1200. doi: 10.1021/bm501849p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones CD, Lyon LA. Dependence of Shell Thickness on Core Compression in Acrylic Acid Modified Poly(N-isopropylacrylamide) Core/Shell Microgels. Langmuir. 2003;19:4544–4547. [Google Scholar]
- 41.Gua J, Xiab F, Wub Y, Qua X, Yanga Z, Jiang L. Programmable delivery of hydrophilic drug using dually responsive hydrogel cages. J Cont Rel. 2007;117:396–402. doi: 10.1016/j.jconrel.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 42.Ayala-Lopez W, Xia W, Varghese B, Low PS. Imaging of atherosclerosis in apoliprotein e knockout mice: targeting of a folate-conjugated radiopharmaceutical to activated macrophages. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2010;51:768–774. doi: 10.2967/jnumed.109.071324. [DOI] [PubMed] [Google Scholar]
- 43.Dubbert J, Honold T, Pedersen JS, Radulescu A, Drechsler M, Karg M, Richtering W. How Hollow Are Thermoresponsive Hollow Nanogels? Macromolecules. 2014 [Google Scholar]
- 44.Dubbert J, Nothdurft K, Karg M, Richtering W. Core–Shell–Shell and Hollow Double- Shell Microgels with Advanced Temperature Responsiveness. Macromolecular rapid communications. 2014 doi: 10.1002/marc.201400495. [DOI] [PubMed] [Google Scholar]
- 45.Tripathi BP, Dubey NC, Stamm M. Hollow Microgel Based Ultrathin Thermoresponsive Membranes for Separation, Synthesis, and Catalytic Applications. ACS applied materials & interfaces. 2014;6:17702–17712. doi: 10.1021/am504120c. [DOI] [PubMed] [Google Scholar]
- 46.Liu R, Fraylich M, Saunders BR. Thermoresponsive copolymers: from fundamental studies to applications. Colloid and Polymer Sci. 2009;287:627–643. [Google Scholar]
- 47.Poole AR, Pidoux I, Reiner A, Tang LH, Choi H, Rosenberg L. Localization of proteoglycan monomer and link protein in the matrix of bovine articular cartilage: An immunohistochemical study. J Histochem Cytochem. 1980;28:621–635. doi: 10.1177/28.7.6156200. [DOI] [PubMed] [Google Scholar]
- 48.Pratta MA, Yao W, Decicco C, Tortorella MD, Liu RQ, Copeland RA, Magolda R, Newton RC, Trzaskos JM, Arner EC. Aggrecan protects cartilage collagen from proteolytic cleavage. J Biol Chem. 2003;278:45539–45545. doi: 10.1074/jbc.M303737200. [DOI] [PubMed] [Google Scholar]
- 49.Feil H, Bae YH, Feijen J, Kim SW. Effect of comonomer hydrophilicity and ionization on the lower critical solution temperature of N-isopropylacrylamide copolymers. Macromolecules. 1993;26:2496–2500. [Google Scholar]
- 50.Bartlett R, Medow M, Panitch A, Seal B. Hemocompatible Poly(NIPAm-MBA-AMPS) Colloidal Nanoparticles as Carriers of Anti-inflammatory Cell Penetrating Peptides. Biomacromolecules. 2012;13:1204–1211. doi: 10.1021/bm300173x. [DOI] [PubMed] [Google Scholar]
- 51.Bartlett RL, Panitch A. Thermosensitive Nanoparticles with pH-Triggered Degradation and Release of Anti-inflammatory Cell-Penetrating Peptides. Biomacromolecules. 2012;13:2578–2584. doi: 10.1021/bm300826v. [DOI] [PubMed] [Google Scholar]