

Abstract
Recent advances show that insulin may affect β adrenergic receptor (βAR) signaling in heart, to modulate cardiac function in clinically relevant states such as diabetes and heart failure. Conversely, activation of βAR regulates cardiac glucose uptake and promotes insulin resistance in HF. We discussed recent characterization on the interaction between cardiac insulin receptor and βAR in myocardium, in which insulin stimulation cross talk with cardiac βAR via insulin receptor substrate-dependent and G protein receptor kinase 2-mediated phosphorylation of β2AR. The insulin-induced phosphorylation promotes β2AR coupling to Gi and expression of phosphodiesterase 4, which inhibit cardiac adrenergic signaling and compromise cardiac contractile function. These progresses might support new approaches for effectively prevention or treatment of obesity-or diabetes-related heart failure.
Keywords: Insulin, adrenergic receptor, diabetes, heart failure, GRK2
Sympathetic Nervous Activity and Insulin Resistance in Heart Failure
Insulin regulates a broad range of function in the heart including cardiac metabolism, myocyte survival, and cardiac growth (Figure 1 and Box 1). Insulin promotes glucose uptake in cardiomyocytes through activation of an InsR-mediated phosphatidylinositol 3-kinase (PI3K)-Akt pathway, which mobilizes glucose transporter 4 (GLUT4) vesicles to the cell surface for glucose uptake [1]. Interestingly, stimulation of βAR in heart also induces an additive effect on insulin-induced glucose uptake; and this effect is mediated by phosphorylation of Akt in threonine 308 through protein kinase A (PKA)/Ca2+-dependent pathway [2, 3]. On the cellular level, stimulation with either insulin or catecholamines antagonizes the ability of the other to activate glucose transport and to modulate cardiomyocyte survival [4, 5]. These observations suggest a counter-regulation between insulin and βAR signaling in heart. Recently, a functional membrane complex consisting of insulin receptor (InsR) and β2AR has been characterized [6, 7], providing a biochemical basis for intimate cross talk between cardiac insulin and β adrenergic regulatory systems. In this review, this novel complex and other progresses in understanding of the connection between cardiac insulin and βAR signaling is summarized, and the implications in the pathophysiology of heart diseases such as diabetes-related heart failure (HF) will be discussed.
Figure 1. Insulin signaling and insulin resistance in heart.
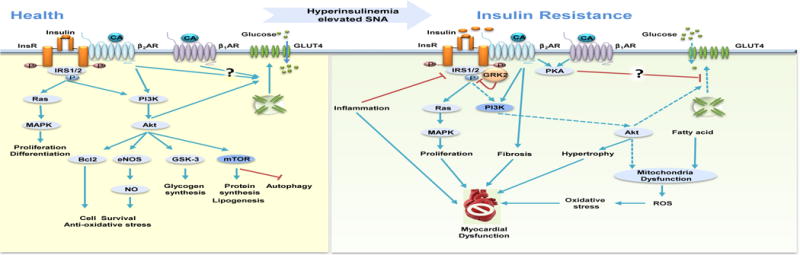
Insulin stimulates metabolic, mitogenic, and anti-apoptotic pathways via activation of insulin receptor (InsR)-insulin receptor substrates 1 and 2 (IRS1/2) cascades. In heart failure, insulin resistance leads to impaired Akt activation and/or transportation of GLUT4 to the plasma membrane for glucose uptake. Abbreviations: βAR, β adrenergic receptor; Bcl-2, B-cell lymphoma 2; CA, catecholamine; eNOS, endothelial nitric oxidase; ERK1/2, extracellular-regulated kinase 1/2; Gi, inhibitory regulative G-protein; GLUT4, glucose transporter 4; GRK2, G protein receptor kinase 2; Gs, stimulatory G protein; GSK, glycogen synthase kinase; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NO, nitric oxide; PI3K, phosphatidyl-inositol 3-kinase; PKA, protein kinase A; Ras, rat sarcoma; ROS, reactive oxygen species; SNA, sympathetic nervous activity. The blue lines indicate stimulatory actions, the blue dash lines indicate impaired stimulation; the red lines indicate inhibitory actions.
BOX 1. Insulin Signaling in the Heart.
Insulin is a pleiotropic hormone with various effects on glucose metabolism, the central nervous system, immune system, and cardiovascular system. InsR are highly abundant in the heart [93]. Activated InsR recruits and phosphorylates insulin receptor substrate (IRS) proteins, which in turn activate a complex signal transduction network, including phosphatidyl-inositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK) pathways such as extracellular-regulated kinase 1/2 (ERK1/2). Akt regulates the translocation of vesicles containing glucose transporter 4 (GLUT4), which enhances glucose uptake, whereas mTOR activation promotes lipogenesis and protein synthesis[93]. In addition, short-term insulin stimulation also activates pro-survival Akt to inhibit apoptotic signaling, and to inhibit autophagy by activating other intracellular signaling intermediates such as mTOR, S6 kinase (S6K), forkhead transcription factors 1/3(FOXO1/3), glycogen synthase kinase 3β (GSK3β) and nitric oxide synthase 2 (NOS2). The MAPK-ERK1/2-dependent branch of insulin signaling pathway increases the expression of genes that modulate biological processes such as differentiation and proliferation [93].
In the absence of systemic metabolic disorders, cardiac insulin resistance (IR) can develop in congestive and ischemic HF most likely due to the elevated sympathetic nervous system (SNS) activity (Table 1) [8]. IR can be classified into two categories: failure of InsR in promoting intracellular signals such as Akt activity or an impaired translocation of GLUT4 to the cell surface despite increases in intracellular Akt activity. In both scenarios, dysregulation of the InsR-Akt signal may compromise the ability of myocardium to cope with stresses or exacerbate cardiac phenotype in HF [9]. The SNS effects on cardiomyocytes are mediated by βAR activation. Chronic β2AR stimulation under the elevated SNS activity leads to sustained activation of Akt through PKA/Ca2+and PI3K-dependent pathways, which decreases GLUT4 expression and GLUT4 translocation to the plasma membrane and compromises the ability of insulin to promote glucose uptake [10, 11]. CHF is associated with IR characterized by both fasting and stimulated hyperinsulinemia, which can drive chronic stimulation of the InsR signaling [12]. Accordingly, expression of constitutive active PI3K and Akt in cardiomyocytes compromises the insulin-induced GLUT4-dependent glucose uptake [13]. Abnormal insulin signaling plays an essential role in the development of HF, including cardiac cell survival, hypertrophy, and fibrosis [14]. In the late stage of HF, IR develops in both cardiac and peripheral tissues such as skeletal muscle, fat, and liver [15], and presents a poor prognosis of HF associated with worsening cardiac function and increasing mortality. Treatment with β-blockers including metoprolol and carvedilol antagonizes isoproterenol-induced IR and cardiac dysfunction, with non-selective βAR antagonist carvedilol displaying a greater efficiency [10, 16, 17]. Meanwhile, restoration of cardiac output after left ventricular assist devices improves glycemic control and decreases insulin requirements in patients with advanced CHF [18]. Together, these studies indicate that IR emerges as an important index along with βAR desensitization in management of HF with metabolic disorders.
Table 1.
βAR and Insulin regulate cardiac metabolic, structure, and function in HF
| Glucose Uptake and IR | Cardiac Contractility | Cardiac Hypertrophy | Cardiac Fibrosis | |
|---|---|---|---|---|
| βAR (acute) | Insulin-induced glucose uptake ↑ (PKA/Ca2+-dependent Akt308 phosphorylation↓ 2) | ↑ (cAMP/PKA↑) | ||
| ßAR (chronic) | IR↑ (PKA/Ca2+ and PI3K-dependent Akt phosphorylation↑ 10, 11, 16, 17) | ↓ (βAR endocytosis, 44–47) | ↑ (Akt/GSK-3β signaling↑, Gβγ/ERK↑, fibroblast-derived IGF-1↑ 58, 61–63) | |
| GRK2 | IR↑ (IRS-1 phosphorylation↑ 4, 51, 52) | ↓ (βAR endocytosis, 44–47) | ||
| Insulin | IR↑ (persistent PI3K-Akt↑, GLUT4 function↓ 12, 13, 14) | ↓ (β2AR phosphorylation mediated Gi-biased β2-adrenergic signaling↑, β2AR-dependent PDE4 expression↑ 6, 32–34) | ↑ (Akt phosphorylation↑, PPARγ-PI3K/Akt-NO signaling pathway↑ β2AR and Akt independent? 33, 53, 55, 57) | ↑ (β2AR dependent? 33) |
Desensitization of Cardiac βARs in Heart Failure
In congestive and failing ischemic hearts, loss of cardiac output leads to compensatory increase in the SNS activity that stimulates cardiac βARs, which enhance cardiac contractility. Persistent and excessive stimulation with catecholamines promotes phosphorylation of βARs by PKA and GRK that leads to receptor desensitization and endocytosis in cardiomyocytes (Table 1). In a classic paradigm, HF displays a selective downregulation of β1AR that is often associated with an upregulation of GRK2 and Gi. In contrast, the expression of β2AR is not altered (Figure 2 and Box 2). The mechanism for selective downregulation of β1AR is not clear; though it is generally considered a mechanism that removes excessive and persistent toxic pro-apoptotic β1AR signaling that is detrimental to myocardium [19]. Thus, selective β-blocker against β1AR such as metaprolol has been developed to block the receptor activity in HF. One of clinical outcomes in an effective β-blocker therapy is to restore β1AR density in myocardium, which may play a role in restoring cardiac contractility and cardiac reserve in response to catecholamines. The apparent paradox between toxic β1AR signal and the recovered β1AR density after therapy is not fully understood. Recent evidence suggests that GPCR including β2AR can continue to signal from the endosome after agonist-induced endocytosis [20]; and the signal induced by endosomal β1AR is shown to promote hypertrophy in cardiomyocytes [21]. Studies show that in adaptive HF cardiac β1AR is shifted from the cell surface to the endosome without significant decrease in receptor density [22]. Therefore, one can argue that endosomal β1AR potentially transduces detrimental signaling to promote cardiac remodeling during the development of HF; and β-blocker therapy is to eliminate toxic endosomal β1AR signaling and restore normal β1AR signaling from the plasma membrane.
Figure 2. βAR signaling and βAR desensitization in heart failure.
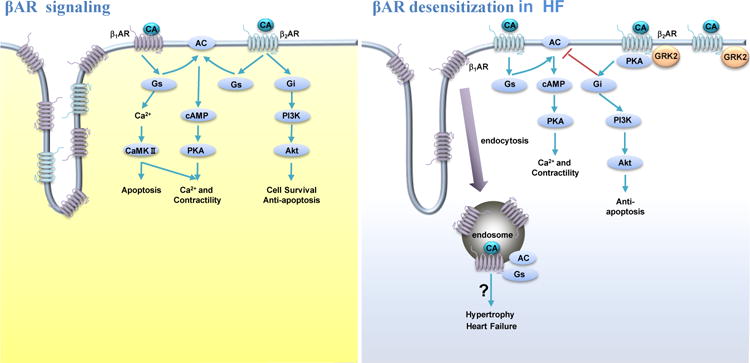
Activation of βARs by catecholamines induces production of cAMP through the GS-AC cascade, which promotes PKA activity for cellular responses in cardiomyocytes. In heart failure, β1ΑΚ undergoes endocytosis whereas β2ΑΚ is relocated from t-tubular to crest membrane. The enhanced β2AR-Gi coupling attenuates βAR-induced cAMP signal in cardiomyocytes. Abbreviations: AC, adenylyl cyclase; βAR, β adrenergic receptor; CA, catecholamine; CaMKII, calmodulin-dependent kinase; cAMP, cyclic monophosphate adenosine; Gi, inhibitory regulative G-protein; GRK2, G protein receptor kinase 2; Gs, stimulatory G protein; PI3K, phosphatidyl-inositol 3-kinase; PKA, protein kinase A; GRK2, G protein receptor kinase 2.
BOX 2. β-Adrenergic Receptor Signaling in the Heart.
Sympathetic activation increases cardiac output through the release of catecholamines. The effects of catecholamines on myocardium are primarily mediated by βAR activation. There are three subtypes of βARs: β1AR, β2AR, and β3AR. In the heart, nonselective βAR stimulation activates the Gs-AC-cAMP cascade, leading to PKA-dependent phosphorylation of a set of regulatory proteins involved in cardiac excitation-contraction coupling and energy metabolism, and resulting in greater contractility. However, activation of β2AR can also promote a coupling switch from Gs to Gi pathway [27]. The coupling of β2AR to Gi is under the influence of GRK-and PKA-and/or PKC-mediated phosphorylation [85]. The β2AR-Gi signaling pathway plays a crucial role in the regulation of cell proliferation and protection against cardiomyocyte apoptosis via transactivation of a PI3K-Akt signaling pathway. The β2AR-Gi signaling pathway also attenuates the βAR-Gs-mediated inotropic response via inhibition of AC activity [86]. Meanwhile, adrenergic signaling also activates PKA and Akt to promote glucose uptake in heart[10, 11]. The shared cellular functions suggest insulin signaling and adrenergic signaling can potentially converge together in the heart.
In comparison, the expression of cardiac β2AR is not decreased during HF. A loss of caveolae leads to redistribution of β2AR from t-tubular membrane to crest membrane [23]. In HF, stimulation of β2AR promotes a redistributed cAMP-PKA signaling in cardiomyocytes for contractile response [23–25]. Meanwhile, an upregulation of Gi is often associated with enhanced β2AR-Gi signaling in HF. The enhanced β2AR-Gi signaling leading to Akt activation can protect against myocyte apoptosis, and consequently slow down the progression of cardiomyopathy [26]. Therefore, a combined therapy of β1AR blocker and β2AR agonist has been proposed for treatment of HF. Despite its beneficial effects in myocardium, the same β2AR-Gi coupling could blunt the Gs-mediated stimulation of contractile support, contributing to contractile defect in failing hearts [27]. Thus, the status of β2AR-Gi coupling can potentially shape the overall output of cardiac βAR signaling under different pathological circumstances.
Hyperinsulinemia Promotes Desensitization of Cardiac βAR
Diabetes mellitus (DM) is an important predictor of HF independent of concomitant hypertension or coronary artery diseases [28]. One of the hallmarks in DM is insulin resistance and associated hyperinsulinemia. In metabolic syndromes including obesity and diabetes, InsR is no longer responsive to insulin stimulation that promotes glucose uptake in skeletal muscle, fat, and liver. General IR is often associated with nutrition overload such as free fatty acids and cholesterol, together with systemic inflammation. General IR is accompanied by compensatory increases in insulin released from the pancreas, which leads to high levels of insulin in the plasma [29]. In diabetes, metabolic stress can lead to cardiomyopathy via depressing Akt signaling and activate Foxo1-mediated gene expression [9]. Meanwhile, inflammation, oxidative stress, and mitochondria dysfunction also contribute to the development of cardiomyopathy [30]. However, whether hyperinsulinemia play a role in diabetic cardiomyopathy and heart failure is not well established. Previous studies have shown a direct relationship between insulin signaling and cardiac contractility [31]. The newly characterized InsR and β2AR complex in myocardium brings new prospects to the issue [32–34]. In this new paradigm, insulin stimulates recruitment of GRK2 to InsR in an IRS2-dependent manner, and promotes GRK2-mediated phosphorylation of β2AR [32], through which insulin inhibits cardiac contractility by promoting a Gi-biased β2-adrenergic signaling (Table 1 and Figure 3). Insulin also promotes dissociation of the InsR-β2AR complex and β2AR internalization to attenuate β-adrenergic stimulation of contractile responses in cardiomyocytes [6].
Figure 3. Insulin induces βAR desensitization in heart.
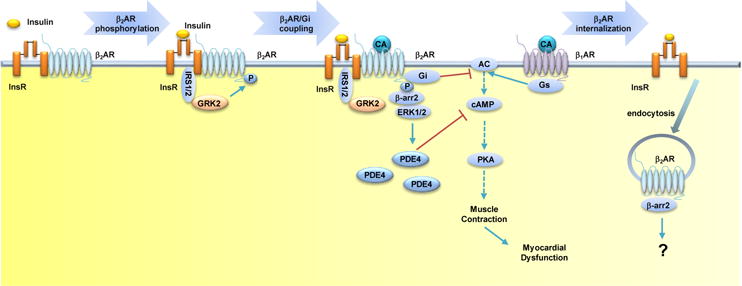
InsR and β2AR form a complex in the heart. Insulin stimulates recruitment of GRK2 to InsR in an IRS-dependent manner, which promotes β2AR phosphorylation. The phosphorylated β2AR couples to Gi and induces expression of PDE4D, both of which attenuates βAR signaling in cardiomyocytes. In addition, the phosphorylated β2AR also undergoes internalization. Together, hyperinsulinemia contributes to cardiac dysfunction via desensitization of βAR signaling. Abbreviations: AC, Gs-adenylyl cyclase; βAR, β adrenergic receptor; β-arr2, β-arrestin2; CA, catecholamine; cAMP, cyclic monophosphate adenosine; ERK1/2, extracellular-regulated kinase 1/2; Gi, inhibitory regulative G-protein; GRK2, G protein receptor kinase 2; Gs, stimulatory G protein; InsR, insulin receptor; IRS1/2, insulin receptor substrates 1 and 2; PKA, protein kinase A; PDE4D, phosphodiesterase 4D.
It’s noteworthy that short-term HFD feeding leads to decreases in cardiac functional reserve and mitochondrial ROS production in response to β-adrenergic stimulation without significant alteration in β1AR and β2AR expression, cardiac structure, and cardiac function [34, 35]. This decrease in cardiac functional reserve is associated with significant increases in the levels of phosphorylation of β2AR at PKA and GRK sites. Genetic deletion of the β2AR preserves adrenergic response and cardiac contractile reserve in HFD-fed mice [34]. These observations suggest that hyperinsulinemia drives phosphorylation of the β2AR for coupling to Gi protein to compromises cardiac adrenergic stimulation. Clinically, diabetic patients show decreases in exercise tolerance and blunted inotropic responses to dobutamine stimulation before the onset of overt cardiac dysfunction, suggesting that the cardiac β-adrenergic system is compromised [36]. Therefore, exercise intolerance and blunted inotropic response to adrenergic stimulation can be considered as early signs of diabetic cardiomyopathy, and the insulin-adrenergic signaling network can offer potential targets for early intervention to prevent cardiac complications.
Meanwhile, the PDE-mediated degradation of cAMP recently emerged as a previously underappreciated mechanism in the desensitization of cardiac βAR signaling [37, 38]. Insulin promotes an InsR-IRS-GRK2-and β-arrestin2-dependent transactivation of a β2AR-ERK signaling cascade to induce PDE4 expression in cardiomyocytes. An increased PDE4 expression is also observed in atrial biopsies from patients with cardiac diseases and diabetes relative to those patients without diabetes [33]. The PDE4-dependent cAMP hydrolysis counterbalances the βAR-induced cAMP signal. Inhibition of PDE4 activity recovers the receptor-induced cAMP signal and rescues contractile response to adrenergic stimulation in HFD heart [33]. Pharmacological inhibition of β2AR or GRK2, or genetic deletion of β2AR or β-arrestin2 significantly attenuate insulin-induced phosphorylation of ERK and induction of PDE4D, and prevent diabetes-related contractile dysfunction [33]. Thus, hyperinsulinemia may contribute to the impairment of adrenergic signaling via the increasing expression of PDE4D in heart. Accordingly, aggressive glycemic control with insulin secretagogues displays adverse outcomes in comorbidity of HF and diabetes [39]. Together, these observations suggest that hyperinsulinemia can directly act on myocardium and promote desensitization of adrenergic signaling by inducing PKA and GRK phosphorylation of β2AR to increase Gi coupling and the expression of PDE4D.
Hyperinsulinemia also leads to increase in activity of the sympathetic nervous system (SNS) [40]. This could be a compensatory mechanism in response to the inhibitory effects of insulin on cardiac βAR signal and reduced cardiac contractility and output. Alternatively, increased free fatty acid in metabolic disorders is also associated with elevated SNS activity that drives cardiac βARs. The elevated levels of catecholamines further antagonize insulin’s action, which leads to worsen cardiac IR and energy metabolism in CHF [10]. Chronic hyperinsulinemia can also work with hyperlipidemia and inflammation to drive IR in heart. Together, hyperinsulinemia and SNS activity are integrated via activation of the InsR-β2AR signaling network to promote the development of HF. The relationship between DM and congestive HF (CHF) are bidirectional since CHF and DM often coexist with overlapping pathophysiological processes [41–43].
GRK2 Serves as a Nexus linking IR and βAR Desensitization
GRK2 emerges as a critical nexus to connect cardiac InsR and adrenergic signaling (Table 1). Following stimulation by catecholamines or insulin, GRK2 shuttles from the cytosol to the plasma membrane to desensitize βAR and InsR, respectively [4, 32].
After catecholamine stimulation, GRK2 is activated and recruited to βARs through binding to Gβγ subunits on the plasma membrane, which allows GRK2 to phosphorylate receptor and other substrates such as PI3K to promote receptor endocytosis [44]. Chronic adrenergic stimulation is a signaling abnormality that also leads to the upregulation of GRK2 in HF [45]. Disruption of GRK2 binding to Gβγ or deletion of GRK2 prevents βAR endocytosis induced by catecholamines [46]. Thus, inhibition of GRK2 has been proposed for HF therapy. A peptide that disrupts GRK2 binding to Gβγ has been effective in treatment of various models of HF, including rodents and pigs [46]. More recently, a small molecule paroxetine, which is a FDA approved selective serotonin reuptake inhibitor, has been characterized as a selective GRK2 inhibitor [47]. Inhibition of GRK2 with paroxetine has been effective in modulation of βAR signaling pathway in cardiomyocytes, and in treatment of HF after transverse aortic constriction [47, 48].
Biochemical evidence suggests that GRK2 binds to IRS1/2 upon acute insulin stimulation, which facilitates phosphorylation of both InsR and IRS1/2, and prevents excessive insulin stimulation [49]. However, in HFD feeding model, the elevated expression of GRK2 promotes IR via inhibition of tyrosine phosphorylation and activation of IRS1/2 [33, 49]. The upregulated GRK2 also impairs myocardial glucose uptake before cardiac dilation, and the reduced function is evident (Table 1), indicating a metabolic remodeling at early stages of HF [50, 51]. Accordingly, overexpression of GRK2 has been shown to reduce myocardial insulin signaling. In germline GRK2 heterozygous knockout mice, insulin sensitivity is preserved in aging mice or when these animals are placed on a high fat diet (HFD) [52]. Genetic ablation of GRK2 is effective in ameliorating IR and glucose intolerance in an animal model of HFD-induced diabetes [53]. Thus, GRK2 serves as a crucial node in integrating cardiac metabolic and contractile regulation.
Early data implicates that GRK2 mediates adrenergic-induced IR and that inhibition of GRK2 leads to increased insulin sensitivity both in cells and in animal model of IR [45]. On the other hands, treatment with insulin promotes GRK2-mediated phosphorylation of β2AR, which leads to β2AR-Gi coupling to inhibit cardiac adrenergic signal. Both insulin and catecholamines also promote expression of GRK2. Depending on clinical onset conditions, GRK2 can be activated by catecholamines and/or hyperinsulinemia, and acts as a critical node linking insulin and adrenergic signal in control of contractility and cardiac metabolism, and promotes IR and βAR desensitization in heart. Genetic and pharmacological inhibition of GRK2 displays beneficial effects including ameliorating hyperglycemia and glucose intolerance, and improving cardiac function in HFD mice [33, 53, 54]. It’s worth mentioning that pharmacological inhibition of β2AR with carvedilol partially attenuates the expression levels of cardiac GRK2, which may contribute to the improved cardiac function and glucose tolerance [33]. Moreover, the GRK2 inhibitor paroxetine largely reverses cardiac fibrosis and apoptosis, suggesting a necessary role of GRK2 in the adverse remodeling in diabetic cardiomyopathy. Therefore, targeting GRK2 may provide dual benefits by normalizing adrenergic signaling and metabolic function in HF, particularly in HF with comorbidity of metabolic disorders such as obesity and diabetes.
Hyperinsulinemia Promotes Cardiac Remodeling: Hypertrophy and Fibrosis
Cardiac hypertrophy is defined as an increment of ventricular mass resulting from increased cardiomyocyte size, and is an adaptive response of the heart to increased hemodynamic load due to either physiological stresses or pathological states [55]. Insulin is an anabolic hormone that promotes protein synthesis, cell growth, and hypertrophy. Insulin and insulin-like growth factor 1 (IGF-1) receptors differentially modulate cardiac growth in an IRS1/2-dependent manner at resting conditions and following exercise training [56]. Genetic deletion of InsR in cardiomyocytes leads to a 30% reduction in heart size associated with diminished Akt phosphorylation [57].
Chronic hyperinsulinemia is shown to increase myocardial mass and reduce cardiac output in rats [58]. Recent evidence shows that cardiac insulin signaling is hyperactive in a pathological hypertrophic model induced by pressure overload [55]. Genetic inhibition of the InsR-Akt1 signaling pathway substantially attenuates hypertrophy, adverse remodeling, and dysfunction following transverse aortic constriction [55, 59–62]. Thus, activation of InsR signaling in pressure overload model appears to promote detrimental pathological hypertrophy. The underlying mechanism of distinct roles of InsR in physiological (exercise) and pathological cardiac hypertrophy is not well understood. One of the potential factors is co-activation of cardiac ARs in pathological conditions, Stimulation of both α1ARs and βARs promote pathological cardiac hypertrophy [63]; and βAR stimulation promotes hypertrophic growth via an Akt-dependent mechanism [60, 64]. In a TAC model, while the elevation of SNS activity drives the cardiac βAR to increase cardiac contractility and compensatory cardiac output, it also promotes persistent activation of PI3K and Akt via stimulation of β2AR [65]. This signaling cascade may be involved and/or dependent on co-activation of InsR in the InsR-β2AR complex. Activation of InsR, PI3K, and Akt is necessary for cardiac hypertrophy, indicating an early compensatory role of InsR activation in a TAC heart. Meanwhile, persistent activation of PI3K and Akt also promotes cardiac IR [13], which compromises cardiac metabolism and the ability of myocyte survival in responding to stress, contributing to maladaptation in HF development. While both IR and hyperactive InsR occur in a TAC heart, the mechanisms underlying the development of these two connected features are not understood. In addition, in pathological cardiac hypertrophy, cytotoxic β1AR signal, the renin-angiotensin-aldosterone system (RAAS), reactive oxygen species (ROS), and inflammation all promote cardiac maladaptive remodeling (e.g. cardiac apoptosis and fibrosis) [66]. These factors can also promote cardiac and general IR to exacerbate HF failure. For example, Angiotensin II abolishes insulin-induced tyrosine phosphorylation of IRS-1, activation of Akt, and GLUT4 translocation to the plasma membrane [67]. Multiple serine phosphorylation events are involving in the negative modulation of insulin signaling by Angiotensin II, which is consistent with the observation of the association of insulin resistance with cardiac hypertrophy [68].
Interestingly, in a recent study with HFD feeding, deletion of the β2AR gene prevents myocardial fibrosis and apoptosis, further supporting the involvement of adrenergic signaling in adverse cardiac remodeling in pathological hypertrophy [33]. In HFD heart, hyperinsulinemia promotes activation of cardiac InsR to induce Akt and ERK1/2 activity. Deletion of β2AR gene blocks Akt and ERK1/2 activity and cardiac fibrosis in HFD heart, indicating that hyperinsulinemia may promote β2AR-dependent Akt and ERK1/2 activity through activation of the newly characterized InsR-β2AR complex. However, genetic and pharmacological inhibition of the β2AR does not prevent pathological cardiac hypertrophy in HFD mice; thus pathological cardiac hypertrophy in this model is not dependent on β2AR, InsR, and Akt signaling [33]. Besides the SNS, the RAAS is activated in obese animals with IR; and an activated RAAS can also promote the development of pathological cardiac hypertrophy [69]. It is noteworthy that the detrimental role of β2AR involved in fibrosis in diabetic hearts is distinct from the traditional beneficial role of β2AR relative to cardiac toxicity induced by the β1AR [27, 70]. Thus, one should be cautious when designing therapeutic strategies that target the cardiac insulin-adrenergic signaling network.
Together, in TAC heart with elevated SNS activity, stimulation of β2AR induces the InsR-dependent Akt activity to promote cardiac hypertrophy probably as an early adaptation whereas the development of IR may contribute to maladaptive response. In contrast, in HFD heart with hyperinsulinemia, insulin-induced β2AR-dependent ERK and Akt signaling does not contribute to cardiac hypertrophy; instead, the insulin-induced activation of InsR-β2AR signaling may promote fibrosis in HFD heart. Further dissection of signaling processes of the InsR-β2AR complex induced by SNS activity or hyperinsulinemia will help us to understand distinct consequences in myocardium in pathological conditions in different animal models.
Chronic βAR Stimulation and IR Promotes Oxidative Stress
Oxidative stress represents another common insult to promote cardiac remodeling in HF (Table 2). Stimulation of adrenergic signaling promotes mitochondria fission via increased PKA phosphorylation of Drp1, a key enzyme involved in mitofusion [71]. The fragmented mitochondria usually lead to less effective respiratory chain reaction [72]. Thus, acute β-adrenergic stimulation increases the production of ROS[35]; and transgenic activation of βAR leads to an elevation of NADPH oxidase activity with greater ROS production [73]. Meanwhile, cardiac glycogen utility involves a complex interplay between multiple signaling pathways including insulin-dependent glycogen synthesis [74], βAR-dependent glycogen breakdown [75], and AMPK [76]. In HF, chronic activation of the SNS promotes cardiac IR, which leads to metabolic switch from glucose to fatty acids due to reduced usage of glucose in myocardium [8]. Increased burning of fatty acids increases production of ROS in mitochondria, contributing to damage of myocytes (Table 2).
Table 2.
βAR and insulin regulate cardiac oxidative stress in I/R.
| Oxidative Stress | Cardioprotective Effect in I/R | |
|---|---|---|
| βAR | ↑ (mitochondria fissiont↑, NADPH oxidase activity↑, ROS↑ 35, 69, 71) | ↓ β1AR detrimental (cAMP and PKA, metabolism disorder 79); ↑ β2AR protective (Akt phosphorylationt↑, apoptosis↓, β1AR signaling↓, eNOS↑ 82–85) |
| βAR blocker | ↓(Akt and eNOS↑ 78) | ↑ (Akt phosphorylationt↑, miR-199a-3p and miR-214 ↑ 80) |
| insulin | ↓ (mitochondria integrity↑, fatty acid oxidation capacity↑, ROS↓ 75) | ↑ (fatty acid oxidation↓, glucose consumption↑, oxidative stress↓, Gi-biased β2AR signaling? 86–90) |
On the other side, insulin signaling is essential to maintain mitochondria integrity via regulating mitophagy, a process that clears dysfunctional mitochondria. In cardiac muscle, activation of PI3K increases myocardial fatty acid oxidation capacity, whereas impaired PI3K signaling leads to cardiac mitochondrial dysfunction and prevents mitochondrial adaptations in response to physiological hypertrophic stimuli [77]. IR can potentially lead to accumulation of dysfunctional mitochondria and further exacerbate production and accumulation of ROS in myocytes [77]. Therefore, the subjects with metabolic syndrome that is associated with prolonged metabolic stress and SNS activity are prone to develop cardiovascular diseases due in part to increased ROS production [78]. Consequently, an increasing insulin sensitivity by Sirt1 is able to counteract oxidative stress [79]. Blocking βAR by nebivolol in IR transgenic rats significantly promotes Akt activation and endothelial NO synthase (eNOS), resulting in reduction of oxidant stress [80].
Insulin and βAR Signaling in Ischemia and Reperfusion
In myocardial I/R, the elevated SNS activity drives β1AR signaling that plays a detrimental role in cardiac injury (Table 2) [81]. β-blockers have favorable effects on the outcome of ischemic heart diseases [82, 83]. However, the cardiac β2AR-Gi-Akt signaling is traditionally considered beneficial to myocardium relative to cardiac toxicity induced by β1AR signaling [27]. β2AR agonist promotes Akt activity to protect against myocyte apoptosis induced by oxidative stress or overactivated β1AR signaling in I/R [84–86]. Meanwhile, stimulation of β2AR with formoterol also activates eNOS and augments NO bioavailability, which attenuates myocardial cell death after I/R [87].
It’s well known that insulin possesses cardioprotective effects in I/R injury (Table 2) [88–90]. In an isolated rat heart, insulin attenuates isoproterenol-induced cardiac dysfunction and cell injury in I/R [90]. Recent studies show that insulin promotes βAR phosphorylation and attenuates catecholamine-sensitive adenylyl cyclases [6, 34]. Thus, the cardioprotective effects of insulin in I/R might be due in part to the insulin-induced Gi-biased β2AR signaling, which counters cardiac toxicity induced by β1AR. In agreement, administration of glucose insulin potassium (GIK) in acute ischemia improves hemodynamic performance and reduces mortality. These effects are also associated with suppression of myocardial fatty acid oxidation, greater glucose consumption, increased SERCA2a and phospholamban mRNA expression [91, 92]. In contrary, long-term βAR stimulation inhibits insulin-induced phosphorylation of ERK1/2 and JNK [2], which are required for insulin to exert its protective effect against the hypoxia-induced activation of pro-apoptotic caspase-3 [2]. Together, these data suggests that the correlation between cardiac sympathetic function and insulin sensitivity contributes to IR and subsequently outcomes in failing ischemic human hearts, supporting the use of insulin and/or a combination of selective β1AR-blocker and β2AR agonist in treatment of acute I/R injury.
Concluding Remarks and future perspectives
Both insulin and β2AR signaling may play either detrimental or protective roles depending on the pathological settings in heart. The elevated SNS activity resulting in enhanced stimulation of βARs is a typical feature of HF. Altered myocardial insulin signaling has also emerged as a potential pathophysiological mechanism that contributes to dysregulation of cardiac metabolism and the development of HF. Modification of cardiac metabolism is a fascinating field of investigation for the development of new treatments of myocardial dysfunction. The recent observation of direct interaction between InsR and βAR provides new insight into InsR-and β2AR-mediated signaling induced by DM-associated hyperinsulinemia and/or the elevated SNS activity. Thus, focusing on the interactions between insulin and adrenergic signaling systems might help in the understanding of the relationship between IR and the SNS activity in type 2 diabetes-associated HF. For example, what roles do the insulin-adrenergic signaling network play in cardiac hypertrophy, fibrosis, and metabolism in HF? It is effective to target insulin receptor or β2AR in comorbidity of heart diseases and metabolic disorders? Moreover, GRK2 acts as a central node that links both insulin and adrenergic regulation; can we target GRK2 as an effective treatment of heart diseases associated with metabolic disorders such as type 2 DM?
Trends.
Insulin plays bidirectional roles in cardiac metabolism and contractile function depending on pathophysiological states.
Chronic hyperinsulinemia impairs cardiac contractile function by inducing a Gi-biased β2AR signaling and upregulation of phosphodiesterase 4D (PDE4D) expression in heart.
Inhibition of β2AR is effective in rescuing cardiac dysfunction but not in preventing cardiac hypertrophy in diabetic mice.
GRK2acts as a node, linking insulin and βAR signaling in cardiac metabolism and cardiac contractile function, thus serving as a potential target for treatment of diabetes-related HF.
Early intervention by targeting the insulin-adrenergic signaling network may be effective in preventing cardiac complications in diabetes.
Outstanding Questions.
What roles do the insulin-adrenergic signaling network play in cardiac hypertrophy, fibrosis, and metabolism in HF?
What roles do βAR agonists and β-blockers have in cardiac insulin sensitivity in diabetes?
Is insulin treatment detrimental in HF with type 2 diabetes?
Will non-selective βAR antagonists be more effective than selective β1AR antagonists in treatment of HF with diabetes?
Can inhibition of GRK2 be an effective therapeutic strategy in controlling diabetic cardiac dysfunction?
Acknowledgments
This study is supported by China NSFC grants 81473212 (QF), 81202541 (QW), and 81428022 (YKX), by Hubei Natural Science Foundation of China 2016CFB417 (QF), and by NIH grants HL127764 and HL112413 (YKX). YKX is an established investigator of the American Heart Association and a Shanghai Eastern Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Qin Fu, Department of Pharmacology, School of Basic Medicine, Tongji Medical College, Hubei Key Laboratory of Drug Target Research and Pharmacodynamic Evaluation, Huazhong University of Science and Technology, Wuhan, China.
Qingtong Wang, Institute of Clinical Pharmacology, Key Laboratory of Anti-inflammatory and Immune Medicine, Ministry of Education, Collaborative Innovation Center of Anti-inflammatory and Immune Medicine, Anhui Medical University, Hefei, China.
Yang K. Xiang, Department of Pharmacology, University of California, Davis, CA, USA, and VA northern California healthcare system, Mather, CA, USA
References
- 1.Contreras-Ferrat AE, et al. An inositol 1,4,5-triphosphate (IP3)-IP3 receptor pathway is required for insulin-stimulated glucose transporter 4 translocation and glucose uptake in cardiomyocytes. Endocrinology. 2010;151(10):4665–77. doi: 10.1210/en.2010-0116. [DOI] [PubMed] [Google Scholar]
- 2.Mangmool S, et al. beta-Adrenergic Receptor and Insulin Resistance in the Heart. Biomol Ther (Seoul) 2017;25(1):44–56. doi: 10.4062/biomolther.2016.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shao D, Tian R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr Physiol. 2015;6(1):331–51. doi: 10.1002/cphy.c150016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciccarelli M, et al. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation. 2011;123(18):1953–62. doi: 10.1161/CIRCULATIONAHA.110.988642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rane S, et al. An antagonism between the AKT and beta-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p. Cellular signalling. 2010;22(7):1054–62. doi: 10.1016/j.cellsig.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu Q, et al. Insulin inhibits cardiac contractility by inducing a Gi-biased beta2-adrenergic signaling in hearts. Diabetes. 2014;63(8):2676–89. doi: 10.2337/db13-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mandic M, et al. Demonstration of a direct interaction between beta2-adrenergic receptor and insulin receptor by BRET and bioinformatics. PLoS One. 2014;9(11):e112664. doi: 10.1371/journal.pone.0112664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nestel PJ, et al. Markers of sympathetic nervous system activity associate with complex plasma lipids in metabolic syndrome subjects. Atherosclerosis. 2016;256:21–28. doi: 10.1016/j.atherosclerosis.2016.11.032. [DOI] [PubMed] [Google Scholar]
- 9.Battiprolu PK, et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012;122(3):1109–18. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mangmool S, et al. Sustained betaAR Stimulation Mediates Cardiac Insulin Resistance in a PKA-Dependent Manner. Molecular endocrinology. 2016;30(1):118–32. doi: 10.1210/me.2015-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morisco C, et al. Akt mediates the cross-talk between beta-adrenergic and insulin receptors in neonatal cardiomyocytes. Circ Res. 2005;96(2):180–8. doi: 10.1161/01.RES.0000152968.71868.c3. [DOI] [PubMed] [Google Scholar]
- 12.Inoue T, et al. Hyperinsulinemia and sulfonylurea use are independently associated with left ventricular diastolic dysfunction in patients with type 2 diabetes mellitus with suboptimal blood glucose control. BMJ Open Diabetes Res Care. 2016;4(1):e000223. doi: 10.1136/bmjdrc-2016-000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Y, et al. Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Molecular endocrinology. 2013;27(1):172–84. doi: 10.1210/me.2012-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harmancey R, et al. Chronic Hyperinsulinemia Causes Selective Insulin Resistance and Down-regulates Uncoupling Protein 3 (UCP3) through the Activation of Sterol Regulatory Element-binding Protein (SREBP)-1 Transcription Factor in the Mouse Heart. J Biol Chem. 2015;290(52):30947–61. doi: 10.1074/jbc.M115.673988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimizu I, et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab. 2012;15(1):51–64. doi: 10.1016/j.cmet.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 16.Alves-Wagner AB, et al. Beta-adrenergic blockade increases GLUT4 and improves glycemic control in insulin-treated diabetic Wistar rats. Auton Neurosci. 2015;193:108–16. doi: 10.1016/j.autneu.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Ayers K, et al. Differential effects of nebivolol and metoprolol on insulin sensitivity and plasminogen activator inhibitor in the metabolic syndrome. Hypertension. 2012;59(4):893–8. doi: 10.1161/HYPERTENSIONAHA.111.189589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uriel N, et al. Improved diabetic control in advanced heart failure patients treated with left ventricular assist devices. European journal of heart failure. 2011;13(2):195–9. doi: 10.1093/eurjhf/hfq204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu W, et al. Interaction of beta1-adrenoceptor with RAGE mediates cardiomyopathy via CaMKII signaling. JCI Insight. 2016;1(1):e84969. doi: 10.1172/jci.insight.84969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irannejad R, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495(7442):534–8. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morisco C, et al. Endocytosis machinery is required for beta1-adrenergic receptor-induced hypertrophy in neonatal rat cardiac myocytes. Cardiovasc Res. 2008;78(1):36–44. doi: 10.1093/cvr/cvn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu Q, Xiang YK. Trafficking of beta-Adrenergic Receptors: Implications in Intracellular Receptor Signaling. Prog Mol Biol Transl Sci. 2015;132:151–88. doi: 10.1016/bs.pmbts.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikolaev VO, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327(5973):1653–7. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 24.Barbagallo F, et al. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ Res. 2016;119(8):931–43. doi: 10.1161/CIRCRESAHA.116.308964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ Res. 2011;109(2):231–44. doi: 10.1161/CIRCRESAHA.110.231340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu MP, et al. Higenamine protects ischemia/reperfusion induced cardiac injury and myocyte apoptosis through activation of beta2-AR/PI3K/AKT signaling pathway. Pharmacol Res. 2016;104:115–23. doi: 10.1016/j.phrs.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo AY, et al. Biased beta2-adrenoceptor signalling in heart failure: pathophysiology and drug discovery. Br J Pharmacol. 2015;172(23):5444–56. doi: 10.1111/bph.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Echouffo-Tcheugui JB, et al. Temporal trends and factors associated with diabetes mellitus among patients hospitalized with heart failure: Findings from Get With The Guidelines-Heart Failure registry. Am Heart J. 2016;182:9–20. doi: 10.1016/j.ahj.2016.07.025. [DOI] [PubMed] [Google Scholar]
- 29.Lorenzo C, et al. Novel Protein Glycan-Derived Markers of Systemic Inflammation and C-Reactive Protein in Relation to Glycemia, Insulin Resistance, and Insulin Secretion. Diabetes Care. 2016 doi: 10.2337/dc16-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okonko DO, Shah AM. Heart failure: mitochondrial dysfunction and oxidative stress in CHF. Nat Rev Cardiol. 2015;12(1):6–8. doi: 10.1038/nrcardio.2014.189. [DOI] [PubMed] [Google Scholar]
- 31.Boly CA, et al. The effect of perioperative insulin treatment on cardiodepression in mild adiposity in mice. Cardiovasc Diabetol. 2016;15(1):135. doi: 10.1186/s12933-016-0453-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Q, et al. Insulin induces IRS2-dependent and GRK2-mediated beta2AR internalization to attenuate betaAR signaling in cardiomyocytes. Cellular signalling. 2015;27(3):707–15. doi: 10.1016/j.cellsig.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q, et al. Inhibiting Insulin-Mediated beta2AR Activation Prevents Diabetes-Associated Cardiac Dysfunction. Circulation. 2016 doi: 10.1161/CIRCULATIONAHA.116.022281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu Q, et al. High fat diet induces PKA and GRK phosphorylation of beta2 -adrenergic receptor and impairs cardiac adrenergic reserve in animal hearts. J Physiol. 2016 doi: 10.1113/JP273314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Llano-Diez M, et al. The Role of Reactive Oxygen Species in beta-Adrenergic Signaling in Cardiomyocytes from Mice with the Metabolic Syndrome. PloS one. 2016;11(12):e0167090. doi: 10.1371/journal.pone.0167090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson GA, et al. beta-adrenergic Responsiveness in the Type 2 Diabetic Heart: Effects on Cardiac Reserve. Med Sci Sports Exerc. 2016 doi: 10.1249/MSS.0000000000001184. [DOI] [PubMed] [Google Scholar]
- 37.Fu Q, et al. A long lasting beta1 adrenergic receptor stimulation of cAMP/protein kinase A (PKA) signal in cardiac myocytes. The Journal of biological chemistry. 2014;289(21):14771–81. doi: 10.1074/jbc.M113.542589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao CY, Greenstein JL, Winslow RL. Interaction between phosphodiesterases in the regulation of the cardiac beta-adrenergic pathway. J Mol Cell Cardiol. 2015;88:29–38. doi: 10.1016/j.yjmcc.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pantalone KM, et al. Increase in overall mortality risk in patients with type 2 diabetes receiving glipizide, glyburide or glimepiride monotherapy versus metformin: a retrospective analysis. Diabetes, obesity & metabolism. 2012;14(9):803–9. doi: 10.1111/j.1463-1326.2012.01604.x. [DOI] [PubMed] [Google Scholar]
- 40.Straznicky NE, et al. Comparable Attenuation of Sympathetic Nervous System Activity in Obese Subjects with Normal Glucose Tolerance, Impaired Glucose Tolerance, and Treatment Naive Type 2 Diabetes following Equivalent Weight Loss. Front Physiol. 2016;7:516. doi: 10.3389/fphys.2016.00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmager P, et al. Galectin-3 and fibulin-1 in systolic heart failure -relation to glucose metabolism and left ventricular contractile reserve. BMC Cardiovasc Disord. 2017;17(1):22. doi: 10.1186/s12872-016-0437-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Utrera-Lagunas M, et al. Abnormal myocardial perfusion and risk of heart failure in patients with type 2 diabetes mellitus. Exp Clin Cardiol. 2013;18(1):e44–6. [PMC free article] [PubMed] [Google Scholar]
- 43.Kishi S, et al. Association of Insulin Resistance and Glycemic Metabolic Abnormalities With LV Structure and Function in Middle Age: The CARDIA Study. JACC Cardiovasc Imaging. 2016 doi: 10.1016/j.jcmg.2016.02.033. [DOI] [PubMed] [Google Scholar]
- 44.Kamal FA, Smrcka AV, Blaxall BC. Taking the heart failure battle inside the cell: small molecule targeting of Gbetagamma subunits. J Mol Cell Cardiol. 2011;51(4):462–7. doi: 10.1016/j.yjmcc.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato PY, et al. The evolving impact of g protein-coupled receptor kinases in cardiac health and disease. Physiol Rev. 2015;95(2):377–404. doi: 10.1152/physrev.00015.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casey LM, et al. Small molecule disruption of G beta gamma signaling inhibits the progression of heart failure. Circ Res. 2010;107(4):532–9. doi: 10.1161/CIRCRESAHA.110.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thal DM, et al. Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem Biol. 2012;7(11):1830–9. doi: 10.1021/cb3003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schumacher SM, et al. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci Transl Med. 2015;7(277):277ra31. doi: 10.1126/scitranslmed.aaa0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horinouchi T, et al. Endothelin-1 suppresses insulin-stimulated Akt phosphorylation and glucose uptake via GPCR kinase 2 in skeletal muscle cells. Br J Pharmacol. 2016;173(6):1018–32. doi: 10.1111/bph.13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lucas E, et al. Obesity-induced cardiac lipid accumulation in adult mice is modulated by G protein-coupled receptor kinase 2 levels. Cardiovasc Diabetol. 2016;15(1):155. doi: 10.1186/s12933-016-0474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rengo G, et al. Impact of diabetes mellitus on lymphocyte GRK2 protein levels in patients with heart failure. European journal of clinical investigation. 2015;45(2):187–95. doi: 10.1111/eci.12395. [DOI] [PubMed] [Google Scholar]
- 52.Lucas E, et al. Downregulation of G protein-coupled receptor kinase 2 levels enhances cardiac insulin sensitivity and switches on cardioprotective gene expression patterns. Biochimica et biophysica acta. 2014;1842(12 Pt A):2448–56. doi: 10.1016/j.bbadis.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 53.Vila-Bedmar R, et al. Reversal of diet-induced obesity and insulin resistance by inducible genetic ablation of GRK2. Sci Signal. 2015;8(386):ra73. doi: 10.1126/scisignal.aaa4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ciccarelli M, Cipolletta E, Iaccarino G. GRK2 at the control shaft of cellular metabolism. Curr Pharm Des. 2012;18(2):121–7. doi: 10.2174/138161212799040493. [DOI] [PubMed] [Google Scholar]
- 55.Shimizu I, et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. The Journal of clinical investigation. 2010;120(5):1506–14. doi: 10.1172/JCI40096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riehle C, et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Molecular and cellular biology. 2014;34(18):3450–60. doi: 10.1128/MCB.00426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Dijk CG, et al. Distinct Endothelial Cell Responses in the Heart and Kidney Microvasculature Characterize the Progression of Heart Failure With Preserved Ejection Fraction in the Obese ZSF1 Rat With Cardiorenal Metabolic Syndrome. Circ Heart Fail. 2016;9(4):e002760. doi: 10.1161/CIRCHEARTFAILURE.115.002760. [DOI] [PubMed] [Google Scholar]
- 59.Peng X, et al. PPARgamma-PI3K/AKT-NO signal pathway is involved in cardiomyocyte hypertrophy induced by high glucose and insulin. J Diabetes Complications. 2015;29(6):755–60. doi: 10.1016/j.jdiacomp.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 60.Chowdhury D, et al. Prohibitin confers cytoprotection against ISO-induced hypertrophy in H9c2 cells via attenuation of oxidative stress and modulation of Akt/Gsk-3beta signaling. Mol Cell Biochem. 2017;425(1–2):155–168. doi: 10.1007/s11010-016-2870-3. [DOI] [PubMed] [Google Scholar]
- 61.Yang B, et al. TWEAK protects cardiomyocyte against apoptosis in a PI3K/AKT pathway dependent manner. Am J Transl Res. 2016;8(9):3848–3860. [PMC free article] [PubMed] [Google Scholar]
- 62.Mei L, et al. Increased cardiac remodeling in cardiac-specific Flt-1 receptor knockout mice with pressure overload. Cell Tissue Res. 2015;362(2):389–98. doi: 10.1007/s00441-015-2209-5. [DOI] [PubMed] [Google Scholar]
- 63.Vidal M, et al. beta-Adrenergic receptor stimulation causes cardiac hypertrophy via a Gbetagamma/Erk-dependent pathway. Cardiovasc Res. 2012;96(2):255–64. doi: 10.1093/cvr/cvs249. [DOI] [PubMed] [Google Scholar]
- 64.Bhavsar PK, et al. Clenbuterol induces cardiac myocyte hypertrophy via paracrine signalling and fibroblast-derived IGF-1. Journal of cardiovascular translational research. 2010;3(6):688–95. doi: 10.1007/s12265-010-9199-1. [DOI] [PubMed] [Google Scholar]
- 65.Murray DR, et al. beta2 adrenergic activation induces the expression of IL-18 binding protein, a potent inhibitor of isoproterenol induced cardiomyocyte hypertrophy in vitro and myocardial hypertrophy in vivo. J Mol Cell Cardiol. 2012;52(1):206–18. doi: 10.1016/j.yjmcc.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koitabashi N, Kass DA. Reverse remodeling in heart failure–mechanisms and therapeutic opportunities. Nat Rev Cardiol. 2011;9(3):147–57. doi: 10.1038/nrcardio.2011.172. [DOI] [PubMed] [Google Scholar]
- 67.Ramalingam L, et al. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbadis.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 68.Muniyappa R, Yavuz S. Metabolic actions of angiotensin II and insulin: a microvascular endothelial balancing act. Mol Cell Endocrinol. 2013;378(1–2):59–69. doi: 10.1016/j.mce.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang JP, et al. High-fructose and high-fat feeding correspondingly lead to the development of lysoPC-associated apoptotic cardiomyopathy and adrenergic signaling-related cardiac hypertrophy. International journal of cardiology. 2016;215:65–76. doi: 10.1016/j.ijcard.2016.03.239. [DOI] [PubMed] [Google Scholar]
- 70.Pagano F, et al. Beta2-adrenergic signaling affects the phenotype of human cardiac progenitor cells through EMT modulation. Pharmacol Res. 2017 doi: 10.1016/j.phrs.2017.01.016. [DOI] [PubMed] [Google Scholar]
- 71.Xu S, et al. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat Commun. 2016;7:13189. doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ambrose LJ, et al. Investigating mitochondrial metabolism in contracting HL-1 cardiomyocytes following hypoxia and pharmacological HIF activation identifies HIF-dependent and independent mechanisms of regulation. J Cardiovasc Pharmacol Ther. 2014;19(6):574–85. doi: 10.1177/1074248414524480. [DOI] [PubMed] [Google Scholar]
- 73.Bovo E, et al. Increased Energy Demand during Adrenergic Receptor Stimulation Contributes to Ca(2+) Wave Generation. Biophys J. 2015;109(8):1583–91. doi: 10.1016/j.bpj.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Landis J, Shaw LM. Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3beta to regulate aerobic glycolysis. J Biol Chem. 2014;289(26):18603–13. doi: 10.1074/jbc.M114.564070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelsall IR, et al. R3F, a novel membrane-associated glycogen targeting subunit of protein phosphatase 1 regulates glycogen synthase in astrocytoma cells in response to glucose and extracellular signals. J Neurochem. 2011;118(4):596–610. doi: 10.1111/j.1471-4159.2011.07345.x. [DOI] [PubMed] [Google Scholar]
- 76.Chandramouli C, et al. Myocardial glycogen dynamics: new perspectives on disease mechanisms. Clinical and experimental pharmacology & physiology. 2015;42(4):415–25. doi: 10.1111/1440-1681.12370. [DOI] [PubMed] [Google Scholar]
- 77.Martin SD, et al. Mitochondrial dysfunction has divergent, cell type-dependent effects on insulin action. Mol Metab. 2014;3(4):408–18. doi: 10.1016/j.molmet.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ji L, et al. The antioxidant edaravone prevents cardiac dysfunction by suppressing oxidative stress in type 1 diabetic rats and in high-glucose-induced injured H9c2 cardiomyoblasts. Can J Physiol Pharmacol. 2016;94(9):996–1006. doi: 10.1139/cjpp-2015-0587. [DOI] [PubMed] [Google Scholar]
- 79.Manna P, Achari AE, Jain SK. Vitamin D supplementation inhibits oxidative stress and upregulate SIRT1/AMPK/GLUT4 cascade in high glucose-treated 3T3L1 adipocytes and in adipose tissue of high fat diet-fed diabetic mice. Arch Biochem Biophys. 2017;615:22–34. doi: 10.1016/j.abb.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 80.Ma L, et al. Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the transgenic (mRen2) rat. American journal of physiology. Heart and circulatory physiology. 2012;302(11):H2341–51. doi: 10.1152/ajpheart.01126.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xue H, et al. Rutaecarpine and evodiamine selected as beta1-AR inhibitor candidates using beta1-AR/CMC-offline-UPLC/MS prevent cardiac ischemia-reperfusion injury via energy modulation. J Pharm Biomed Anal. 2015;115:307–14. doi: 10.1016/j.jpba.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 82.Park KM, et al. Carvedilol-responsive microRNAs, miR-199a-3p and -214 protect cardiomyocytes from simulated ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2016;311(2):H371–83. doi: 10.1152/ajpheart.00807.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huang MH, et al. Therapeutic synergy and complementarity for ischemia/reperfusion injury: beta1-adrenergic blockade and phosphodiesterase-3 inhibition. Int J Cardiol. 2016;214:374–80. doi: 10.1016/j.ijcard.2016.03.200. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Q, et al. Beta(2)-adrenoceptor agonist clenbuterol reduces infarct size and myocardial apoptosis after myocardial ischaemia/reperfusion in anaesthetized rats. British journal of pharmacology. 2010;160(6):1561–72. doi: 10.1111/j.1476-5381.2010.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fajardo G, et al. beta2-adrenergic receptors mediate cardioprotection through crosstalk with mitochondrial cell death pathways. J Mol Cell Cardiol. 2011;51(5):781–9. doi: 10.1016/j.yjmcc.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carr R, 3rd, et al. beta-arrestin-biased signaling through the beta2-adrenergic receptor promotes cardiomyocyte contraction. Proc Natl Acad Sci U S A. 2016;113(28):E4107–16. doi: 10.1073/pnas.1606267113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bhushan S, et al. Selective beta2-adrenoreceptor stimulation attenuates myocardial cell death and preserves cardiac function after ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2012;32(8):1865–74. doi: 10.1161/ATVBAHA.112.251769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liepinsh E, et al. The heart is better protected against myocardial infarction in the fed state compared to the fasted state. Metabolism: clinical and experimental. 2014;63(1):127–36. doi: 10.1016/j.metabol.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 89.Ji L, et al. Insulin attenuates myocardial ischemia/reperfusion injury via reducing oxidative/nitrative stress. American journal of physiology. Endocrinology and metabolism. 2010;298(4):E871–80. doi: 10.1152/ajpendo.00623.2009. [DOI] [PubMed] [Google Scholar]
- 90.Salie R, Huisamen B, Lochner A. High carbohydrate and high fat diets protect the heart against ischaemia/reperfusion injury. Cardiovasc Diabetol. 2014;13:109. doi: 10.1186/s12933-014-0109-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Straus S, et al. Glucosa-Insulin-Potassium (GIK) solution used with diabetic patients provides better recovery after coronary bypass operations. Med Arch. 2013;67(2):84–7. doi: 10.5455/medarh.2013.67.84-87. [DOI] [PubMed] [Google Scholar]
- 92.Ellis KL, et al. Common variants associated with changes in levels of circulating free fatty acids after administration of glucose-insulin-potassium (GIK) therapy in the IMMEDIATE trial. Pharmacogenomics J. 2015 doi: 10.1038/tpj.2015.84. [DOI] [PubMed] [Google Scholar]
- 93.Riehle C, Abel ED. Insulin Signaling and Heart Failure. Circulation research. 2016;118(7):1151–69. doi: 10.1161/CIRCRESAHA.116.306206. [DOI] [PMC free article] [PubMed] [Google Scholar]