

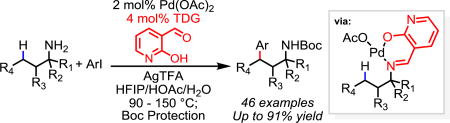
Abstract
Pd(II)-catalyzed γ-C(sp3)–H arylation of primary amines is realized by using 2-hydroxynicotinaldehyde as a catalytic transient directing group. Importantly, the catalyst and the directing group loading can be lowered to 2% and 4% respectively, thus demonstrating high efficiency of this newly designed transient directing group. Heterocyclic aryl iodides are also compatible with this reaction. Furthermore, swift synthesis of 1,2,3,4-tetrahydronaphthyridine derivatives is accomplished using this reaction.
In the past decade, directed C–H activations have been extended to a wide range of synthetically useful substrates and transformations.1 Nevertheless, the covalent installation and removal of directing groups often poses a major obstacle for their synthetic applications. In addition to adding two steps to the synthetic sequence, the reaction conditions for these steps are sometimes incompatible with labile functionalities on advanced synthetic intermediates. Thus, developing catalytic directing groups that can transiently bond to the substrate for C–H activation and subsequently dissociate reversibly is highly desirable. This strategy has been successfully applied in a number of Rh(I)-catalyzed C(sp2)–H activations.2 Recently, our group discovered that simple amino acids can serve as an effective transient directing group for Pd(II)-catalyzed C(sp3)–H functionalization of aldehydes and ketones via a reversible imine linkage, thus demonstrating the feasibility of using transient directing groups for Pd(II) catalysts, although the loading of directing groups is still high (40%).3 Notably, the bidentate coordination system provided by the imine moiety and the weakly coordinating carboxylic acid points to a new direction for our long search of transient directing groups that assist the activation of C(sp3)–H bonds (Eq 1).
Following the success of identifying a transient directing group for ketones, we wondered whether similar approach could be applied for the activation of free amines. Amines are ubiquitous structural motifs in compounds with pharmaceutical, agrochemical and agricultural importance.4 While site-selective C–H functionalization of free aliphatic amine is highly desirable as it enables rapid late-stage modifications and derivations, this process is traditionally difficult due to the formation of the unreactive Pd(RNH2)2X2 complexes,5 as well as the vulnerability of amines towards α-oxidation and electrophiles.6 Nevertheless, numerous methods have been developed for the C–H functionalization of amines with various protecting groups.7–9 An interesting β-C–H functionalization of free secondary amines (R2NH) has also been reported, although bulky α-quaternary center is required for this reaction.10 Herein, we report an efficient Pd-catalyzed C–H arylation reaction of free primary amines with aryl iodide as coupling partners under air using a catalytic transient directing group. This development, in combination with
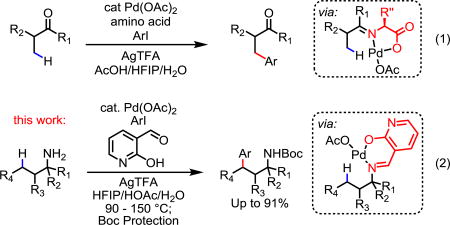 |
our previous transient directing group for ketone substrates,3 identifies imine and weakly coordinating carboxylate or its surrogate as privileged structural motifs for efficient transient directing groups.
Inspired by Jun’s Rh(I)-catalyzed aldehydic C–H activation using a reversible imine/pyridine directing group,2a we have previously investigated the development of imine/chiral oxazoline11 transient directing groups for asymmetric C–H activation of ketones and amines without success. Recently, Dong’s group reported an interesting example of γ-arylation of free primary amines via in situ generation of an imine directing group with stoichiometric 8-formylquinoline.12 However, this method is mostly limited to amines containing α-substituents and requires using highly active aryl iodonium Ar2IBF4 salts as coupling partners. The use of glovebox is also necessary, presumably to prolong the lifetime of the stoichiometric imines. The use of a transient directing group to effect γ-arylation of amines with ArI has also appeared in literature recently, albeit limited to primary C–H bonds.13 From our previous studies on imine/oxazoline and imine/pyridine transient directing groups, we believe that the strongly coordinating transient directing groups have two fundamental disadvantages for rendering the directing group catalytic: undesired strong bischelation of amino/oxazoline or pyridine with Pd(II) prevents the required formation of the imine linkage; the imine/oxazoline or pyridine bischelation with Pd(II), even if generated, is not sufficiently reactive for developing highly effective catalytic directing groups.
Since the combination of the imine moiety and the weakly coordinating carboxyl group constitutes an efficient transient directing group for ketones (Eq 1), we envisioned α-keto acids could form similar transient intermediates upon condensation with amine substrates (Table 1). Cyclohexylamine was chosen as model substrate due to its abundancy and relative high boiling point. Boc protection of the arylated amine was performed for ease of separation and analysis. Our initial experimental exploration was largely based on our previous reaction conditions.3 10 eq of H2O was added to facilitate the imine hydrolysis after C–H activation. We were encouraged to find that trace amount of arylated product 2a19 was detected with glyoxylic acid (TDG1).13 Slightly improved yield was obtained when glyoxylic acid was replaced with the more stable phenylglyoxylic acid (TDG2). Gratifyingly, replacing the carboxyl group with an acidic phenol afforded the desired product in 62% yield (TDG3). Since 2-hydroxypyridine (pyridone) has been used as carboxyl surrogate in our previous ligand design for meta-C–H activation,14 we tested commercially available 2-hydroxynicotinaldehyde as the catalytic transient directing group. To our delight, the reaction proceeded to completion and afforded 2a19 in 94% yield (TDG4). Introducing an electron-withdrawing CF3 group to the hydroxypyridine directing group reduced the yield (TDG5). TDG6 was completely unreactive, which suggests that the 7-membered ring bischelation is not reactive. Other bidentate coordination systems such as the imine/pyridine and imine/quinoline systems provided negligible yields regardless the involvement of the 5-membered or the 6-membered ring bischelation (TDG7–8), confirming the importance of the weakly coordinating anionic hydroxyl group.
Table 1.

|
Conditions: 1a (0.2 mmol), 4-CO2MePhI (0.4 mmol), Pd(OAc)2 (10 mol%), TDG (20 mol%), AgTFA (0.4 mmol), HFIP/HOAc = 19/1 (1.0 mL), H2O (2.0 mmol), 120 °C, 12 h.
Yields were determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard.
A series of control experiments were also conducted. The reaction did not proceed in the absence of the transient directing group. Simple 2-hydroxypyridine did not give any product, which confirms the importance of the imine generation. Furthermore, the bidentate chelation mode of the imine and hydroxyl moieties in TDG4 was also shown to be crucial in this reaction as changing their spatial arrangements provided trace products (TDG 9).
With the optimized conditions in hand, we next investigated the scope of the aryl iodide coupling partners. We were pleased to find that γ-C(sp3)–H arylation of 1a with a vast variety of aryl iodides proceeded smoothly to provide an efficient access of 3-aryl cyclohexylamines with good to excellent yields (Table 2), which were found to be applicable in the synthesis of potent antitumor reagents.15 Simple iodobenzene and various other methyl and phenyl substituted aryl iodides are well tolerated, affording the desired products in excellent yields (2a1–2a5). Electron rich aryl iodides with alkoxy substituents afforded the corresponding products in good yields (2a6–2a8). Halogenated aryl iodides containing fluoro, chloro and bromo substituents are also tolerated (2a9–2a11, 2a13–2a14). When 1,3-diiodobenzene was employed as the coupling partner, only one iodide was activated (2a12). Electron deficient aryl iodides bearing trifluoromethyl, nitro, methyl ketone and ester substituents are all well tolerated, providing consistently good to excellent yields (2a15–2a17, 2a19). Notably, reactive aldehyde functionality on the aryl iodide remained intact during the reaction (2a18). Furthermore, sterically demanding aryl iodides bearing substitutions at the ortho position are also compatible with this protocol (2a7, 2a9, 2a11). While arylation of cyclohexylamine with heterocyclic aryl iodide gave less than 10% yield, acyclic alkyl amine displayed excellent compatibility with a range of heteroaryl iodides. Pyridine based aryl iodides with different substitutions such as fluoro, chloro, bromo and trifluoromethyl groups at different positions are well tolerated, providing 50–70% yields (2b1–2b6, 2b8). Even electron donating methoxy group is also compatible (2b7). Various other thiophene, quinoline and quinoxaline based heterocyclic aryl iodides are also tolerated, providing moderate yields (2b9–2b12).
Table 2.

|
Conditions: 1a–b (0.2 mmol), ArI (0.4 mmol), Pd(OAc)2 (10 mol%), TDG4 (20 mol%), AgTFA (0.4 mmol), HFIP/HOAc = 19/1 (1.0 mL), H2O (2.0 mmol), 120 °C, 12 h.
Isolated yields.
130 °C, 48h.
Next we surveyed the amine scope of this γ-C(sp3)–H arylation. We were pleased to find that our protocol was applicable to a variety of free aliphatic amines. The arylation of methyl C–H bonds in simple free aliphatic amines such as propylamine, isobutylamine, and 2-methylbutylamine proceeded selectively at the γ position with good yields (2c–e). Aliphatic amines bearing one or two methyl groups at the α-substitution are also well tolerated (2f–g). In comparison to aliphatic amines with α-substituent, those without α-substituent are usually more difficult substrates in C–H activations,16 which presumably attributes to both the lack of the Thorpe-Ingold effect and the increased susceptibility to oxidation and electrophiles. However, our system is effective in both α-substituted and non-substituted substrates. Various functionalities such as phenyl, ether and even additional free amine (the di-free amine was used in 2j and subsequently di-Boc protected) are compatible with this catalytic system, providing the corresponding products in moderate yields (2h–j). Furthermore, methylene C–H arylations of both cyclic and acyclic substrates are achieved. Simple acyclic aliphatic amines of different lengths and bearing methyl α-substitution proceeded with moderate to good efficiencies (2k–l, 2m). Cyclic amines such as cyclopentyl- and cyclooctyl- amines are also tolerated, affording the desired products as a single disastereomer (2n–o).
To demonstrate the synthetic utility of this reaction, we were pleased to find that the catalyst and directing group loading can be lowered to sub 5%, thus rendering the transient directing group catalytic (Scheme 1a). When the reaction was scaled up to 2 mmol, the catalyst and template could be further lowered to 2% and 4% respectively. The desired pure arylated free amine product could be obtained in 61% isolated yield following simple acid-base extraction protocols without any further purification (Scheme 1b). Furthermore, the C–H arylation of 1b with 2-fluoro-4-iodopyridine provides a facile access to 1,2,3,4-tetrahydronaphthyridine derivatives, which exhibit important biological activities.17 SNAr of amine to the pyridyl fluoride spontaneously took place in a one-pot fashion without any reaction work up, affording 2q in 70% isolated yield (Scheme 1c).
Scheme 1.

Scale up Reaction and Synthetic Applications
In conclusion, we have developed an unprecedented Pd(II)-catalyzed γ-C(sp3)−H arylation of aliphatic amines with aryl iodides as the coupling partners using a commercially available catalytic transient directing group. Methyl as well as cyclic and acyclic methylene C−H bonds were functionalized with good efficiencies. Notably, this catalytic system works well with aliphatic amines bearing no substituent and one or two substituents at the α position. In addition, straightforward ring closure by amine nucleophilic addition provides convenient access to valuable heterocyclic motif of 1,2,3,4-tetrahydronaphthyridine.
Supplementary Material
Table 3.

|
Conditions: 1c–o (0.2 mmol), 4-CO2MePhI (0.4 mmol), Pd(OAc)2 (10 mol%), TDG4 (20 mol%), AgTFA (0.4 mmol), HFIP/HOAc = 19/1 (1.0 mL), H2O (2.0 mmol), 120 °C, 12 h.
Isolated yields.
90 °C.
50 mol% TDG4.
150 °C, HFIP/HOAc = 2:1, 30 eq H2O.
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, the NIH (NIGMS, 2R01 GM084019) and Bristol-Myers Squibb for financial support.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.For selected reviews, see: Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273.Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058.Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e.Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n.Arockiam PB, Bruneau C, Dixneuf PH. Chem. Rev. 2012;112:5879. doi: 10.1021/cr300153j.Huang Z, Lim HN, Mo F, Young MC, Dong G. Chem. Soc. Rev. 2015;44:7764. doi: 10.1039/c5cs00272a.
- 2.For leading references, see: Jun C-H, Lee H, Hong J-B. J. Org. Chem. 1997;62:1200.Bedford RB, Coles SJ, Hursthouse MB, Limmert ME. Angew. Chem., Int. Ed. 2003;42:112. doi: 10.1002/anie.200390037.Grünanger CU, Breit B. Angew. Chem., Int. Ed. 2008;47:7346. doi: 10.1002/anie.200802296.Lightburn TE, Dombrowski MT, Tan KL. J. Am. Chem. Soc. 2008;130:9210. doi: 10.1021/ja803011d.Mo F, Dong G. Science. 2014;345:68. doi: 10.1126/science.1254465.
- 3.Zhang F-L, Hong K, Li T-J, Park H, Yu J-Q. Science. 2016;351:252. doi: 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Nugent TC. Chiral Amine Synthesis: Methods, Developments and Applications. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2010. [Google Scholar]; (b) Vitaku E, Smith DT, Njardarson JT. J. Med. Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]; (c) Jeanmart S, Edmunds AJF, Lamberth C, Pouliot M. Bioorg. Med. Chem. 2016;24:317. doi: 10.1016/j.bmc.2015.12.014. [DOI] [PubMed] [Google Scholar]
- 5.Vicente J, Saura-Llamas I, Palin MG, Jones PG, Ramírez de Arellano MC. Organometallics. 1997;16:826. [Google Scholar]
- 6.Corey EJ, Achiwa K. J. Am. Chem. Soc. 1969;91:1429.Hartwig JF, Richards S, Barañano D, Paul F. J. Am. Chem. Soc. 1996;118:3626.Wolfe JP, Wagaw S, Buchwald SL. J. Am. Chem. Soc. 1996;118:7215.Sigman MS, Eaton BE. Tetrahedron Lett. 1993;34:5367. Ryland, B.L.Stahl SS. Angew. Chem., Int. Ed. 2014;53:8824. doi: 10.1002/anie.201403110.For a review on oxidation of amine to imine, see: Largeron M. Eur. J. Org. Chem. 2013:5225.
- 7.For examples with amide protecting group, see: Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f.Neumann JJ, Rakshit S, Dröge T, Glorius F. Angew. Chem., Int. Ed. 2009;48:6892. doi: 10.1002/anie.200903035.Shabashov D, Daugulis O. J. Am. Chem. Soc. 2010;132:3965. doi: 10.1021/ja910900p.He G, Chen G. Angew. Chem., Int. Ed. 2011;50:5192. doi: 10.1002/anie.201100984.He G, Zhao Y, Zhang S, Lu C, Chen G. J. Am. Chem. Soc. 2012;134:3. doi: 10.1021/ja210660g.Nadres ET, Daugulis O. J. Am. Chem. Soc. 2012;134:7. doi: 10.1021/ja210959p.Zhang S-Y, He G, Zhao Y, Wright K, Nack WA, Chen G. J. Am. Chem. Soc. 2012;134:7313. doi: 10.1021/ja3023972.Zhang S-Y, He G, Nack WA, Zhao Y, Li Q, Chen G. J. Am. Chem. Soc. 2013;135:2124. doi: 10.1021/ja312277g.Ye X, He Z, Ahmed T, Weise K, Akhmedov NG, Petersen JL, Shi X. Chem. Sci. 2013;4:3712.Fan M, Ma D. Angew. Chem., Int. Ed. 2013;52:12152. doi: 10.1002/anie.201306583.Zhang L-S, Chen G, Wang X, Guo Q-Y, Zhang X-S, Pan F, Chen K, Shi Z-J. Angew. Chem., Int. Ed. 2014;53:3899. doi: 10.1002/anie.201310000.Ling P-X, Fang S-L, Yin X-S, Chen K, Sun B-Z, Shi B-F. Chem.–Eur. J. 2015;21:17503. doi: 10.1002/chem.201502621.Xu J-W, Zhang Z-Z, Rao W-H, Shi B-F. J. Am. Chem. Soc. 2016;138:10750. doi: 10.1021/jacs.6b05978.
- 8.For examples with sulfonamide protecting group, see: Rodriguez N, Romero-Revilla JA, Fernandez-Ibanez MA, Carretero JC. Chem. Sci. 2013;4:175.Chu L, Wang X-C, Moore CE, Rheingold AL, Yu J-Q. J. Am. Chem. Soc. 2013;135:16344. doi: 10.1021/ja408864c.Chan KSL, Wasa M, Chu L, Laforteza BN, Miura M, Yu J-Q. Nat Chem. 2014;6:146. doi: 10.1038/nchem.1836.Chu L, Xiao K-J, Yu J-Q. Science. 2014;346:451. doi: 10.1126/science.1258538.Chan KSL, Fu H-Y, Yu J-Q. J. Am. Chem. Soc. 2015;137:2042. doi: 10.1021/ja512529e.Jiang H, He J, Liu T, Yu J-Q. J. Am. Chem. Soc. 2016;138:2055. doi: 10.1021/jacs.5b13462.
- 9.For examples with other protecting groups, see: Espino CG, Fiori KW, Kim M, Du Bois J. J. Am. Chem. Soc. 2004;126:15378. doi: 10.1021/ja0446294.Kim M, Mulcahy JV, Espino CG, Du Bois J. Org. Lett. 2006;8:1073. doi: 10.1021/ol052920y.Spangler JE, Kobayashi Y, Verma P, Wang D-H, Yu J-Q. J. Am. Chem. Soc. 2015;137:11876. doi: 10.1021/jacs.5b06740.Huang Z, Wang C, Dong G. Angew. Chem., Int. Ed. 2016;55:5299. doi: 10.1002/anie.201600912.Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS. Nature. 2016;531:220. doi: 10.1038/nature16957.
- 10.(a) McNally A, Haffemayer B, Collins BSL, Gaunt MJ. Nature. 2014;510:129. doi: 10.1038/nature13389. [DOI] [PubMed] [Google Scholar]; (b) He C, Gaunt MJ. Angew. Chem., Int. Ed. 2015;54:15840. doi: 10.1002/anie.201508912. [DOI] [PubMed] [Google Scholar]
- 11.Giri R, Chen X, Yu J-Q. Angew. Chem. 2005;117:2150–2153. Angew. Chem. Int. Ed. 2005, 44, 2112 – 2115. [Google Scholar]
- 12.Xu Y, Young MC, Wang C, Magness DM, Dong G. Angew. Chem., Int. Ed. 2016;55:9084. doi: 10.1002/anie.201604268. [DOI] [PubMed] [Google Scholar]
- 13.While we are submitting this manuscript, γ-arylation of alkyl amines using transient directing group TDG1 was reported: Liu Y, Ge H. Nature Chem. 2016 advance online publication.This transient directing group is largely limited to amines containing α-quaternary centers and not compatible with acyclic methylene C–H bonds.
- 14.Wang P, Farmer ME, Huo X, Jain P, Shen P-X, Ishoey M, Bradner JE, Wisniewski SR, Eastgate MD, Yu J-Q. J. Am. Chem. Soc. 2016;138:9269. doi: 10.1021/jacs.6b04966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siliphaivanh P, Methot J, Lipford KA, Molinari D, Sloman DL, Witter D, Zhou H, Boyce C, Huang X, Lim J, Guerin D, Karunakaran GB, Bakshi RK, Liu Z, Fu J, Wan Z, Liu W. PCT Int. 2016:100050. [Google Scholar]
- 16.In most cases, directed C(sp3)-H functionalization only applies for amines bearing α-substituent, see ref 7–8. Reduced yields were obtained with amines bearing no α-substituent in ref. 7f, 7g, 7l, 8c, 11 & 13.
- 17.For selected examples, see: Wang J, Breslin MJ, Coleman PJ, Duggan ME, Hunt CA, Hutchinson JH, Leu C-T, Rodan SB, Rodan GA, Duong LT, Hartman GD. Bioorg. Med. Chem. Lett. 2004;14:1049. doi: 10.1016/j.bmcl.2003.11.036.Manley PJ, Miller WH, Uzinskas IN. PCT Int. 2001:017959.Nagarajan SR, Khanna IK, Clare M, Gasiecki A, Rogers T, Chen B, Russell M, Lu H-F, Yi Y, Huff RM. 0092538. US Patent. 2004
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.