

Abstract
Introduction
The consequences of chronic disease are vast and unremitting; hence, understanding the pathogenic mechanisms mediating such disorders holds promise to identify therapeutics and diminish the consequences. The ligands of the receptor for advanced glycation end products (RAGE) accumulate in chronic diseases, particularly those characterized by inflammation and metabolic dysfunction. Although first discovered and reported as a receptor for advanced glycation end products (AGEs), the expansion of the repertoire of RAGE ligands implicates the receptor in diverse milieus, such as autoimmunity, chronic inflammation, obesity, diabetes, and neurodegeneration.
Areas covered
This review summarizes current knowledge regarding the ligand families of RAGE and data from human subjects and animal models on the role of the RAGE axis in chronic diseases. The recent discovery that the cytoplasmic domain of RAGE binds to the formin homology 1 (FH1) domain, DIAPH1, and that this interaction is essential for RAGE ligand-stimulated signal transduction, is discussed. Finally, we review therapeutic opportunities targeting the RAGE axis as a means to mitigate chronic diseases.
Expert commentary
With the aging of the population and the epidemic of cardiometabolic disease, therapeutic strategies to target molecular pathways that contribute to the sequelae of these chronic diseases are urgently needed. In this review, we propose that the ligand/RAGE axis and its signaling nexus is a key factor in the pathogenesis of chronic disease and that therapeutic interruption of this pathway may improve quality and duration of life.
Keywords: glycation, RAGE, receptor for AGE, DIAPH1, diabetes, obesity, neurodegeneration, inflammation
1.0 Introduction
The receptor for advanced glycation endproducts (RAGE) is implicated in the pathogenesis of chronic diseases and evidence from human subjects and animal models localizes RAGE and its ligand to the cell types implicated in the pathogenesis of these diseases. RAGE is expressed at low levels in most tissues in the human and the mouse in homeostasis. The exception to this is in lung tissue, in which RAGE is more highly expressed in the basal state in the alveolar type 1 epithelial cell [1]. In most other tissues, the expression of RAGE is low in the absence of stress and is upregulated in disease settings such as obesity [2], diabetes, neurodegeneration such as Alzheimer’s disease and amyotrophic lateral sclerosis (ALS) [3], malignant disorders [4] and autoimmune/inflammatory conditions [5], as examples. Of note, the production and accumulation of RAGE ligands also increases in these disease milieus, thereby implicating the ligand-RAGE axis in the pathogenesis of these disorders and their complications. Interestingly, in lung cancer, however, expression of RAGE is actually lower than in normal lung tissue [1]; the reasons for this apparent paradox are not yet clear. In the sections to follow, we detail the ligand families of RAGE, the evidence from human subjects and animal models linking RAGE to the pathogenesis of chronic diseases and novel insights into RAGE signal transduction. We conclude with the array of strategies under way to therapeutically target the RAGE axis.
2.0 RAGE binds to a diverse repertoire of ligands
2.1. Advanced Glycation Endproducts: The First Ligands of RAGE to be Described
Advanced glycation endproducts (AGEs) form in diverse settings through endogenous mechanisms such as hyperglycemia, aging, oxidative stress, and renal failure. Exogenous sources of AGEs have also been reported, such as food-derived AGEs and AGEs found in tobacco products [6]. Central intermediates in the pathway to AGE formation include the reactive carbonyl species, glyoxal, methylglyoxal and deoxyglucosone. These key species are the precursors to the AGEs, a heterogeneous group of structures, which includes specific AGEs such as carboxymethyl lysine (CML), carboxyethyl lysine (CEL), methylglyoxal lysine dimer (MOLD), glyoxal lysine dimer (GOLD), and glycolic acid lysine amide (GALA) [7]. In the pathways of pre-AGE and AGE regulation, the enzymes glyoxalase (GLO) 1 and 2 play key roles, in that they detoxify key AGE precursors such as methylglyoxal, thereby reducing AGE burden [8]. We discovered in studies performed in murine diabetic kidneys expressing or devoid of Ager (gene encoding RAGE), that despite equal degrees of hyperglycemia, diabetic mice devoid of Ager display lower levels of methylglyoxal and AGEs; this was likely accounted for, at least in part, by significantly higher levels of GLO1 [9]. At this time, the precise mechanism(s) by which RAGE downregulates Glo1 remain to be elucidated.
2.2 Non-AGE ligands of RAGE
In addition to AGEs, RAGE is also a signal transduction receptor for distinct classes of ligands (Table 1). RAGE transduces the signals of pro-inflammatory and pro-migration molecules, the S100/calgranulins and high mobility group box 1 (HMGB1) [10]. These ligand families of RAGE are involved in acute and chronic inflammation and in cancer, thus suggesting plausible mechanistic links between the development of cancers from sites of antecedent inflammation, at least in part through RAGE and its ligand families [11].
Table 1.
Examples of non-AGE ligands of RAGE.
| S100/calgranulins (e.g., S100A12, S100B, S100P, S100A8/A9) |
| High Mobility Group Box 1 (HMGB1) |
| Lysophosphatidic acid (LPA) |
| Amyloid-β peptide |
| Complement 1q |
| Phosphatidylserine |
| Leukocyte β2 integrin, Mac1 |
RAGE is also a receptor for such ligands as oligomeric forms of amyloid β-peptide, phosphatidylserine, lysophosphatidic acid (LPA) and complement 1q [10]. Importantly, AGEs may stimulate and advance amyloidogenesis mechanisms, thus pinpointing glycation as a possible central nexus in the generation of the RAGE ligand families [12]. Others reported that glycation exacerbates the neuronal toxicity of amyloid-β-peptide [13].
3.0 Soluble Forms of RAGE – Tracking Receptor Activity In Vivo
In addition to the cell-surface and membrane-embedded form of RAGE that is capable of binding ligand and triggering signal transduction, soluble forms of RAGE have been detected in the circulation (Table 2) [14]. There are two detectable forms of soluble RAGE; the first is a cell surface cleaved form of RAGE that is released by the action of molecules such as A Disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and matrix metalloproteinases (MMPs). This form of sRAGE appears to represent about 80% of the total circulating sRAGE. The other form is known as endogenous secretory, es RAGE (or RAGEv1), and is produced through alternative splicing of the AGER gene. Less is currently known about the mechanisms that regulate the production of esRAGE.
Table 2.
Soluble RAGEs.
| Form | Source | Method of Detection |
|---|---|---|
| Soluble RAGE | Cell surface cleavage by ADAM10 and MMPs | ELISA |
| Es RAGE | Alternatively spliced mRNA form | ELISA |
The circulating forms of RAGE are likely not to be inert; many studies have reported on the detection of soluble RAGEs using commercially-available ELISAs. Although these studies report relationships between levels of sRAGE and esRAGE and disease propensity or extent in a range of RAGE ligand-linked disorders, the overall conclusions are not congruent. For example, Moriya and colleagues showed that low levels of sRAGE and esRAGE were associated with high carotid intima media thickness scores; only low sRAGE, however, was associated with calcification of carotid plaques [15]. In a distinct study, Basta and colleagues showed that higher levels of circulating sRAGE were associated with symptomatic carotid atherosclerosis [16]. What are the reasons that underlie this apparent discrepancy? Possible confounding factors not often taken into account in the reporting of such studies include renal function, status of AGER polymorphisms, confounding illness and disease, and sample acquisition and storage (including the number of freeze-thaw cycles). Also, it is not always clarified if the age of the subjects is factored into the reporting of the sRAGE levels.
Despite these caveats, research has illustrated that levels of sRAGE and esRAGE may be mutable upon introduction of distinct forms of intervention, such as pharmacological agents. Two studies reported, for example, that subjects treated with statins demonstrated increases in the levels of soluble RAGE [17, 18] and that exercise training resulted in increased circulating levels of esRAGE [19].
Taken together, more research is needed to determine the sensitivity and specificity of tracking levels of sRAGE in human disease. If and by what mechanisms and conditions sRAGEs play beneficial vs. deleterious roles (or neither) in homeostasis and response to disease remains to be dissected. Notwithstanding these caveats, studies testing administration of soluble RAGE in vivo in animal models suggested its beneficial role as a means to sequester ligands and block their pathological interactions with the cell surface receptor.
4.0 Studies in animal models
Studies in animal models support that pharmacological antagonism or genetic deletion of Ager in mice is protective in disease models in which the family of RAGE ligands is known to accumulate. In the sections to follow, we review some of the recent studies buttressing the notion that RAGE may be a tractable target for therapeutic intervention in chronic diseases.
4.1 Diabetic complications
Recent studies focused on the microvascular complications of diabetes have underscored roles for RAGE in the pathogenesis of diabetes-associated retinopathy [20]. In mice rendered insulin-deficient with streptozotocin, retinopathic changes occur in the eye over time. In diabetic mice, increased expression of RAGE in the retina was demonstrated particularly after 12 weeks of hyperglycemia. In diabetic mice devoid of Ager, compared to the wild type control animals, despite equal degrees of hyperglycemia, less accumulation of the pre-AGE methylglyoxal resulted, in parallel with significant protection against increased vascular permeability, leukostasis, activation of microglia and the formation of acellular capillaries. In contrast, no protection against pericyte loss was observed in this model [20].
In diabetic kidney disease, experiments using homozygous Ager null mice bred into the OVE26 background revealed significant reduction in nephromegaly, mesangial sclerosis, thickening of the glomerular basement membrane, podocyte effacement, albuminuria and reduced inulin clearance [9]. Contributions of myeloid Ager in the pathogenesis of diabetes-associated nephropathy were noted in wild type animals lethally irradiated and reconstituted with Ager null vs. wild type bone marrow. Streptozotocin-treated diabetic mice that received Ager null bone marrow displayed partial protection against nephropathic changes, with reduced excretion of urinary albumin and podocyte loss and less tubulointerstitial injury and fibrosis. Functionally, reconstitution with Ager null bone marrow was associated with less impairment in creatinine clearance vs. the mice receiving wild type bone marrow [21]. However, in that model, it was interesting to note that despite the significant evidence of protection against nephropathic pathology, no protection against glomerulosclerosis and accumulation of collagen was noted. Such findings suggest distinct contributions of parenchymal RAGE vs. RAGE-expressing infiltrating myeloid cells in the pathogenesis of diabetes-associated nephropathy.
Roles for RAGE in diabetic cardiomyopathy have been suggested by a number of studies in which either pharmacological or genetic approaches were taken to address these concepts [22]. Recent work has suggested that RAGE binds to the β1 adrenergic receptor and that this interaction promotes the activation of Ca2+/calmodulin-dependent kinase II (CaMKII), which led to loss of cardiomyocytes and aberrant myocardial remodeling [23]. Hence, such findings reinforce that the full repertoire of RAGE-binding species may yet to be uncovered.
A critical test of the RAGE hypothesis was the extent to which RAGE blockade or Ager deletion might impact diabetic complications in which repair mechanisms were seminal. In this context, both wound healing and peripheral neuropathy are logical complications in which to test these concepts. In the diabetic peripheral nerve, it is known that in human subjects, expression of RAGE in the skin is increased, in parallel with the degree of neuropathic changes [24]. In a murine model, crush injury to the diabetic sciatic nerve was associated with an impaired regenerative process, likely due to a multitude of perturbations in the vascular, neuronal and immune cells in the affected tissue. Deletion of Ager resulted in significantly higher myelinated fiber densities and conduction velocities (motor and sensory) compared to diabetic wild type mice [25]. Key roles for myeloid RAGE were illustrated, as lethally irradiated wild type mice reconstituted with Ager null, not wild type bone marrow, resulted in similar degrees of protection. Interestingly, this work indicated that one consequence of Ager deletion was the increased “M2”-type polarization of macrophages infiltrating the regenerating diabetic sciatic nerve after crush [25]. Taken together, this work suggests that RAGE does not appear to be innately required for regenerative mechanisms in the diabetic sciatic nerve, but, rather, that RAGE activity aggravates and disengages optimal repair mechanisms.
We recently reviewed the evidence of underlying mechanisms linking RAGE to the pathogenesis of vascular disease; the reader is referred to that work for further details [10].
4.2 Neurodegeneration
In human subjects, compared to age-matched non-diseased brain and spinal cord, RAGE is expressed to higher degrees in neurodegenerative disorders such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS) and Parkinson’s disease [3, 26, 27]. RAGE is expressed in multiple distinct cell types in these tissues. Studies in animal models support pathogenic roles for RAGE in neurons, microglia and/or vascular cells in the pathogenesis of these disorders.
In murine models of AD, in addition to accumulation of RAGE ligand Aβ, recent work has suggested that diets high in AGEs may exacerbate Aβ42 deposition and impairments in spatial learning in the transgenic (Tg) 2576 mouse model, in parallel with increased expression of RAGE in the brain [28]. Cell-specific pathogenic roles for RAGE in AD were suggested by studies in which deletion of Ager in neurons was accomplished and in other studies, deletion of RAGE signaling in microglia (using a dominant negative construct strategy) also revealed protection in key functional and pathological endpoints in the AD mouse models [29, 30].
Roles for vascular cell RAGE in the pathogenesis of AD in the mouse models were reinforced by studies in which FPS-ZMI was used; FPS-ZMI is a specific inhibitor of the binding of Aβ to the V-type immunoglobulin domain of RAGE. Treatment of transgenic APPswe/0 mice with this agent resulted in reduced levels of both Aβ40 and Aβ42 in the brain, improvements in cognitive performance and cerebral blood flow, and reduced microglial activation and its consequent neuroinflammation [31]. These considerations indicate that RAGE is expressed in multiple key cell types in the human and mouse AD brain and strongly suggest pathogenic roles for the RAGE pathway in the pathogenesis of AD.
The accumulation of RAGE ligands and upregulation of RAGE expression in the human ALS spinal cord also suggested possible pathogenic roles for this pathway in ALS. In a recent study, Juranek and colleagues showed that administration of the soluble form of RAGE, sRAGE, to SOD1G93A mice significantly prolonged life span and delayed disease progression compared to vehicle treatment [32]. At the end stage of disease, spinal cord tissue revealed higher numbers of neurons and less astrogliosis in the sRAGE-treated group vs. the mice receiving vehicle. These findings support the further study of RAGE in ALS, particularly as very few therapies are currently available for this disease.
4.3 Immune/inflammatory disorders
The discovery of S100/calgranulins (initially S100A12 and S100B) and high mobility group box 1 (HMGB1) as ligands of RAGE transformed the understanding of this receptor pathway from one exclusively linked to AGEs and diabetes to a broader role in the immune/inflammatory response [33, 34]. RAGE also binds to distinct ligands such as the leukocyte β2 integrin, Mac1, thereby implicating this receptor pathway directly to immune cell behavior [35]. In addition to RAGE expression on macrophages, the receptor is also expressed on human and murine T and B lymphocytes. Herold’s group provided substantial evidence of roles for RAGE in the adaptive immune response; his work showed that RAGE played important roles in T cell proliferation and production of inflammatory cytokines and differentiation, with implications for such settings as type 1 diabetes and ovalbumin (OVA)-induced asthma [36].
Other conditions in which the ligand-RAGE pathway appears to play roles in propagation of immune/inflammatory stimulation is the settings such as arthritis, inflammatory bowel disorders, and sepsis [37–39]. Recent reviews have covered a broader range of immune/inflammatory conditions in which RAGE expression appears to be modulated and contributory to the underlying disease process [35, 40].
Finally, it is important to consider that although ligands of RAGE such as HMGB1 and certain of the S100/calgranulins may also interact with the toll like receptors (TLRs), there are many open questions in this area. It appears that depending on the cell type, the presence of homeostasis vs. distinct forms of pathological cellular stress, the ligand concentration and/or the concentration of TLRs vs. RAGE, that these ligands may preferentially bind to TLR or RAGE [41]. In our first attempts to address the interplay between RAGE and TLR in vivo, we employed a mouse model of severe stress. In mice subjected to massive liver injury, whereas Myd88 null mice displayed high levels of mortality after massive (85%) hepatectomy vs. the wild type controls, mice devoid of Ager displayed significantly higher survival versus the wild type controls. The key test of the potential interdependence of these pathways was tested in mice doubly devoid of Myd88 and Ager; the double null mice displayed comparable mortality to mice solely devoid of Myd88, suggesting that RAGE did not play critical roles in the innate and immediate response to massive liver injury [42]. In that work, we demonstrated that six hours after massive hepatectomy in mice devoid of Myd88 vs. the wild type controls, there were no differences in the expression of Ager mRNA transcripts; in both sets of mice after massive injury, Ager was upregulated vs the sham controls. Similarly, over that same time course in mice subjected to massive liver injury, expression of Tlr4 mRNA transcripts did not differ between Ager-expressing or null mice; in both sets of mice however, Tlr4 was significantly upregulated after massive hepatectomy vs. the wild type controls. These data further support the concept that at least in massive hepatectomy, RAGE and TLR4/MYD88 do not appear to directly interact. In future work, mice singly or doubly devoid of Tlr2, Tlr4 or Myd88 vs. Ager should be studied for their response to distinct severe stresses such as sepsis, massive infection or ischemia/reperfusion injury, as examples, to discern the full scope of RAGE/TLR dependence and independence.
4.4 Obesity
A burgeoning body of literature suggests strong links between the RAGE pathway and human obesity. First, in the context of expression patterns, RAGE has been shown to be highly expressed in human adipose tissue, in adipocytes, vascular cells, and in macrophages [2]. In that work, it was shown that a key RAGE ligand, carboxy methyl lysine (CML) AGE was upregulated in obese human adipose tissue, with lower circulating levels of AGEs observed in obese vs. lean individuals, suggesting that the adipose tissue RAGE might “trap” the CML-AGE ligand. Interestingly, recent studies suggest links between the Ser allele of the Gly82Ser AGER polymorphism to obesity risk and that the Ser allele was associated with higher levels of sRAGE in children [43]. A distinct study demonstrated that the Ser allele was more frequently observed in obese adolescents vs. the lean [44]. In that study, however, lower levels of sRAGE were linked to obesity [44]. In obesity and bariatric surgery, higher baseline levels of sRAGE were found to be associated with better weight loss outcomes [45]. Taken together, these considerations suggest that genetic predisposition, expression of cell surface RAGE and levels of sRAGE may be linked to the risk of obesity. What is the evidence linking RAGE to obesity in animal models?
Mice fed a high fat diet (HFD) (60% kcal/fat) are an excellent model for the study of the effect of high dietary intake of fat. In the C57BL/6 background, mice fed a high fat diet develop a time dependent increase in body mass that exceeds that of the chow-fed control animals. Insulin resistance and glucose intolerance, along with frank hyperglycemia eventually occur in this model. Levels of RAGE ligands HMGB1 and AGEs increase in the metabolic organs of mice fed HFD even before the development of diabetes, suggesting that excess dietary fat ingestion is sufficient to drive increases in RAGE ligands and engagement of the RAGE pathway [46].
To test the role of RAGE, male homozygous Ager null mice and their littermate controls were weight-matched and fed either a low fat diet (LFD) (13% fat) or HFD (60%). Compared to LFD-fed mice, HFD-fed mice displayed significantly higher body mass after three months of diet [46]. In parallel, the leaner Ager null mice were more glucose- and insulin tolerant and displayed lower levels of adipose inflammation, with lower macrophage content (both F4/80+/CD11B+ (FB) and F4/80+/CD/11B+/CD11C+ (FBC) compartments [46].
Myeloid-derived RAGE-expressing cells contributed importantly to the overall phenotype, as lethally-irradiated wild-type mice reconstituted with Ager null vs. wild type bone marrow displayed partial protection against diet induced obesity. The same profile of lower numbers of overall macrophage content (FB and FBC) was observed in these chimeric mice recipients of Ager null bone marrow [46]. In both sets of studies, global- or bone marrow-Ager deletion, food consumption was identical to that observed in the wild type control cohorts.
Finally, adult wild-type mice were treated with sRAGE either at the onset of HFD or three weeks later. Compared to vehicle-treated wild-type mice, those mice receiving sRAGE displayed significantly lower body mass despite consumption of equal caloric intake [46]. Although the mice in the intervention study did not lose weight on the sRAGE treatment, they failed to gain weight at the same rate as that observed in the animals treated with vehicle.
Taken together, these findings greatly expanded the understanding of RAGE roles in metabolism and underscored that the breadth of RAGE effects in high fat feeding extends not only to the complications of type 2 diabetes but to the origins of obesity and metabolic dysfunction.
5.0 RAGE Signal Transduction: the discovery of the Diaph1 interaction with the RAGE cytoplasmic domain
Previous studies by many laboratory groups indicated that RAGE ligands stimulated signal transduction through pathways such as MAP kinases, PI3K/Akt, and Rho GTPases and that a common factor in many cell types and conditions was RAGE-dependent activation of NF-kB and its downstream effectors linked to oxidative and proinflammatory stress. It was known that the cytoplasmic domain of RAGE was essential for RAGE-mediated signaling, as its deletion, in vitro and in vivo, protected from RAGE ligand cellular activation [47]. Yet, definitive evidence of the proximate mechanisms within the cytoplasmic domain that activated RAGE signaling were yet to be identified.
5.1 RAGE/DIAPH1: cellular and in vivo biology
To address this critical question, a yeast two hybrid assay was employed using the RAGE cytoplasmic domain as “bait” for probing within a lung library (tissue type from which RAGE was first discovered). This strategy identified the formin molecule, mammalian diaphanous-1 or DIAPH1, as a potential candidate, particularly its FH1 or formin homology 1 domain [48]. Formins have diverse functions; they are effectors of Rho GTPase signaling and they modulate actin and microtubule remodeling of the cytoskeleton, functions that are linked to cellular migration and invasion [49, 50]. These characteristics and roles of the formin molecules suggested plausible and testable links to the RAGE signal transduction pathways.
First, however, it was essential to affirm the yeast two hybrid assay result and demonstrate the interaction of the RAGE cytoplasmic domain with DIAPH1. Co-immunoprecipitation and immunofluorescence microscopy studies confirmed the interaction [48]. Next, the key test of these concepts was whether modulation of DIAPH1 expression affected RAGE ligand-stimulated signaling. In cultured cells, RAGE ligands failed to stimulate activation of Cdc42 or Rac1 in the presence of knockdown of DIAPH1 expression; in contrast, experiments using control siRNAs showed typical robust activation of these Rho GTPase signaling molecules by RAGE ligands [48]. Of note, siRNA-knockdown of DIAPH1 expression had no effect on expression of RAGE. Further, siRNA-knockdown of DIAPH1 but not control siRNAs blocked RAGE ligand-stimulated cellular migration, but had no effect on the migration-provoking effects of fetal bovine serum, which is not known to contain functionally-relevant levels of any RAGE ligands [48].
Distinct studies linked RAGE ligands to upregulation of Egr1, a key transcription factor linked to upregulation of proinflammatory and prothrombotic molecules in stresses such as hypoxia. Vascular endothelial cells devoid of Ager failed to upregulate Egr1 in hypoxic stress; the link to RAGE was revealed by experiments demonstrating that hypoxia generated AGEs, as detected by time-dependent generation of AGE epitopes in the supernatants of hypoxia- but not normoxia-exposed cells [51]. The potential role of DIAPH1 in transducing RAGE ligand signaling in regulation of Egr1 was addressed in macrophages. Bone marrow derived macrophages devoid of Ager or Diaph1 failed to upregulate Egr1 in hypoxia; the mechanisms underlying these findings were traced to signaling via protein kinase C-βII, ERK1/2 MAP kinases and c-Jun NH(2) terminal kinase [52]. Recent studies have linked the RAGE ligand signaling pathway (S100A4 and S100B) to DIAPH1 in thyroid cancer cells and murine microglia, respectively [53, 54].
The in vivo test of these concepts was an essential next step in defining the relationship between RAGE and DIAPH1. To address this concept, wild type or mice devoid of Diaph1 were subjected to femoral artery endothelial denudation injury. In this model, it was previously shown that mice devoid of Ager displayed significant protection against pathological neointimal expansion. In an analogous manner, mice devoid of Diaph1 were protected against expansion of the neointima [55]. Critically, the mechanisms were traced to smooth muscle cell (SMC) signaling in that RAGE ligands were shown to require DIAPH1 to stimulate development of lamellopodia, a key proximal step in cellular migration. The signaling mechanisms in SMCs were traced directly to RAGE ligand S100B – RAGE – DIAPH1-dependent membrane translocation of c-Src, activation of Rac1, a process that generated oxidative stress and consequent activation of PI3K/Akt and serine 9 phosphorylation of GSK-3β and consequent regulation of SMC migration [55].
5.2 RAGE/DIAPH1: biophysical studies
Shekhtman and colleagues addressed the mechanisms by which the cytoplasmic domain of RAGE bound the FH1 domain of DIAPH1 [56]. By using 15N HSQC (heteronuclear single quantum coherence) NMR (Nuclear Magnetic Resonance) spectroscopy, these authors showed that the residues in the cytoplasmic domain of RAGE displayed limited chemical shift dispersion and were mostly disordered [56]. Using heteronuclear 15N{1H} NOE (nuclear Overhauser effect) NMR, they reported that amino acids 2–15 of the RAGE cytoplasmic domain were partially ordered and that residues 16–42 were disordered. Further analysis focused on identifying putative regions in which protein-protein interactions might be feasible. This led to the suggestion that a hydrophobic patch formed by the methylene groups of Arg-5 and Gln-6 in the RAGE cytoplasmic domain might be important for protein-protein interaction [56]. NMR spectroscopic analysis revealed that residues Gln-3, Arg-4, Arg-5 and Gln-6 of the RAGE cytoplasmic domain participated in the interaction with the FH1 domain of DIAPH1. The suggested dissociation constant was Kd < 10 μM) [56]. Mutation of RAGE cytoplasmic domain Arg-5 and Gln-6 to alanine residues supported the relationship to the FH1 of DIAPH1, as the mutant showed only weak interaction with Kd =1 mM [56]. Finally, in cellular studies of primary murine aortic SMCs, introduction of the mutant (Arg-5 and Gln-6 to alanine residues) blocked RAGE ligand-evoked signal transduction, cellular migration and proliferation. Critically, however, there was no effect of the mutant on the biological responses to PDGF, which is not a RAGE ligand [56].
The structural mechanism of RAGE/DIAPH1 signal transduction was studied by Xue and colleagues [57]. Using a combination of NMR spectroscopy and protein cross-linking analyzed by mass spectrometry, the authors showed that the ligation of RAGE by its ligand, S100B, results in extensive oligomerization and subsequent increases in the molecular dimension of RAGE. DIAPH1 is a dimer [58]. Molecular modeling suggested that the increase in the RAGE dimension facilitates RAGE binding to the DIAPH1 dimer (Figure 1). Indeed, addition of S100B to HEK293 cells, which expressed fluorescently labeled RAGE and DIAPH1, resulted in the enhanced fluorescence energy transfer (FRET) between RAGE and DIAPH1; this is a strong indication of the RAGE/DIAPH1 binding. Importantly, cellular studies in primary murine aortic SMCs showed that the fluorescently-labeled RAGE and DIAPH1 are functional [57].
Figure 1.
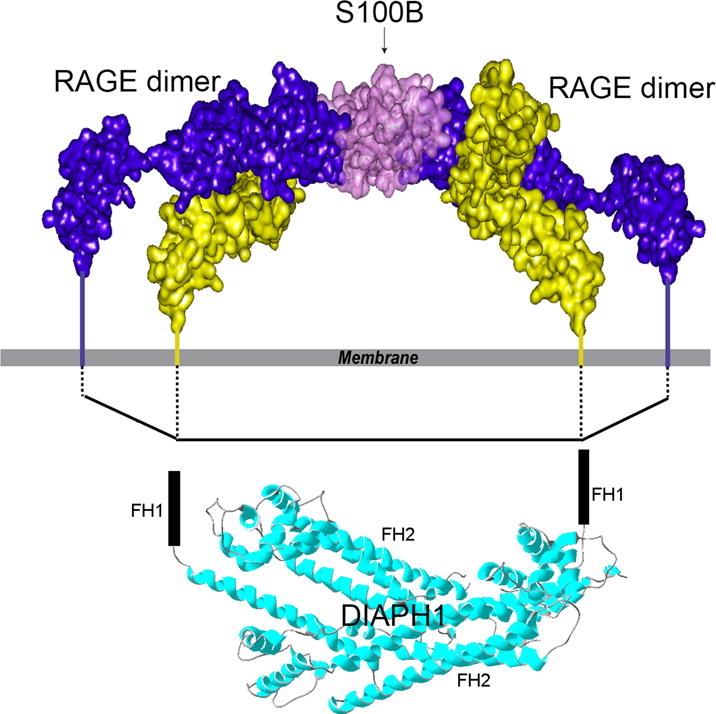
Model of S100B bound to two RAGE dimers showing the proposed interaction between RAGE cytoplasmic domains and FH1 domains of DIAPH1. Due to S100 ligation, the spacing between the RAGE cytoplasmic domains is comparable to the distance between the FH1 domains. The FH1 domains are unstructured; FH2 domains of DIAPH1 determine the distance between the FH1 domains. Reprinted and modified from Structure, 21/9, Xue et al., Change in the Molecular Dimension of a RAGE-Ligand Complex Triggers RAGE Signaling, 1509-1522, Copyright (2016), with permission from Elsevier.
5.3 RAGE/DIAPH1: small molecule antagonism
In order to refine understanding of the nature of the interaction of the RAGE cytoplasmic domain with DIAPH1 and the implications for signal transduction, a high throughput binding assay was developed in which anti-DIAPH1 IgG was used to immunoprecipitate DIAPH1 from cultured cells and then binding of GFP (green fluorescent protein)-labeled RAGE cytoplasmic domain was verified [59]. This assay was used to screen the approximately 58,000 compound DiVERSet library, CT488. Ultimately, 13 compounds were identified that bound specifically to the cytoplasmic domain of RAGE and blocked the interaction with DIAPH1. Based on NMR spectroscopy, the inhibition of RAGE-DIAPH1 binding is direct, since the interaction surface of the RAGE cytoplasmic domain with these 13 compounds overlaps with the RAGE surface required for binding to DIAPH1 [59]. Native tryptophan fluorescence titration (quenching) assays showed that the 13 compounds display nM affinity for the RAGE cytoplasmic domain [59]. Many of these compounds (1 μM) blocked the effects of RAGE ligands on stimulation of phosphorylation of Akt and ERK1/2 in murine aortic SMCs [59].
In vitro cell based assays employed primary murine and human SMCs, primary murine endothelial cells and cultured macrophage-like human THP1 cells. In primary murine and human SMCs, all 13 compounds blocked the effects of RAGE ligands on SMC migration, but had no inhibitory effect on migration provoked by a non-RAGE ligand, PDGF. In murine endothelial cells and human THP1 cells, many of the 13 compounds blocked the effects of RAGE ligands on upregulated of inflammatory Tnfa and Il6 (by real time quantitative PCR) [59].
In vivo, wild type mice were injected with RAGE ligands alone or with compounds 1–13. Four doses of compound (5 mg/kg q 12 h for four total doses by intraperitoneal route) were administered prior to RAGE ligand; six hours later, kidney was retrieved and mRNA prepared. Compared to vehicle treatment, compounds 1, 2, 3, 5, 6, 8, 9, 10, 11, 12 and 13 suppressed RAGE ligand-stimulated upregulation of Tnfa transcripts and all 13 compounds suppressed upregulation of Il6 mRNA transcripts in the kidney [59].
Finally, ex vivo, in the diabetic isolated perfused mouse heart subjected to hypoxia and reoxygenation, treatment of the isolated diabetic hearts with compounds 3, 7, 9 and 11 improved the percent functional recovery (left ventricular developed pressure, or LVDP) compared to the vehicle treatment [59].
6.0 Expert Commentary
Ongoing research throughout the world on the RAGE pathway continues to unveil the consequences of engagement of this receptor in a diverse array of metabolic, inflammatory and degenerative disorders in which the families of RAGE ligands accumulate and contribute to mediation of chronic and often unremitting diseases. The viability and normal reproductive capacity of the Ager null mouse, along with the plethora of published studies suggesting benefit and not detriment of genetic deletion in animals subjected to RAGE ligand-associated disorders, has provided provisional assurance that chronic antagonism of the receptor is likely to be safe, tolerated and efficacious. Although definitive studies from human subjects treated with RAGE antagonists are either underway or planned, many studies have reported expression levels of RAGE (and its ligands) in human diseased vs. normal tissue and have assessed single nucleotide polymorphisms (SNPs) of AGER, and levels of soluble RAGEs in multiple types of biological fluids, as biomarkers for RAGE activity in the human subject.
Despite the lack of detriment of RAGE antagonism or Ager deletion in diabetic wound healing and in the response to sciatic nerve crush in diabetes [25, 60], some studies have suggested adaptive roles for RAGE in the effective handling of Klebsiella pneumonia (pneumonia) [61], protection of the lower airways from human respiratory syncytial virus [62], the response to Mycobacterium tuberculosis pneumonia [63], and host defense in peritonitis and sepsis due to Escherichia coli [64]. In these instances, the studies used animal models (mice) and therefore the extrapolation to the human’s handling of these pathogens under conditions of partial pharmacological antagonism initiated after embryonic development remains to be tested. Further, the means and circumstances by which the animals were infected with these pathogens might not fully reflect the human’s mode of infection and biological response.
Given the research evidence that blockade/deletion of RAGE may be beneficial in chronic disease, it is not surprising that these bodies of work have stimulated efforts to antagonize RAGE and its biological actions (Table 3). For example, small molecule antagonism of the RAGE extracellular domains (to block ligand binding) [31, 65], blocking antibodies to RAGE [66, 67], soluble RAGEs [14], and RAGE peptide aptamers [68] are reported. A series of 4,6-bis(4-chlorophenyl)pyrimidine analogs was reported to bind to RAGE and block its interaction with Aβ [69]. Further, efforts to block RAGE-DIAPH1 interaction may represent a novel strategy to block RAGE actions [59].
Table 3.
Examples of approaches to RAGE antagonism.
| Small molecule antagonists of the RAGE extracellular domains |
| Anti-RAGE antibodies |
| Soluble RAGE |
| RAGE peptide aptamers |
| 4,6-bis(4-chlorophenyl)pyrimidine analogs |
| Antagonists of RAGE-DIAPH1 interaction |
| Small interfering RNAs |
In summary, as clinical trials targeting RAGE are designed, executed and the results analyzed, the RAGE hypothesis, from the origins of the RAGE discovery in bovine lung extract, to the bedside of patients with RAGE-related chronic diseases, will finally be rigorously tested.
7.0 Five- year view
Much has been learned in the course of the study of RAGE biology in the past two decades since its discovery. It is tempting to postulate that pharmacological targeting of RAGE may present a unique and feasible strategy to block the complications of RAGE ligand accumulation in chronic disease; yet, there are challenges ahead.
First, a chief challenge is to identify the optimal means and timing of intervention in the RAGE signal transduction pathway. Multiple strategies are possible; which of these holds the greatest promise to target the receptor and in what clinical conditions is not yet established. It is also possible that combination anti-RAGE strategies may be required to fully antagonize the RAGE pathway. In the next five years, at least some of the answers to these questions will likely become available as the RAGE therapeutic strategies begin to take shape in advanced clinical trials.
Second, a chief issue that arises in chronic diseases and clinical trials is identification of the best “start time” for a proposed new RAGE-directed therapy. Important questions include, will it be optimal to initiate intervention upon disease diagnosis or upon the first hint that “complications” of the disorder are evident? Will RAGE antagonism be safe in children, especially those with newly-diagnosed type 1 diabetes, for example? Will local anti-RAGE therapies be possible, particularly in settings such as the diabetic eye or inflamed skin in psoriasis or the non-healing diabetic wound? Ongoing work by many laboratories exploring the fundamental biology of RAGE and its signal transduction should uncover the answers to these questions and aid in the design of the best possible clinical trials.
Finally, in great part, the diseases associated with RAGE ligand engagement are largely chronic and therefore this impacts the logistics of clinical trials that will be required for full phase testing. In this context, a major challenge ahead is the identification of bona fide target engagement biomarkers [70, 71] to identify in short-term and in readily-accessible tissues, the status of blockade of the RAGE pathway. It is plausible that measures of the various classes of RAGE ligands may be specific to different stages of the disease process. For example, in diabetes, early and increased accumulation of AGEs may be followed by the release and local accumulation of proinflammatory ligands, such as the S100/calgranulins and HMGB1. Hence, the pattern and timing of RAGE ligand appearance may offer the opportunity to track the state of RAGE-mediated disease.
Furthermore, plasma/serum biomarkers and altered gene expression in peripheral blood mononuclear cells may be key sites for detection of RAGE activities. We predict that markers of inflammation will emerge in the next five years as the chief means to track the modulation of the RAGE signaling pathway. Such efforts are essential for feasible approaches to targeting this pathway in chronic disease trials. Experiments employed unbiased “omic” approaches may be a logical means to biomark the status of RAGE signaling activities in response to its ligands. Using diverse tissues and disease models, from diabetes to inflammation, it is plausible that a unique panel of biomarkers may emerge. Such efforts are thoroughly essential to secure the most logical and economically feasible means to advance RAGE antagonism through clinical trials and, if successful, eventually, to the local pharmacy.
8.0 Key Issues
RAGE is a multi-ligand receptor of the immunoglobulin superfamily. Its ligands accumulate in multiple acute and chronic diseases, such as obesity, diabetes and its complications, immune/inflammatory disorders, tumors and neurodegeneration.
Soluble forms of RAGE have been detected in human subject plasma/serum and are generated either by cell surface cleavage of the full length receptor or through alternative splicing and the production of endogenous secretory (es) RAGE. Many studies in the literature report associations of the levels of soluble RAGEs with disease risk or activities.
Studies in animal models support that blockade of or genetic deletion of RAGE is protective in multiple animal models in which the ligands of RAGE accumulate, such as in diabetes complications, neurodegeneration, immune/inflammatory disorders and obesity.
Signal transduction triggered by RAGE ligands requires the interaction of its cytoplasmic domain with the formin, DIAPH1. Biophysical and cellular studies support that mutation of the key amino acids in the cytoplasmic domain of RAGE (R5/Q6) to alanine residues blocks the physical interaction with DIAPH1 (FH1 domain) and suppresses RAGE ligand-stimulated signal transduction.
Multiple strategies to target / antagonize RAGE signaling are in development and early clinical trials. The pinpointing of target engagement biomarkers is a key next and critical step in efforts to deliver RAGE-based treatments to the local pharmacy.
Acknowledgments
Funding
The authors were supported by grants from the U.S. Department of Health and Human Services, National Institutes of Health (grant numbers 1R01DK109675, 1R01HL118565, 1R24DK103032 and P01HL60901).
Footnotes
Declaration of Interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Reference annotations
*Of interest
**Of considerable interest
- 1.Marinakis E, Bagkos G, Piperi C, Roussou P, Diamanti-Kandarakis E. Critical role of RAGE in lung physiology and tumorigenesis: a potential target of therapeutic intervention? Clin Chem Lab Med. 2014;52:189–200. doi: 10.1515/cclm-2013-0578. [DOI] [PubMed] [Google Scholar]
- 2*.Gaens KH, Goossens GH, Niessen PM, et al. Nepsilon-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler Thromb Vasc Biol. 2014;34:1199–1208. doi: 10.1161/ATVBAHA.113.302281. This paper describes for the first time the differential expression of RAGE protein in obese and lean human subject adipose tissue. The authors showed that RAGE expression was higher in obese vs. lean human adipose tissue and localized to adipocytes, vascular and immune cells. This work links RAGE to the pathogenesis of obesity. [DOI] [PubMed] [Google Scholar]
- 3*.Juranek JK, Daffu GK, Wojtkiewicz J, Lacomis D, Kofler J, Schmidt AM. Receptor for Advanced Glycation End Products and its Inflammatory Ligands are Upregulated in Amyotrophic Lateral Sclerosis. Front Cell Neurosci. 2015;9:485. doi: 10.3389/fncel.2015.00485. This paper reports that RAGE and its ligands are highly expressed in the spinal cord of human subjects affected with ALS compared to more basal expression in the normal spinal cord. These studies suggest that the ligands of RAGE are upregulated in chronic disease and lead to the hypothesis that RAGE might contribute to neuronal dysfunction and loss in ALS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malik P, Chaudhry N, Mittal R, Mukherjee TK. Role of receptor for advanced glycation end products in the complication and progression of various types of cancers. Biochim Biophys Acta. 2015;1850:1898–1904. doi: 10.1016/j.bbagen.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 5.Nienhuis HL, Westra J, Smit AJ, Limburg PC, Kallenberg CG, Bijl M. AGE and their receptor RAGE in systemic autoimmune diseases: an inflammation propagating factor contributing to accelerated atherosclerosis. Autoimmunity. 2009;42:302–304. doi: 10.1080/08916930902831746. [DOI] [PubMed] [Google Scholar]
- 6.Uribarri J, del Castillo MD, de la Maza MP, et al. Dietary advanced glycation end products and their role in health and disease. Adv Nutr. 2015;6:461–473. doi: 10.3945/an.115.008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henning C, Glomb MA. Pathways of the Maillard reaction under physiological conditions. Glycoconj J. 2016 doi: 10.1007/s10719-016-9694-y. published online 2016/06/14. [DOI] [PubMed] [Google Scholar]
- 8.Maessen DE, Stehouwer CD, Schalkwijk CG. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin Sci (Lond) 2015;128:839–861. doi: 10.1042/CS20140683. [DOI] [PubMed] [Google Scholar]
- 9**.Reiniger N, Lau K, McCalla D, et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes. 2010;59:2043–2054. doi: 10.2337/db09-1766. This paper reports that diabetic OVE26 mice devoid of Ager are significanlty protected from podocyte loss and glomerular sclerosis that typifies long-term diabtes. Further, this work was among the first to show that the ligand-RAGE axis downregulates Glo1, thereby leading to increased levels of methylglyoxal, which is a major AGE precursor. Thus, this work established roles for RAGE in regulation of AGE levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Diez R, Shekhtman A, Ramasamy R, Schmidt AM. Cellular mechanisms and consequences of glycation in atherosclerosis and obesity. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbadis.2016.05.005. published online 2016/05/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moravkova P, Kohoutova D, Rejchrt S, Cyrany J, Bures J. Role of S100 Proteins in Colorectal Carcinogenesis. Gastroenterol Res Pract. 2016;2016:2632703. doi: 10.1155/2016/2632703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iannuzzi C, Maritato R, Irace G, Sirangelo I. Glycation accelerates fibrillization of the amyloidogenic W7FW14F apomyoglobin. PLoS One. 2013;8:e80768. doi: 10.1371/journal.pone.0080768. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Li XH, Du LL, Cheng XS, et al. Glycation exacerbates the neuronal toxicity of beta-amyloid. Cell Death Dis. 2013;4:e673. doi: 10.1038/cddis.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt AM. Soluble RAGEs – Prospects for treating & tracking metabolic and inflammatory disease. Vascul Pharmacol. 2015;72:1–8. doi: 10.1016/j.vph.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moriya S, Yamazaki M, Murakami H, Maruyama K, Uchiyama S. Two soluble isoforms of receptors for advanced glycation end products (RAGE) in carotid atherosclerosis: the difference of soluble and endogenous secretory RAGE. J Stroke Cerebrovasc Dis. 2014;23:2540–2546. doi: 10.1016/j.jstrokecerebrovasdis.2014.05.037. [DOI] [PubMed] [Google Scholar]
- 16.Basta G, Castagnini M, Del Turco S, et al. High plasma levels of the soluble receptor for advanced glycation endproducts in patients with symptomatic carotid atherosclerosis. Eur J Clin Invest. 2009;39:1065–1072. doi: 10.1111/j.1365-2362.2009.02212.x. [DOI] [PubMed] [Google Scholar]
- 17.Tam HL, Shiu SW, Wong Y, Chow WS, Betteridge DJ, Tan KC. Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis. 2010;209:173–177. doi: 10.1016/j.atherosclerosis.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 18.Santilli F, Bucciarelli L, Noto D, et al. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med. 2007;43:1255–1262. doi: 10.1016/j.freeradbiomed.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 19.Santilli F, Vazzana N, Iodice P, et al. Effects of high-amount-high-intensity exercise on in vivo platelet activation: modulation by lipid peroxidation and AGE/RAGE axis. Thromb Haemost. 2013;110:1232–1240. doi: 10.1160/TH13-04-0295. [DOI] [PubMed] [Google Scholar]
- 20.McVicar CM, Ward M, Colhoun LM, et al. Role of the receptor for advanced glycation endproducts (RAGE) in retinal vasodegenerative pathology during diabetes in mice. Diabetologia. 2015;58:1129–1137. doi: 10.1007/s00125-015-3523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tesch G, Sourris KC, Summers SA, et al. Deletion of bone-marrow-derived receptor for AGEs (RAGE) improves renal function in an experimental mouse model of diabetes. Diabetologia. 2014;57:1977–1985. doi: 10.1007/s00125-014-3291-z. [DOI] [PubMed] [Google Scholar]
- 22.Bucciarelli LG, Ananthakrishnan R, Hwang YC, et al. RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes. 2008;57:1941–1951. doi: 10.2337/db07-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu W, Tsang S, Browe DM, et al. Interaction of beta1-adrenoceptor with RAGE mediates cardiomyopathy via CaMKII signaling. JCI Insight. 2016;1:e84969. doi: 10.1172/jci.insight.84969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park SY, Kim YA, Hong YH, Moon MK, Koo BK, Kim TW. Up-regulation of the receptor for advanced glycation end products in the skin biopsy specimens of patients with severe diabetic neuropathy. J Clin Neurol. 2014;10:334–341. doi: 10.3988/jcn.2014.10.4.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Juranek JK, Geddis MS, Song F, et al. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes. 2013;62:931–943. doi: 10.2337/db12-0632. This paper showed that in the setting of chronic diabetes, acute crush of the sciatic nerve in mice induced upregulation of RAGE and its ligands. Deletion of Ager, either globally or in the myeloid compartment, resulted in improvement in regeneration. Therefore, these key findings indicated that RAGE-dependent processes were not reqired for effetive repair of the crushed sciatic nerve. Rather, this work showed that RAGE blocked effective regeneraiton in this setting. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt AM, Sahagan B, Nelson RB, Selmer J, Rothlein R, Bell JM. The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer’s disease. Curr Opin Investig Drugs. 2009;10:672–680. [PubMed] [Google Scholar]
- 27.Vicente Miranda H, Outeiro TF. The sour side of neurodegenerative disorders: the effects of protein glycation. J Pathol. 2010;221:13–25. doi: 10.1002/path.2682. [DOI] [PubMed] [Google Scholar]
- 28.Lubitz I, Ricny J, Atrakchi-Baranes D, et al. High dietary advanced glycation end products are associated with poorer spatial learning and accelerated Abeta deposition in an Alzheimer mouse model. Aging Cell. 2016;15:309–316. doi: 10.1111/acel.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Wang Y, Yan S, et al. Genetic deficiency of neuronal RAGE protects against AGE-induced synaptic injury. Cell Death Dis. 2014;5:e1288. doi: 10.1038/cddis.2014.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang F, Lue LF, Yan S, et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deane R, Singh I, Sagare AP, et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juranek JK, Daffu GK, Geddis MS, et al. Soluble RAGE Treatment Delays Progression of Amyotrophic Lateral Sclerosis in SOD1 Mice. Front Cell Neurosci. 2016;10:117. doi: 10.3389/fncel.2016.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33**.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. This paper was the first to report that the extracellular S100/calgranulins were signal transduction ligands of RAGE and that this axis contributed to a sustained inflammatory response in vivo. [DOI] [PubMed] [Google Scholar]
- 34.Taguchi A, Blood DC, del Toro G, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 35.Chuah YK, Basir R, Talib H, Tie TH, Nordin N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int J Inflam. 2013;2013:403460. doi: 10.1155/2013/403460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akirav EM, Henegariu O, Preston-Hurlburt P, Schmidt AM, Clynes R, Herold KC. The receptor for advanced glycation end products (RAGE) affects T cell differentiation in OVA induced asthma. PLoS One. 2014;9:e95678. doi: 10.1371/journal.pone.0095678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofmann MA, Drury S, Hudson BI, et al. RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002;3:123–135. doi: 10.1038/sj.gene.6363861. [DOI] [PubMed] [Google Scholar]
- 38.Ciccocioppo R, Vanoli A, Klersy C, et al. Role of the advanced glycation end products receptor in Crohn’s disease inflammation. World J Gastroenterol. 2013;19:8269–8281. doi: 10.3748/wjg.v19.i45.8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto Y, Harashima A, Saito H, et al. Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol. 2011;186:3248–3257. doi: 10.4049/jimmunol.1002253. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez I, Romero J, Rodriguez BL, Perez-Castro R, Rojas A. The immunobiology of the receptor of advanced glycation end-products: trends and challenges. Immunobiology. 2013;218:790–797. doi: 10.1016/j.imbio.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Ibrahim ZA, Armour CL, Phipps S, Sukkar MB. RAGE and TLRs: relatives, friends or neighbours? Mol Immunol. 2013;56:739–744. doi: 10.1016/j.molimm.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 42.Zeng S, Zhang QY, Huang J, et al. Opposing roles of RAGE and Myd88 signaling in extensive liver resection. FASEB J. 2012;26:882–893. doi: 10.1096/fj.11-192997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kucukhuseyin O, Ozgen T, Karagedik EH, et al. The effects of Advanced Glycation End Products (RAGE)-374T/A and Gly82Ser variants and soluble-RAGE levels to obesity in children. Cell Mol Biol (Noisy-le-grand) 2016;62:9–14. [PubMed] [Google Scholar]
- 44.Rowisha M, El-Batch M, El Shikh T, El Melegy S, Aly H. Soluble receptor and gene polymorphism for AGE: relationship with obesity and cardiovascular risks. Pediatr Res. 2016;80:67–71. doi: 10.1038/pr.2016.55. [DOI] [PubMed] [Google Scholar]
- 45*.Parikh M, Chung M, Sheth S, et al. Randomized pilot trial of bariatric surgery versus intensive medical weight management on diabetes remission in type 2 diabetic patients who do NOT meet NIH criteria for surgery and the role of soluble RAGE as a novel biomarker of success. Ann Surg. 2014;260:617–622. doi: 10.1097/SLA.0000000000000919. discussion 622–614. This paper showed that in obese human subjects undergoing bariatric surgery, higher baseline levels of soluble RAGE were predictors of weight loss and metabolic improvement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46**.Song F, Hurtado del Pozo C, Rosario R, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. 2014;63:1948–1965. doi: 10.2337/db13-1636. This work illustrated that mice devoid of Ager – either globally or in the myeloid compartment – were protected from high fat diet induced obesity. In parallel with protection from diet induced obesity, Ager null mice fed high fat diet were protected from insulin resistance and adipose inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harja E, Bu DX, Hudson BI, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hudson BI, Kalea AZ, Del Mar Arriero M, et al. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2008;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeWard AD, Eisenmann KM, Matheson SF, Alberts AS. The role of formins in human disease. Biochim Biophys Acta. 2010;1803:226–233. doi: 10.1016/j.bbamcr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 50.Kuhn S, Geyer M. Formins as effector proteins of Rho GTPases. Small GTPases. 2014;5:e29513. doi: 10.4161/sgtp.29513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang JS, Wendt T, Qu W, et al. Oxygen deprivation triggers upregulation of early growth response-1 by the receptor for advanced glycation end products. Circ Res. 2008;102:905–913. doi: 10.1161/CIRCRESAHA.107.165308. [DOI] [PubMed] [Google Scholar]
- 52.Xu Y, Toure F, Qu W, et al. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J Biol Chem. 2010;285:23233–23240. doi: 10.1074/jbc.M110.117457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bianchi R, Kastrisianaki E, Giambanco I, Donato R. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286:7214–7226. doi: 10.1074/jbc.M110.169342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Medapati MR, Dahlmann M, Ghavami S, et al. RAGE Mediates the Pro-Migratory Response of Extracellular S100A4 in Human Thyroid Cancer Cells. Thyroid. 2015;25:514–527. doi: 10.1089/thy.2014.0257. [DOI] [PubMed] [Google Scholar]
- 55.Toure F, Fritz G, Li Q, et al. Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ Res. 2012;110:1279–1293. doi: 10.1161/CIRCRESAHA.111.262519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rai V, Maldonado AY, Burz DS, et al. Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J Biol Chem. 2012;287:5133–5144. doi: 10.1074/jbc.M111.277731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xue J, Manigrasso M, Scalabrin M, et al. Change in the Molecular Dimension of a RAGE-ligand complex triggers RAGE Signaling. Structure. 2016 doi: 10.1016/j.str.2016.06.021. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maiti S, Michelot A, Gould C, Blanchoin L, Sokolova O, Goode BL. Structure and activity of full-length formin mDia1. Cytoskeleton (Hoboken) 2012;69:393–405. doi: 10.1002/cm.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59**.Manigrasso MB, Pan J, Rai V, et al. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci Rep. 2016;6:22450. doi: 10.1038/srep22450. This work identified novel small molecule antagonists that block the interaction of the RAGE cytoplasmic domain with the formin DIAPH1 (its FH1 domain). These early stage molecules block RAGE ligand induced signaling, smooth muscle migration and immune cell activation. These molecules form the basis of an entirely new class of small molecules for testing in chronic disease states in which RAGE ligands accumulate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goova MT, Li J, Kislinger T, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Achouiti A, de Vos AF, van ‘t Veer C, et al. Receptor for Advanced Glycation End Products (RAGE) Serves a Protective Role during Klebsiella pneumoniae – Induced Pneumonia. PLoS One. 2016;11:e0141000. doi: 10.1371/journal.pone.0141000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tian J, Huang K, Krishnan S, et al. RAGE inhibits human respiratory syncytial virus syncytium formation by interfering with F-protein function. J Gen Virol. 2013;94:1691–1700. doi: 10.1099/vir.0.049254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Zoelen MA, Wieland CW, van der Windt GJ, et al. Receptor for advanced glycation end products is protective during murine tuberculosis. Mol Immunol. 2012;52:183–189. doi: 10.1016/j.molimm.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 64.van Zoelen MA, Schmidt AM, Florquin S, et al. Receptor for advanced glycation end products facilitates host defense during Escherichia coli-induced abdominal sepsis in mice. J Infect Dis. 2009;200:765–773. doi: 10.1086/604730. [DOI] [PubMed] [Google Scholar]
- 65.Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M, Mjalli AM. Effect of TTP488 in patients with mild to moderate Alzheimer’s disease. BMC Neurol. 2014;14:12. doi: 10.1186/1471-2377-14-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bro S, Flyvbjerg A, Binder CJ, et al. A neutralizing antibody against receptor for advanced glycation end products (RAGE) reduces atherosclerosis in uremic mice. Atherosclerosis. 2008;201:274–280. doi: 10.1016/j.atherosclerosis.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 67.Finlay WJ, Cunningham O, Lambert MA, et al. Affinity maturation of a humanized rat antibody for anti-RAGE therapy: comprehensive mutagenesis reveals a high level of mutational plasticity both inside and outside the complementarity-determining regions. J Mol Biol. 2009;388:541–558. doi: 10.1016/j.jmb.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 68.Reverdatto S, Rai V, Xue J, Burz DS, Schmidt AM, Shekhtman A. Combinatorial library of improved peptide aptamers, CLIPs to inhibit RAGE signal transduction in mammalian cells. PLoS One. 2013;8:e65180. doi: 10.1371/journal.pone.0065180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han YT, Choi GI, Son D, et al. Ligand-based design, synthesis, and biological evaluation of 2-aminopyrimidines, a novel series of receptor for advanced glycation end products (RAGE) inhibitors. J Med Chem. 2012;55:9120–9135. doi: 10.1021/jm300172z. [DOI] [PubMed] [Google Scholar]
- 70.Lippert J, Burghaus R, Kuepfer L, et al. Modeling and Simulation of In Vivo Drug Effects. Handb Exp Pharmacol. 2016;232:313–329. doi: 10.1007/164_2015_21. [DOI] [PubMed] [Google Scholar]
- 71.Zhao X, Modur V, Carayannopoulos LN, Laterza OF. Biomarkers in Pharmaceutical Research. Clin Chem. 2015;61:1343–1353. doi: 10.1373/clinchem.2014.231712. [DOI] [PubMed] [Google Scholar]