

Abstract
As the obesity epidemic worsens, the prevalence of maternal obesity is expected to rise. Both high-fat and high-sucrose diets are known to promote maternal obesity and several studies have elucidated the molecular influence of high-fat feeding on female reproduction. However, to date, the molecular impact of a high-sucrose diet on maternal obesity remains to be investigated. Using our previously reported Drosophila high-sucrose maternal obesity model, we sought to determine how excess dietary sucrose impacted the ovary. High-sucrose diet (HSD) fed adult females developed systemic insulin resistance and exhibited an ovarian phenotype characterized by excess accumulation of lipids and cholesterol in the ovary, decreased ovary size, and impaired egg maturation. We also observed decreased expression of antioxidant genes and increased protein carbonylation in the ovaries of HSD females. HSD females laid fewer eggs; however, the overall survival of offspring was unchanged relative to lean control females. Ovaries of HSD females had increased mitochondrial DNA copy number and decreased expression of key mitochondrial regulators, suggestive of an ineffective compensatory response to mitochondrial dysfunction. Mitochondrial alterations were also observed in male offspring of obese females. This study demonstrates that high-sucrose-induced maternal obesity promotes insulin resistance, while disrupting ovarian metabolism and function.
Keywords: Obesity, Drosophila, ovary, mitochondria, pregnancy, nutritional programming
1. Introduction
In recent years, maternal obesity has emerged as a major health concern not only for the mother but also for the offspring. In the United States alone, 19-38% of pregnant women are obese and thus at increased risk for gestational diabetes mellitus, pre-eclampsia, miscarriage, caesarian delivery, hypertension, and preterm birth [1–5]. The offspring of obese mothers exhibit a higher incidence of neural tube defects, fetal macrosomia, shoulder dystocia, orofacial clefts, high birth weight, and, in later childhood, type 2 diabetes mellitus, obesity, and cardiovascular disease [1,3,6–10]. There is also a strong association between maternal obesity and offspring congenital heart defects [11].
To understand the molecular mechanisms by which maternal obesity impacts female fertility and offspring health, several reports have employed the use of high-fat or high-fat-high-sucrose diet rodent models. From these studies it is clear that high-fat and high-fat-high-sucrose feedings contribute to a disrupted ovarian phenotype that includes endoplasmic reticulum stress, mitochondrial dysfunction, intracellular lipid accumulation, and apoptosis that correlate strongly with the decreased fertilization rates, delayed offspring development, and fetal brain abnormalities also observed in these same model systems [12–15]. Despite the strong associations between excess sugar consumption and obesity and its co-morbidities, only a few studies have investigated the specific contribution of sucrose to maternal obesity and offspring health [16–23]. Moreover, to date, the role of excess sucrose consumption on the molecular features of maternal obesity remains to be explored.
Drosophila melanogaster has emerged as an invaluable tool in metabolic disease research in part due to the high conservation of metabolic tissues and pathways in the fly, well developed metabolic methodology, ease of diet manipulation, and rapid life-cycle [24–28]. Additionally, in female flies, the ovary is the largest and one of the most well characterized organs in Drosophila, providing a highly accessible way to study egg maturation under various genetic and environmental conditions that mimic human diseases [29]. The fact that diet and insulin can control ovary size, ovariole stem-cell proliferation, and egg production is well documented and further underscores the benefits of using female Drosophila to investigate the impact of diet on reproduction [30– 34]. For these same reasons, we and others have used the fly to further understand the influence of nutrition on both parental and offspring health [35–41]. We previously described a Drosophila maternal obesity model in which adult females fed a high-sucrose diet (HSD) exhibited an obese-like phenotype characterized by increased levels of whole-body triglycerides and the disaccharide trehalose, the most abundant circulating sugar in the fly [39]. Offspring of high-sucrose fed females showed altered metabolism and disrupted expression of carbohydrate and lipid metabolic pathway genes, and went on to produce progeny with altered body composition — highlighting the transgenerational impact of maternal obesity [39].
We employed our Drosophila model to study the impact of sucrose-induced maternal obesity on fertility and offspring survival. Our data demonstrate that high-sucrose-induced maternal obesity promotes insulin resistance, while disrupting ovarian function and altering maternal and offspring mitochondria.
2. Methods and Materials
2.1 Primers
Primers used for quantitative PCR (qPCR) were: dILP2 F 5′-AGGTGCTGAGTATGG TGT-3′, R 5′-TATCGAGTTATCCTCCTCCT-3′; dILP3 F 5′-AACTCTCTCCAAGCTCTGT-3′, R 5′-GAACCGAACTATCACTCAAC-3′; dILP5 F 5′-CTTGATGGACATGCTGAG-3′, R 5′-AAGTGGTCCTCATAATCGAA-3′; ATP synthase F 5′-TGCATCTAAAGCCGATAAGAATTTG-3′, R 5′-TCATCAAACTGGACATCGACG-3′ Cytochrome c oxidase I F 5′-AAAGTTGACGGTACACCTGG-3′, R 5′-AGGAACACTTTCAATTACAATCGG-3′; mt-TFB1 F 5′-TCCCTAGTTCGTAAGCGTTTTC-3′, R 5′-ACCAGCTCCCGAATTGTG-3′ Spargel F 5′-ATTCGGTGCTGGTGCTTCCTT-3′, R 5′-CGTGCCTTCTGTCGTTCATCTTAC-3′; delg F 5′-CGCGTCCTGCAGCCAAAAC-3′, R 5′-GCGGAGCCGGACGACATC-3′; Parkin F 5′-GGCGGAAGCAGTCGATACAGGTGA-3′, R 5′-CCGGCGGGGAGAAGGATTTTG-3′; Rosy F 5′-GGTGGTGAGCCTGTTCTTCAAG-3′, R 5′- ACTGGTGTGTGGAATGTCTCGG-3′; Cytochrome c oxidase III F 5′- CACGAGAAGGAACATACC-3′, R 5′-GCGGGTGATAAACTTCTG-3′; SOD1 F 5′- CAAGGGCACGGTTTTCTTC-3′, R 5′-CCTCACCGGAGACCTTCAC-3′; SOD2 F 5′- AATTTCGCAAACTGCAAGC-3′, R 5′-TGATGCAGCTCCATGATCTC-3′; GstD1 F 5′- TCGCGAGTTTCACAACAGAA-3′, R 5′-TGAGCAGCTTCTTGTTCAGC-3′; αTubulin 84B F 5′-ACACTTCCAATAAAAACTCAATATGC-3′, R 5′- CCGTGCTCCAAGCAGTAGA-3′
2.2 Fly Stocks
w1118 Drosophila stocks were obtained from the Bloomington Drosophila Stock Center and maintained in vials containing molasses-based fly food (recipe available upon request). All flies were housed at 25°C in a humidified, temperature-controlled incubator with a 12 hour on/off light cycle. Virgin female flies were collected from stocks and placed within 24 hours of eclosion on either a low-sucrose (LSD) or HSD diet for seven days as described previously [39]. Both foods were isocaloric for fat and protein and differed only in the amounts of sucrose added. Low and high sucrose food were composed of 0.15 mol/l and 1.0 mol/l sucrose, respectively.
2.3 Ovarian imaging and electron microscopy
Ovaries were isolated from low- and high-sucrose fed females, immediately placed on glass slides with PBS, and viewed with a Zeiss fluorescent microscope equipped with an Axiocamera. Number of stage 14 eggs were individually counted and total ovary area, length, and width were determined using ImageJ. For electron microscopy, ovaries were isolated from low- and high-sucrose fed females and immediately fixed in Modified Karnovsky's Fixative solution and processed for electron microscopy by the Washington University Research Electron Microscopy Core. Samples were viewed on a JEOL 1200EX electron microscope and images acquired via a high resolution CCD based camera. Mitochondrial size was determined using ImageJ.
2.4 Egg laying capacity and offspring survival studies
After seven days of low- or high-sucrose feeding, females were mated with w1118 males for 18 hours on grape agar plates supplemented with the appropriate low- or high-sucrose food. Eggs were counted and transferred to vials containing molasses-based food. Number of eggs that developed into wL3 larvae, then pupae, and finally adults was determined by counting individual offspring at each developmental stage and dividing by number of eggs laid.
2.5 Insulin sensitivity assay
Insulin sensitivity of adult females (3 flies pooled per biological replicate) was determined as previously described for wL3 larvae [42]. Briefly, live adults were rinsed in PBS, bisected, and pinned open prior to incubation with 0.5μM human insulin (Sigma-Aldrich; catalog# I0259) or control buffer (10mM HEPES) at room temperature for 15 minutes. Animals were transferred to lysis buffer and homogenized. Protein concentrations were quantified from lysates by bicinchoninic acid assay (ThermoFisher Scientific) and phosphorylated S505 AKT, total AKT, and actin were detected by Western blot using antibodies against phospho-Drosophila AKT (Ser505) #4054 and total AKT (pan) #4691 from Cell Signaling and β-actin #A2228 from Sigma-Aldrich. Blots were visualized and bands quantified using the LI-COR Odyssey imaging system.
2.6 Ovarian composition measurements
Measurements for triacylglycerides (TAG) and glucose were performed on ovaries (28 ovaries pooled per biological replicate) isolated from adult females, following methods adapted from a previous study that used whole-body adults and larvae [39]. Briefly, animals were rinsed in PBS, ovaries were dissected and homogenized in PBS + 0.1% Tween-20 and for glucose measurements 2μl of homogenate was added to 98μl of Infinity Glucose Hexokinase Liquid table Reagent (Fischer Scientific), incubated for 15 minutes at 37°C and absorbance measured at 340nm on a Tecan Infinite 200 PRO microplate reader. For ovarian TAG measurements, homogenized ovaries were incubated for 5 minutes at 65°C to inactivate lipases and 2μl added to 198μl of Thermo Infinity Triglyceride Reagent (ThermoFisher Scientific) and absorbance measured at 560nm on a microplate reader. Ovary cholesterol concentration (30 ovaries pooled per biological replicate) was measured as previously described using the Amplex Red Cholesterol Assay Kit (Invitrogen) and microplate reader [43]. Ovarian composition measurements were normalized to total protein.
2.7 Quantitative PCR
Adult female heads (5 heads pooled per biological replicate) and ovaries (6-14 ovaries pooled per biological replicate) or wL3 male larvae (5 larvae pooled per biological replicate) were collected, rinsed in PBS, and homogenized in TRIzol reagent (Invitrogen) followed by subsequent RNA isolation as per manufacturer's instructions. RNA was quantified and used to generated cDNA via the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Synthesized cDNA was used for quantitative PCR (qPCR) using SYBR Green (Applied Biosystems), appropriate primers, and performed in triplicate on a Stratagene Mx3005P qPCR machine. Relative quantification after normalization was calculated using the 2-ΔCT formula. For ovarian gene expression 6-14 ovaries were pooled for each biological replicate.
For mitochondrial DNA quantification, larvae (5 larvae pooled per biological replicate) or ovaries (6-14 ovaries pooled per biological replicate) were homogenized in RNA-Bee (Tel Test) as per manufacturer's instructions to obtain total DNA, which was used in qPCR with the primers cytochrome c oxidase III (CG34074; mitochondrial gene) and Rosy (CG7642; nuclear gene). Relative quantification after normalization was calculated using the 2-ΔCT formula and values for cytochrome c oxidase III were subtracted from Rosy to determine amount of mtDNA per sample.
2.8 Protein Carbonylation Assay
Whole-cell lysates were prepared from homogenized ovaries (10-14 ovaries pooled per biological replicate) in RIPA buffer (25mM Tris-HCl pH 7.6, 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) with EDTA-Free protease complete tablet (Roche), PMSF and 2% 2-mercaptoethanol. Homogenates were run through a 27 gauge needle 20-times followed by protein quantification. A total of 20μg of protein per sample was loaded onto 10% SDS PAGE gels. Carbonylated proteins were detected using the Oxyblot Protein Oxidation Detection Kit (S7150; EMD Millipore) as per the manufacturer's instructions. Blots were visualized and bands quantified using the LI-COR Odyssey imaging system.
2.9 Statistics
Data are expressed as mean ±standard error of the mean (SEM). Comparisons between groups were performed by unpaired, two-tailed Student's t-test.
3. Results
3.1 Insulin sensitivity and production are impaired in high-sucrose fed females
Our previous work demonstrated that female adult flies fed a high-sucrose diet exhibited an obese-like phenotype, characterized by elevated whole-body TAG, glycogen, and trehalose [39]. Because insulin resistance is a comorbidity of obesity and the insulin signaling pathway is highly conserved in Drosophila, we sought to determine whether this high-sucrose feeding also impacted insulin sensitivity. Following 7 days on an LSD or HSD diet, bisected adult females were exposed to human insulin to stimulate the insulin signaling pathway. Pathway activation was assessed by measuring levels of AKT phosphorylation on Serine 505, which is homologous to the mammalian AKT Serine 473 position (Figure 1A). Both HSD and LSD adult females responded to insulin treatment; however, LSD females showed a 9-fold increase in phosphorylated AKT levels compared to HSD females, demonstrating that stimulation of the insulin signaling pathway is impaired in HSD obese-like female flies. These studies are in agreement with previous work by Na, et al. which reported that mixed-sex adult populations fed a high-sucrose diet for 3 weeks are insulin resistant [26]. In the fly, Drosophila insulin-like peptides (dILPs) are homologs of mammalian insulin, and, like insulin, respond to changes in nutrient availability [44]. We next investigated whether insulin production was altered in HSD females by measuring dILP gene expression using qPCR. Compared to LSD flies, HSD females exhibited a decrease in dILP 2 and 5, while dILP 3 levels remained unchanged (Figure 1B). These data demonstrate that, in addition to insulin resistance, insulin production is also altered in HSD females.
Fig. 1. Insulin responsiveness and dILP expression are altered in HSD females.
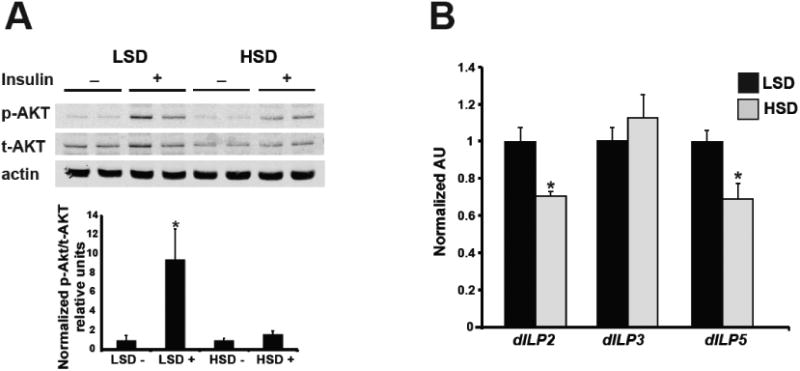
Representative Western blot of total body protein with (+) and without (-) insulin stimulation for phosphorylated (P-AKT) and total (T-AKT) AKT and actin loading control for LSD and HSD adult females (A). Quantification shown is of 5 separate blots using Odyssey imaging software. n=5 pooled samples. qPCR measurement of mRNA expression levels for dILP 2, 3, and 5. n=6 pooled samples (B). All data is expressed as mean + SEM. *p<0.05.
3.2 High-sucrose diet disrupts mature egg production and ovarian lipid and sterol homeostasis
Using our Drosophila model of maternal obesity, we noted that HSD females are quantitatively smaller than LSD controls (Figure 2A-B) and, as our group demonstrated previously, weigh less [39]. This decreased size is also evident in the ovaries, whereby, HSD females had ovaries that were 28% smaller than their LSD counterparts (Figure 2C). Moreover, this decrease in overall ovarian size positively correlated with a decrease in the number of mature (stage 14+) eggs (Figure 2D).
Fig. 2. HSD depletes mature egg production and elevates TAG and cholesterol levels in the ovary.
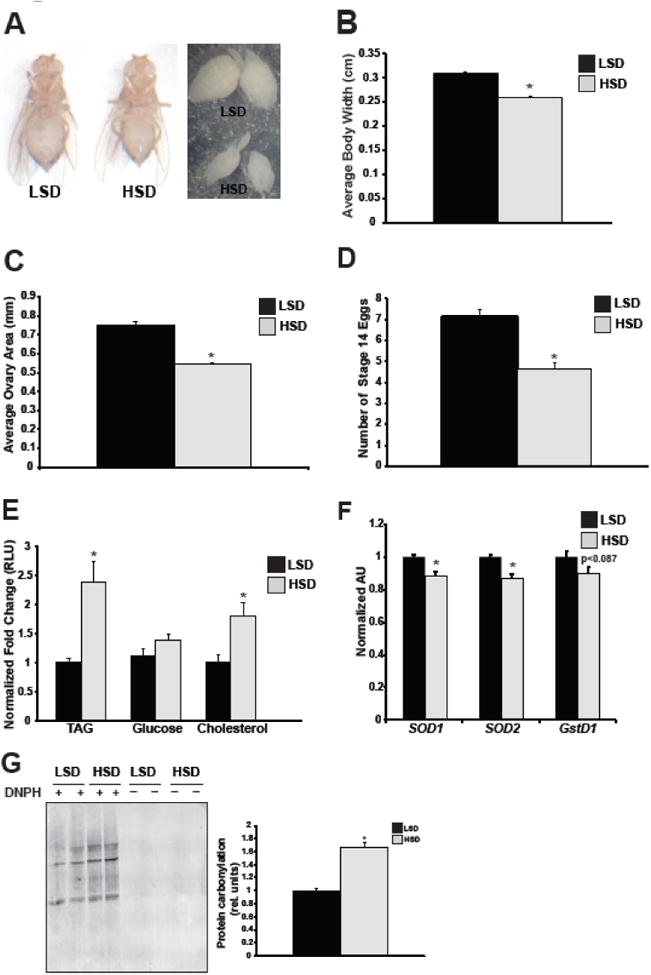
Representative images of LSD and HSD adult females and ovaries are shown after 7 days on diet taken at 10X magnification (A), along with corresponding quantification of average body width (B), ovary area (C), number of stage 14 eggs present in ovaries (D), and ovarian TAG, cholesterol, and glucose levels (E). Body measurements are n=5 flies; ovary area n=83-93 ovaries; stage 14 eggs n=40 ovaries. TAG, cholesterol, and glucose levels were quantified from isolated ovaries of adult females, n=9-14 pooled samples. qPCR measurement of mRNA expression levels of SOD1 (superoxide dismutase 1), SOD2 (superoxide dismutase 2), and gstD1 (glutathione S-transferase D1) from ovaries. n=8 pooled samples (F). Protein carbonylation of ovary whole-cell lysates treated with (+) or without (-) DNPH were visualized by Western blot using Odyssey imaging software. Representative blot is shown (left) as well as quantification (right). n=3 pooled samples (G). All data is expressed as mean ± SEM, *p<0.05.
We previously reported that HSD females exhibited elevated whole-body TAG levels and increased carbohydrates that were indicative of an obese-like phenotype [39]. Recent studies in high-fat fed mice have shown that the ovaries of obese females possess an abundance of lipids [12]. To further characterize the high-sucrose ovarian phenotype, we measured levels of TAG, cholesterol, and glucose specifically in the ovary (Figure 2E). TAG and cholesterol were significantly increased in the ovaries of HSD females relative to the ovaries of LSD controls. There was no observable change in ovarian glucose levels under high-sucrose conditions. These data demonstrate that, as in mice, maternal obesity in the fly causes an abundance of lipids in the ovary; moreover, our data are the first to demonstrate that a high-sucrose obesogenic diet causes excess accumulation of cholesterol in the ovary.
Accumulation of excess lipids in non-adipose tissues is associated with oxidative stress, which can disrupt normal organelle and cellular functions [45]. Recent studies in humans have shown a positive correlation between increasing maternal BMI and oxidative stress biomarkers [46,47]. Moreover, reactive oxygen species levels are elevated in the oocytes of mice fed a high-fat diet [14]. Based on these reports and our observations that high-sucrose feeding increased ovarian TAG levels, we next investigated whether there was evidence of oxidative stress in the ovaries of HSD flies. Protein carbonylation was higher in the ovaries of HSD females compared to LSD controls by 66% (Figure 2G). There was also a modest, yet significant decrease in gene expression of the antioxidant genes SOD1 and SOD2 (Figure 2F). Together these data suggest the presence of oxidative stress in the ovaries of HSD females.
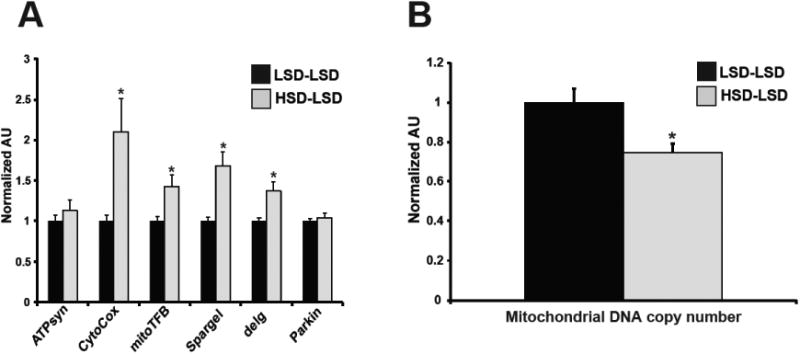
3.3 High-sucrose diet increases ovarian mitochondrial copy number and alters expression of key mitochondrial regulators
Excess cellular lipid and sterol levels are known to disrupt mitochondrial function in vitro [45,48]. Additionally, in high-fat murine models of maternal obesity, there is a strong correlation between oocyte lipid accumulation and mitochondrial abnormalities [12]. Because both TAG and cholesterol accumulation were elevated in the ovaries of HSD females, we investigated whether high-sucrose ovaries exhibited a disrupted mitochondrial phenotype by assessing mitochondrial ultrastructure, number, and gene expression. Unlike in high-fat fed murine maternal obesity models, visualization of mature egg mitochondria by electron microscopy failed to show structural defects or alterations in organelle size in HSD flies (Figure 3A) [13]. However, quantification of mitochondrial DNA copy number demonstrated a 54% higher copy number in the ovaries of HSD females compared to LSD controls (Figure 3B). We next investigated whether sucrose feeding affected expression of genes involved in mitochondrial function and observed that, compared to LSD females, the ovaries of HSD females showed increased expression of ATP synthase and decreased expression of several genes including the mitochondrial gene cytochrome c oxidase I; mt-TFB1, which controls mitochondrial translation; the Drosophila PGC-1α homolog Spargel, a key regulator of mitochondrial biogenesis and mass; the NRF-2α homolog delg, a regulator of mitochondrial mass; and Parkin, a ubiquitin-protein ligase important in mitochondrial remodeling [49–53] (Figure 3C). These data indicate that excess sucrose consumption promotes a disrupted mitochondrial gene profile in the ovary.
Fig. 3. Mitochondrial copy number and gene expression of key mitochondrial regulators are altered in HSD ovaries.
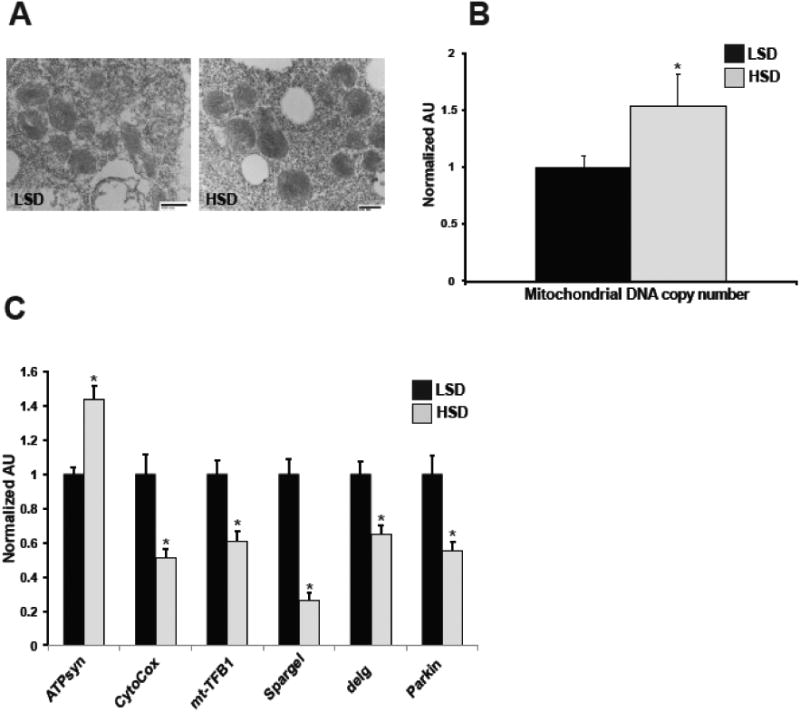
Representative transmission electron microscopy images of LSD and HSD oocytes (A). qPCR measurement of mitochondrial DNA copy number in the ovaries. n=36 pooled samples (B). qPCR measurement of mRNA expression levels of the indicated mitochondrial genes from ovaries. ATPsyn, ATP synthase; CytoCox, cytochrome C oxidase I; mt-TFB1, mitochondrial Transcription Factor B1. n=12-17 pooled samples (C). All data is expressed as mean ± SEM, *p<0.05.
3.4 High-sucrose maternal feeding decreases fertility and alters offspring mitochondria
Based on our observations that high-sucrose diet increased ovarian mitochondrial DNA copy number and altered gene expression and the fact that maternal mitochondria are inherited by the offspring, we next investigated whether excess dietary sucrose impacted fertility and offspring mitochondria. To measure fertility, LSD and HSD adult females were mated with molasses-diet fed males on grape agar (Figure 4). Grape agar plates were supplemented with the appropriate LSD or HSD food, in lieu of yeast paste, in an effort to maintain females on their respective diets. This was done to avoid any dampening of our HSD phenotype by the addition of excess yeast, while still providing necessary nutrients for egg laying. Eggs laid were counted after 18 hours of mating and transferred to molasses food where the number of embryos developing into wL3, pupae, and adults was quantified. Compared to LSD females, high-sucrose feeding reduced the number of eggs laid by 62% (Figure 4A). This decrease in offspring number persisted at the larval, pupal, and adult stages; however, overall survival of offspring from LSD and HSD females remained the same regardless of maternal diet and the developmental stage at which survival was measured (Figure 4B). These data demonstrate that high-sucrose maternal feeding decreases fertility; however, offspring survival is not impaired. We next measured expression of key mitochondrial regulators in the male larval offspring of LSD and HSD females and observed a significant increase in the expression of regulators of mitochondrial translation, biogenesis, and mass (Figure 5A). Larval offspring of HSD females exhibited a decreased mitochondrial DNA copy number compared to controls (Figure 5B). While these data suggest an altered mitochondrial phenotype in the offspring of obese mothers, they also demonstrate that the impact to mitochondria varies from that of HSD female ovaries.
Fig. 4. High-sucrose maternal feeding decreases offspring production, but does not impact offspring survival.
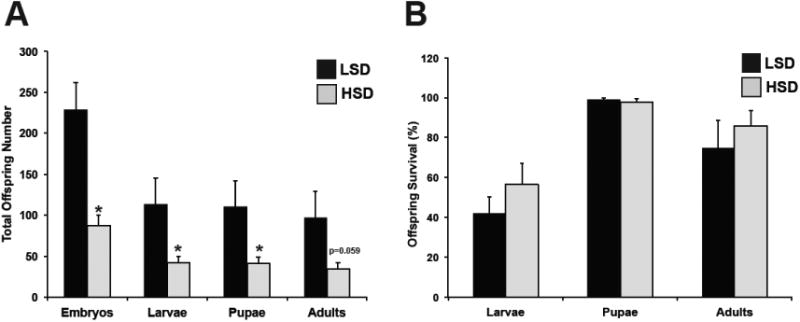
Average number of embryos and number of offspring that subsequently developed into wL3 stage larvae, pupae, and adults from the initial embryonic cohort (A) and percent survival of the offspring at each developmental stage (B). Each developmental group consisted of 9-10 biological replicates. All data is expressed as mean ± SEM, *p<0.05.
Fig. 5. Offspring of HSD females have altered gene expression and mitochondrial copy number.
qPCR analysis of whole-body mitochondrial mRNA expression (A) and whole-body mitochondrial DNA copy number of wL3 stage offspring larvae (B). n=13-27 pooled samples. All data is expressed as mean ± SEM, *p<0.05.
4. Discussion
There is strong evidence supporting a key role for excess sugar consumption in the pathogenesis of obesity and associated co-morbidities [21–23]. As the obesity epidemic worsens, the prevalence of maternal obesity is expected to increase, posing severe health threats not only to the mother and offspring, but to subsequent generations as well [38,39,54]. While the impact of high sucrose consumption on maternal and offspring health has been demonstrated, the molecular mechanisms by which excess sucrose contributes to maternal obesity remain unknown [16–20]. Using a Drosophila melanogaster model of maternal obesity, this study demonstrated that high sucrose consumption in adult females resulted in insulin resistance and a reproductive phenotype characterized by accumulation of TAG and cholesterol in the ovary, decreased egg production and fertility, and increased mitochondrial copy number and decreased expression of key mitochondrial regulators. We also show that this sucrose-driven mitochondrial phenotype extends beyond the mother since mitochondrial copy number and gene expression are also altered in the male offspring of HSD females.
Drosophila has recently emerged as a valuable tool for modeling the metabolic aspects of obesity and diabetes [25,55]. We and others have demonstrated that high-sucrose and high-fat feeding results in metabolically obese larvae and flies, characterized by metabolic abnormalities that include insulin resistance, hyperglycemia, increased whole-body TAG levels, accumulation of lipids in non-adipose tissues, cardiomyopathy, decreased lifespan, and altered gluconeogenic and lipogenic gene expression [26–28,39,42,56–58]. Unlike in mammals, the largest organ in female Drosophila is the ovary, thus under high-sucrose conditions, this organ's size is diminished compared to low-sucrose fed females, which may account for the decrease in overall organismal size we observed (Figure 2A-C) [29]. We report here and previously that HSD-females share many of the same biological and reproductive impairments as rodent maternal obesity models, including excess circulating sugars, TAG accumulation both in whole-body and in the ovaries, decreased fertility, a mitochondrial phenotype, and a metabolic impact on the offspring [12,39,59]. Drosophila obesity models have also uncovered new roles for developmental and metabolic genes in obesity-associated diseases, in addition to exploring the impact of obesity on epigenetic inheritance [26–28,38,57,60–62].
This study demonstrates that HSD adult females have a blunted response to insulin stimulation compared to LSD females. We observed differences in the levels of whole-body phosphorylated AKT between insulin-stimulated LSD versus HSD females that were higher than a recently published report using Drosophila mixed-sex populations (Figure 1A) [26]. Although both studies showed that HSD feeding is associated with insulin resistance, the quantified degree of AKT phosphorylation observed between the two studies may have been affected by several differences including the amounts of exogenous insulin used (our 0.5μM versus Na et. al.'s 1.0μM concentration) [26]. The use of females versus mixed-sex populations in these experiments may also account for the observed differences, especially given the knowledge that in Drosophila the response to dietary manipulation varies by gender and reports suggest that sex may influence insulin-signaling [26,63–65]. Differences in the degree of phosphorylation may have also been influenced by the length of time flies were placed on sucrose diet (our 7 day treatment versus the Na et al. 21 day exposure), since, as has been shown for high-fat fed flies, AKT phosphorylation becomes reduced overtime [26,27]. Despite these experimental differences, both studies demonstrate that excess sucrose consumption is associated with disrupted insulin responsiveness in adult Drosophila [26].
In addition to insulin resistance, HSD adult females also exhibit impaired dILP gene expression. However, work done in HSD fed Drosophila larvae reported increased dILP expression, suggestive of the hyperinsulinemia commonly associated with insulin resistance in humans [42]. Differences in developmental stages (adult versus larvae) may be a factor for this discrepancy since it is known that dILP gene expression, even independent of dietary intervention, changes during development [66]. Additionally, a previous study by Morris et al. showed that, while dILP gene expression increased in adult females fed a 10% (w/v) sucrose diet, at higher sucrose concentrations, dILP gene expression decreased [58]. Interestingly, sucrose concentrations resulted in impaired insulin signaling regardless of the degree of increased dietary sugar content [58]. The current study, in which we report insulin resistance and decreased dILP expression in HSD females, exposed adult female flies to a 72% (w/v) sucrose diet. These studies suggest a threshold may exist whereby excess dietary sucrose levels halt the further promotion of dILP gene expression.
High-fat diet promotes the excess accumulation of lipids in the cumulus-oocyte complexes of female rodents and correlates with decreased fertility and an altered mitochondrial phenotype [12]. The current data demonstrate that in Drosophila, high-sucrose feeding results in excess lipid and cholesterol accumulation in the ovary and decreased egg production. It remains to be known whether, in rodents, high-fat diet promotes cholesterol accumulation. Insects require sterols for growth and survival; however, they lack the enzymes to synthesize sterols and therefore must obtain them through diet [67]. Based on this, we can conclude that the excess cholesterol present in HSD fly ovaries was derived from diet rather than by de novo synthesis. HSD and LSD diets contain identical amounts of lipids and sterols, thus the accumulation of cholesterol in HSD ovaries may be due to increased uptake of sterols and/or decreased sterol utilization. Excess lipid accumulation in non-adipose tissues promotes oxidative stress, which contributes to cellular and organelle dysfunction [45]. In addition to increased lipid levels, ovaries of HSD females exhibit signs of oxidative stress, which is in agreement with previous studies in high-fat fed female rodents and together suggest oxidative stress may serve as one means through which lipid accumulation disrupts normal ovarian function [14].
Our observation that the ovaries of HSD females are smaller compared to LSD controls is in agreement with previous Drosophila studies that have demonstrated overall ovary size, egg production, and proliferation of stem cells in the ovary are highly controlled by nutritional availability, as well as insulin-dependent and independent signaling pathways [30–32,34]. Oogenesis in Drosophila is extremely well studied and occurs in the ovarioles, a group of tubules that make up the fly ovary. Each ovariole consists of a strand of egg chambers that mature in 14 stages as they progress posteriorly down the tubule [29]. We observed fewer mature eggs (stage 14) in HSD female ovaries compared to LSD controls; however, whether this was due to a blunting in egg maturation during oogenesis or other factors is the subject of future studies.
Analysis of ovaries from HSD females showed an increase in mitochondrial DNA copy number and support previous findings in a diabetic mouse model and high-fat fed mouse maternal obesity models, which concluded that increased mitochondrial DNA copy number may be a compensatory response to mitochondrial dysfunction [13,14,68]. Unlike a high-fat diet female rodent study that showed increased expression of PGC-1α, we demonstrate that high-sucrose feeding caused a decrease in the Drosophila PGC-1α homolog Spargel, highlighting the fact that different nutrients cause differential cellular effects [13]. Additionally, in contrast to mitochondria from high-fat diet rodent ovaries, mitochondria from the ovaries of HSD Drosophila failed to show any morphological changes [13]. This observation suggests that, unlike fat, high-sucrose consumption alters mitochondrial number and gene expression, but does not affect mitochondrial morphology under the tested conditions. However, it is possible that increasing the time of exposure to high-sucrose diet may further disrupt mitochondrial function and lead to a change in mitochondrial morphology.
Maternal mitochondria are inherited by the offspring and reports have demonstrated that a high-fat maternal diet causes mitochondrial dysfunction in zygotes and, recently, offspring oocytes and muscle [69,70]. Our work is the first to demonstrate that a high-sucrose maternal diet results in a mitochondrial phenotype in the offspring, characterized by decreased DNA copy numbers and disrupted expression of key mitochondrial regulators. The inheritance of dysfunctional mitochondria past the F1 generation has been shown in high-fat maternal obesity models [70]. While the current study did not examine the transgenerational impact of HSD on mitochondria past F1, we previously reported that F2 generations exhibit increased whole-body glucose and trehalose levels, suggestive of disrupted metabolism. The potential influence the F1 mitochondrial phenotype on metabolism and subsequent impact on the F2 generation is the focus of future studies.
In summary, the current study shows that a maternal diet high in sucrose produces an ovarian phenotype characterized by excess lipid and cholesterol accumulation, altered mitochondria, and reduced egg maturation. These features may play a role in the decreased fertility observed in our maternal obesity model and in the disruptions in offspring mitochondria and metabolism [39]. Whether these features of a high-sucrose diet compound the effects of high-fat consumption on maternal and offspring health remains to be explored and will further our understanding of maternal obesity.
Highlights.
High-sucrose maternal obesity disrupts ovarian lipid and cholesterol homeostasis
Excess sucrose increases mitochondrial DNA copy number while decreasing mitochondrial regulatory genes in the ovary
Mitochondrial disruptions are relayed to the offspring of obese females
Acknowledgments
This work was supported by grants from the American Heart Association (IRG5450013 and GRNT12080056 to J.G. Duncan), the Washington University Diabetes Research Center (P30DK020579 to J.G. Duncan) and the National Institutes of Health (K12HD001459 to R.T. Brookheart).
Abbreviations
- PGC-1α
peroxisome proliferator-activated receptor-ϒ coactivator 1α
- dILPs
Drosophila insulin-like-peptides
- TAG
triacylglycerides
- mt-TFB1
mitochondrial transcription factor binding protein 1
- FSC
follicle stem cells
- HSD
high-sucrose-diet
- LSD
low-sucrose-diet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.a Leddy M, Power ML, Schulkin J. The impact of maternal obesity on maternal and fetal health. Rev Obs Gynecol. 2008;1:170–178. [PMC free article] [PubMed] [Google Scholar]
- 2.O'Brien TE, Ray JG, Chan WS. Maternal body mass index and the risk of preeclampsia: a systematic overview. Epidemiology. 2003;14:368–74. doi: 10.1097/00001648-200305000-00020. [DOI] [PubMed] [Google Scholar]
- 3.Nohr EA, Timpson NJ, Andersen CS, Davey Smith G, Olsen J, Sorensen TI. Severe obesity in young women and reproductive health: the Danish National Birth Cohort. PLoS One. 2009;4:e8444. doi: 10.1371/journal.pone.0008444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yogev Y, Catalano PM, Yogev Y CPM, Yogev Y, Catalano PM. Pregnancy and Obesity. Obstet Gynecol Clin North Am. 2009;36:285–300. doi: 10.1016/j.ogc.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Dokras A, Baredziak L, Blaine J, Syrop C, VanVoorhis BJ, Sparks A. Obstetric outcomes after in vitro fertilization in obese and morbidly obese women. Obstet Gynecol. 2006;108:61–69. doi: 10.1097/01.AOG.0000219768.08249.b6. [DOI] [PubMed] [Google Scholar]
- 6.Stothard KJ, Tennant PW, Bell R, Rankin J. Maternal overweight and obesity and the risk of congenital anomalies: a systematic review and meta-analysis. JAMA. 2009;301:636–650. doi: 10.1001/jama.2009.113. [DOI] [PubMed] [Google Scholar]
- 7.Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L, Damm P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J Clin Endocrinol Metab. 2009;94:2464–70. doi: 10.1210/jc.2009-0305. [DOI] [PubMed] [Google Scholar]
- 8.Diesel JC, Eckhardt CL, Day NL, Brooks MM, Arslanian SA, Bodnar LM. Is gestational weight gain associated with offspring obesity at 36 months? Pediatr Obes. 2015;10:305–10. doi: 10.1111/ijpo.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catalano PM, Presley L, Minium J, De Mouzon SH, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32:1076–80. doi: 10.2337/dc08-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Catalano PM, Farrell K, Thomas A, Huston-Presley L, Mencin P, de Mouzon SH, Amini SB. Perinatal risk factors for childhood obesity and metabolic dysregulation. Am J Clin Nutr. 2009;90:1303–13. doi: 10.3945/ajcn.2008.27416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madsen NL, Schwartz SM, Lewin MB, Mueller BA. Prepregnancy Body Mass Index and Congenital Heart Defects among Offspring: A Population-based Study Congenit. Heart Dis. 2013;8:131–141. doi: 10.1111/j.1747-0803.2012.00714.x. [DOI] [PubMed] [Google Scholar]
- 12.Wu LL, Dunning KR, Yang X, Russell DL, Lane M, Norman RJ, Robker RL. High-fat diet causes lipotoxicity responses in cumulus-oocyte complexes and decreased fertilization rates. Endocrinology. 2010;151:5438–5445. doi: 10.1210/en.2010-0551. [DOI] [PubMed] [Google Scholar]
- 13.Luzzo KM, Wang Q, Purcell SH, Chi M, Jimenez PT, Grindler N, Schedl T, Moley KH. High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS One. 2012;7:e49217. doi: 10.1371/journal.pone.0049217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Igosheva N, Abramov AY, Poston L, Eckert JJ, Fleming TP, Duchen MR, McConnell J. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS One. 2010;5:e10074. doi: 10.1371/journal.pone.0010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu LL, Russell DL, Wong SL, Chen M, Tsai TSS, St John JC, Norman RJ, Febbraio Ma, Carroll J, Robker RL. Mitochondrial dysfunction in oocytes of obese mothers: transmission to offspring and reversal by pharmacological endoplasmic reticulum stress inhibitors. Development. 2015;142:681–691. doi: 10.1242/dev.114850. [DOI] [PubMed] [Google Scholar]
- 16.Kendig MD, Ekayanti W, Stewart H, Boakes RA, Rooney K. Metabolic Effects of Access to Sucrose Drink in Female Rats and Transmission of Some Effects to Their Offspring. PLoS One. 2015;10:e0131107. doi: 10.1371/journal.pone.0131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D'Alessandro ME, Oliva ME, Ferreira MR, Selenscig D, Lombardo YB, Chicco A. Sucrose-rich feeding during rat pregnancy-lactation and/or after weaning alters glucose and lipid metabolism in adult offspring. Clin Exp Pharmacol Physiol. 2012;39:623–9. doi: 10.1111/j.1440-1681.2012.05720.x. [DOI] [PubMed] [Google Scholar]
- 18.Munilla MA, Herrera E. Maternal hypertriglyceridemia during late pregnancy does not affect the increase in circulating triglycerides caused by the long-term consumption of a sucrose-rich diet by rats. J Nutr. 2000;130:2883–8. doi: 10.1093/jn/130.12.2883. [DOI] [PubMed] [Google Scholar]
- 19.Jen KL, Rochon C, Zhong SB, Whitcomb L. Fructose and sucrose feeding during pregnancy and lactation in rats changes maternal and pup fuel metabolism. J Nutr. 1991;121:1999–2005. doi: 10.1093/jn/121.12.1999. [DOI] [PubMed] [Google Scholar]
- 20.Berdanier CD. Effect of maternal sucrose intake on the metabolic patterns of mature rat progeny. Am J Clin Nutr. 1975;28:1416–21. doi: 10.1093/ajcn/28.12.1416. [DOI] [PubMed] [Google Scholar]
- 21.Bes-Rastrollo M, Sayon-Orea C, Ruiz-Canela M, Martinez-Gonzalez MA. Impact of sugars and sugar taxation on body weight control: A comprehensive literature review. Obesity. 2016;24:1410–1426. doi: 10.1002/oby.21535. [DOI] [PubMed] [Google Scholar]
- 22.Yang Q, Zhang Z, Gregg EW, Flanders WD, Merritt R, Hu FB. Added sugar intake and cardiovascular diseases mortality among US adults. JAMA Intern Med. 2014;174:516–24. doi: 10.1001/jamainternmed.2013.13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh GM, Micha R, Khatibzadeh S, Lim S, Ezzati M, Mozaffarian D. Global Burden of Diseases Nutrition and Chronic Diseases Expert Group (NutriCoDE), Estimated Global, Regional, and National Disease Burdens Related to Sugar-Sweetened Beverage Consumption in 2010. Circulation. 2015;132:639–66. doi: 10.1161/CIRCULATIONAHA.114.010636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tennessen JM, Barry WE, Cox J, Thummel CS. Methods for studying metabolism in Drosophila. Methods. 2014;68:105–115. doi: 10.1016/j.ymeth.2014.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker KD, Thummel CS. Diabetic larvae and obese flies-emerging studies of metabolism in Drosophila. Cell Metab. 2007;6:257–266. doi: 10.1016/j.cmet.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Na J, Musselman LP, Pendse J, Baranski TJ, Bodmer R, Ocorr K, Cagan R. A Drosophila model of high sugar diet-induced cardiomyopathy. PLoS Genet. 2013;9:e1003175. doi: 10.1371/journal.pgen.1003175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birse RT, Choi J, Reardon K, Rodriguez J, Graham S, Diop S, Ocorr K, Bodmer R, Oldham S. High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metab. 2010;12:533–544. doi: 10.1016/j.cmet.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diop SBB, Bisharat-Kernizan J, Birse RTT, Oldham S, Ocorr K, Bodmer R. PGC-1/Spargel Counteracts High-Fat-Diet-Induced Obesity and Cardiac Lipotoxicity Downstream of TOR and Brummer ATGL Lipase. Cell Rep. 2015;10:1572–1584. doi: 10.1016/j.celrep.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bastock R, St Johnston D, Huynh JR, Johnston DS, Fuller MT, Spradling AC, Morrison SJ, Spradling AC, Klovstad M, Abdu U, Schupbach T, Klattenhoff C, Theurkauf W, Poulton JS, Deng WM, Steinhauer J, Kalderon D, Palacios IM, Johnston DS, Santos AC, Lehmann R, Starz-Gaiano M, Lehmann R, Bilder D, Montell DJ, Naora H, Montell DJ. Drosophila oogenesis. Curr Biol. 2008;18:R1082-7. doi: 10.1016/j.cub.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Hsu HJ, LaFever L, Drummond-Barbosa D. Diet controls normal and timorous germline stem cells via insulin-dependent and -independent mechanisms in Drosophila. Dev Biol. 2008;313:700–12. doi: 10.1016/j.ydbio.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu HJ, Drummond-Barbosa D. Insulin levels control female germline stem cell maintenance via the niche in Drosophila. Proc Natl Acad Sci U S A. 2009;106:1117–21. doi: 10.1073/pnas.0809144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drummond-Barbosa D, Spradling AC. Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev Biol. 2001;231:265–78. doi: 10.1006/dbio.2000.0135. [DOI] [PubMed] [Google Scholar]
- 33.Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–566. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 34.Mendes CC, Mirth CK. Stage-Specific Plasticity in Ovary Size Is Regulated by Insulin/Insulin-Like Growth Factor and Ecdysone Signaling in Drosophila. Genetics. 2016;202:703–19. doi: 10.1534/genetics.115.179960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valtonen TM, Kangassalo K, Polkki M, Rantala MJ. Transgenerational effects of parental larval diet on offspring development time, adult body size and pathogen resistance in Drosophila melanogaster. PLoS One. 2012;7:e31611. doi: 10.1371/journal.pone.0031611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matzkin LM, Johnson S, Paight C, Markow Ta. Preadult parental diet affects offspring development and metabolism in Drosophila melanogaster. PLoS One. 2013;8:e59530. doi: 10.1371/journal.pone.0059530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ost A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, Pantano L, Boenisch U, Itskov PMM, Stoeckius M, Ruf M, Rajewsky N, Reuter G, Iovino N, Ribeiro C, Alenius M, Heyne S, Vavouri T, Pospisilik JAA, Öst A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, Pantano L, Boenisch U, Itskov PMM, Stoeckius M, Ruf M, Rajewsky N, Reuter G, Iovino N, Ribeiro C, Alenius M, Heyne S, Vavouri T, Pospisilik JAA, Ost A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, Pantano L, Boenisch U, Itskov PMM, Stoeckius M, Ruf M, Rajewsky N, Reuter G, Iovino N, Ribeiro C, Alenius M, Heyne S, Vavouri T, Pospisilik JAA. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell. 2014;159:1352–1364. doi: 10.1016/j.cell.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Aldrich JC, Maggert KA. Transgenerational inheritance of diet-induced genome rearrangements in Drosophila. PLoS Genet. 2015;11:e1005148. doi: 10.1371/journal.pgen.1005148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buescher JL, Musselman LP, a Wilson C, Lang T, Keleher M, Baranski TJ, Duncan JG. Evidence for transgenerational metabolic programming in Drosophila. Dis Model Mech. 2013;6:1123–32. doi: 10.1242/dmm.011924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vijendravarma RK, Narasimha S, Kawecki TJ. Effects of parental larval diet on egg size and offspring traits in Drosophila. Biol Lett. 2010;6:238–241. doi: 10.1098/rsbl.2009.0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prasad MR, Prasad AJNG. Mallikarjun Shakarad, Interaction between the effects of maternal and larval levels of nutrition on pre-adult survival in Drosophila melanogaster. Evol Ecol Res. 2003;5:903–911. [Google Scholar]
- 42.Musselman LP, Fink JL, Narzinski K, Ramachandran PV, Hathiramani SS, Cagan RL, Baranski TJ. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis Model Mech. 2011;4:842–849. doi: 10.1242/dmm.007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horne I, Haritos VS, Oakeshott JG. Comparative and functional genomics of lipases in holometabolous insects. Insect Biochem Mol Biol. 2009;39:547–567. doi: 10.1016/j.ibmb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Brookheart RT, Duncan JG. Modeling dietary influences on offspring metabolic programming in Drosophila melanogaster. Reproduction. 2016;152:R79–90. doi: 10.1530/REP-15-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brookheart RT, Michel CI, Schaffer JE. As a matter of fat. Cell Metab. 2009;10:9–12. doi: 10.1016/j.cmet.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malti N, Merzouk H, Merzouk SA, Loukidi B, Karaouzene N, Malti A, Narce M. Oxidative stress and maternal obesity: feto-placental unit interaction. Placenta. 2014;35:411–416. doi: 10.1016/j.placenta.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 47.Hernández-Trejo M, Montoya-Estrada A, Torres-Ramos Y, Espejel-Núñez A, Guzmán-Grenfell A, Morales-Hernández R, Tolentino-Dolores M, Laresgoiti-Servitje E. Oxidative stress biomarkers and their relationship with cytokine concentrations in overweight/obese pregnant women and their neonates. BMC Immunol. 2017;18:3. doi: 10.1186/s12865-016-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao PM, Tabas I. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J Biol Chem. 2001;276:42468–76. doi: 10.1074/jbc.M101419200. [DOI] [PubMed] [Google Scholar]
- 49.Baltzer C, Tiefenbock SK, Marti M, Frei C, Tiefenböck SK, Marti M, Frei C. Nutrition controls mitochondrial biogenesis in the Drosophila adipose tissue through Delg and cyclin D/Cdk4. PLoS One. 2009;4:e6935. doi: 10.1371/journal.pone.0006935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiefenböck SK, Baltzer C, Egli NA, Frei C. The Drosophila PGC-1 homologue Spargel coordinates mitochondrial activity to insulin signalling. EMBO J. 2010;29:171–83. doi: 10.1038/emboj.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsushima Y, Adán C, Garesse R, Kaguni LS. Drosophila mitochondrial transcription factor B1 modulates mitochondrial translation but not transcription or DNA copy number in Schneider cells. J Biol Chem. 2005;280:16815–20. doi: 10.1074/jbc.M500569200. [DOI] [PubMed] [Google Scholar]
- 52.Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–43. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–83. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, Chi M, Cusumano A, Scheaffer S, Moley KH. Maternal Metabolic Syndrome Programs Mitochondrial Dysfunction via Germline Changes across Three Generations. Cell Rep. 2016;16:1–8. doi: 10.1016/j.celrep.2016.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trinh I, Boulianne GL. Modeling obesity and its associated disorders in Drosophila. Physiology (Bethesda) 2013;28:117–24. doi: 10.1152/physiol.00025.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Musselman LP, Fink JL, Ramachandran PV, Patterson BW, Okunade AL, Maier E, Brent MR, Turk J, Baranski TJ. Role of fat body lipogenesis in protection against the effects of caloric overload in Drosophila. J Biol Chem. 2013;288:8028–42. doi: 10.1074/jbc.M112.371047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasco MY, Leopold P. High sugar-induced insulin resistance in Drosophila relies on the lipocalin Neural Lazarillo. PLoS One. 2012;7:e36583. doi: 10.1371/journal.pone.0036583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris SNS, Coogan C, Chamseddin K, Fernandez-Kim SO, Kolli S, Keller JN, Bauer JH. Development of diet-induced insulin resistance in adult Drosophila melanogaster. Biochim Biophys Acta - Mol Basis Dis. 2012;1822:1230–1237. doi: 10.1016/j.bbadis.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luzzo KM, Wang Q, Purcell SH, Chi M, Jimenez PT, Grindler N, Schedl T, Moley KH. High fat diet induced developmental defects in the mouse: oocyte meiotic aneuploidy and fetal growth retardation/brain defects. PLoS One. 2012;7:e49217. doi: 10.1371/journal.pone.0049217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pospisilik JA, Schramek D, Schnidar H, Cronin SJF, Nehme NT, Zhang X, Knauf C, Cani PD, Aumayr K, Todoric J, Bayer M, Haschemi A, Puviindran V, Tar K, Orthofer M, Neely GG, Dietzl G, Manoukian A, Funovics M, Prager G, Wagner O, Ferrandon D, Aberger F, Hui CC, Esterbauer H, Penninger JM. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140:148–160. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 61.Ost A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, Pantano L, Boenisch U, Itskov PM, Stoeckius M, Ruf M, Rajewsky N, Reuter G, Iovino N, Ribeiro C, Alenius M, Heyne S, Vavouri T, Pospisilik JA. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell. 2014;159:1352–1364. doi: 10.1016/j.cell.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 62.Baumbach J, Hummel P, Bickmeyer I, Kowalczyk KM, Frank M, Knorr K, Hildebrandt A, Riedel D, Jäckle H, Kühnlein RP. A drosophila in vivo screen identifies store-operated calcium entry as a key regulator of adiposity. Cell Metab. 2014;19:331–343. doi: 10.1016/j.cmet.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 63.Castellanos MC, Tang JCY, Allan DW. Female-biased dimorphism underlies a female-specific role for post-embryonic Ilp7 neurons in Drosophila fertility. Development. 2013;140:3915–3926. doi: 10.1242/dev.094714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magwere T, Chapman T, Partridge L. Sex differences in the effect of dietary restriction on life span and mortality rates in female and male Drosophila melanogaster. J Gerontol A Biol Sci Med Sci. 2004;59:3–9. doi: 10.1093/gerona/59.1.b3. [DOI] [PubMed] [Google Scholar]
- 65.Tu MP, Epstein D, Tatar M. The demography of slow aging in male and female Drosophila mutant for the insulin-receptor substrate homologue chico. Aging Cell. 2002;1:75–80. doi: 10.1046/j.1474-9728.2002.00010.x. [DOI] [PubMed] [Google Scholar]
- 66.Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. An evolutionarily conserved function of the drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol. 2001;11:213–221. doi: 10.1016/S0960-9822(01)00068-9. [DOI] [PubMed] [Google Scholar]
- 67.Clark AJ, Block K. The absence of sterol synthesis in insects. J Biol Chem. 1959;234:2578–82. [PubMed] [Google Scholar]
- 68.Wang Q, Ratchford AM, Chi MMY, Schoeller E, Frolova A, Schedl T, Moley KH. Maternal diabetes causes mitochondrial dysfunction and meiotic defects in murine oocytes. Mol Endocrinol. 2009;23:1603–12. doi: 10.1210/me.2009-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Igosheva N, Abramov AY, Poston L, Eckert JJ, Fleming TP, Duchen MR, McConnell J. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS One. 2010;5:e10074. doi: 10.1371/journal.pone.0010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, Chi M, Cusumano A, Scheaffer S, Moley KH. Maternal Metabolic Syndrome Programs Mitochondrial Dysfunction via Germline Changes across Three Generations. 2016 doi: 10.1016/j.celrep.2016.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]