

Abstract
We herein report the Palladium(II)-catalyzed bromination and iodination of a variety of α-hydrogen-containing carboxylic acid and amino acid derived amides. These reactions are exclusively enabled by quinoline-type ligands. The halogenated products obtained in this reaction are highly versatile and rapidly undergo further diversification. Further, we report the first example of a free carboxylic acid-directed Pd(II)-catalyzed C(sp3)–H bromination, enabled by quinoline ligands.
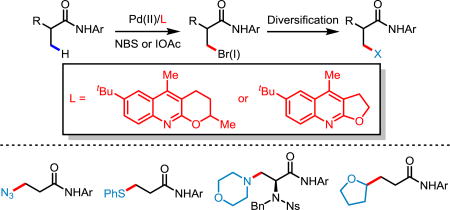
C(sp3)–halogen bonds are widespread motifs in organic chemistry,1 valued for both their ease of functional group interconversion and their presence in biologically-active natural products and pharmaceuticals. Insofar as reactivity is concerned, alkyl halides can behave as both electrophiles and nucleophiles: as electrophiles, alkyl halides can readily undergo nucleophilic substitution and the Fu group has developed invaluable methodology for the cross-coupling of alkyl halides.2a Conversely, alkyl halides may readily undergo lithium-halogen exchange or oxidative addition by magnesium or zinc to form the corresponding organometallic reagents (RLi, RMgX/RZnX, etc.); these organometallic intermediates have been applied in numerous nucleophilic substitution/addition and cross coupling reactions.2
The state-of-the-art in the synthesis of alkyl halides still largely relies on the nucleophilic substitution of alcohol equivalents and the hydrohalogenation of alkenes; these methods are often met with complicated synthetic sequences and regioselectivity issues,1 respectively. Moreover, radical halogenation reactions3 and alkene hydrohalogenation reactions almost exclusively generate secondary or tertiary C(sp3)–halogen bonds; there is presently a dearth of methods for generating primary C(sp3)–halogen bonds. Consequently, an orthogonal method that directly converts C(sp3)–H bonds to C(sp3)–halogen bonds and which can generate primary C(sp3)–halogen bonds is a highly desired transformation. Despite myriad examples of transition-metal-mediated C(sp2)–H halogenation4 and other examples of C(sp3)–H functionalizations, there are sparse examples of C(sp3)–H halogenation.5 In 2004, our group reported the first example of Pd-catalyzed C(sp3)–H iodination with a chiral oxazoline auxiliary (Scheme 1, Eq 1).6a In 2014, Sahoo and coworkers disclosed a Pd-catalyzed C(sp3)–H bromination and chlorination assisted by strong bidentate pyridyl sulfoximine directing group (Scheme 1, Eq 2).6e However, in both examples the transformations were limited to α-quaternary carboxylic acid substrates. We postulate that carboxylic acid substrates containing α-hydrogens are problematic in C(sp3)–H halogenations for three distinct reasons: (A) The α-hydrogen-containing substrate is less reactive in C(sp3)–H activation due to a diminished Thorpe-Ingold effect (compared to α-quaternary substrates); (B) The α-halogenation of the more acidic α-hydrogen may compete with the β-C(sp3)–H halogenation; (C) The β-halogenated product of the α-hydrogen-containing substrate might undergo facile elimination to give the corresponding olefin. To circumvent issue A, we envisioned that the combination of a weakly-coordinating auxiliary and pyridine-type ligands would generate a more reactive catalyst by evading formation of a thermodynamically-favored cyclometallated product based on our previous reports.7 Provided the β-C(sp3)–H activation is accelerated by the pyridine-type ligand, the competing α-halogenation may be suppressed, solving issue B. Finally, issue C may be solved by conducting the reaction in mildly acidic conditions, therefore preventing the deprotonation of the α-hydrogen. As a continuous interest in weak coordination and ligand acceleration for C–H activation, we have shown the ability of pyridine-type ligands to promote various C–H functionalizations of weakly-coordinating directing groups.7 Our recent success in ligand-controlled enantioselective methylene arylation further highlights the unique advantage of using monodentate weakly-coordinating directing groups.8 Recently, the Rao group used a very strong bidentate directing group to achieve the C(sp3)–H chlorination and iodination for α-hydrogen containing substrates (Scheme 1, Eq 3).6f However, β-halogenation of α-halogenated substrates to produce α,β-dihalogenated synthons remain to be demonstrated. Herein, we report a simple weakly-coordinating amide directed C(sp3)–H bromination and iodination enabled by quinoline-based ligands. Further, we also demonstrated for the first time the C(sp3)–H bromination of free carboxylic acid substrates.
Scheme 1.

Relevant Examples of Directed Pd(II)-Catalyzed C(sp3)–H Halogenation
Given our success in ligand-enabled transition-metal-enabled C(sp3)–H functionalization, we began our study of C(sp3)–H bromination by focusing on ligand screening with 1a as the model substrate (Table 1). In the absence of ligand no reactivity was observed. Simple quinoline and phenanthridine ligands (L1, L2, respectively) afforded trace product. Given the ease of substituted quinoline synthesis, we screened quinoline ligands with substitution at C2 and C3; we were pleased to observe that the yield of 2a increased to 21% when L3 was used. We next observed that acridine ligand L4 increased the yield to 27%. Previously, ligand L5 has proven efficient for accelerating many C(sp3)–H activation reactions.7b,7c,7e To our delight, C2/C3-fused quinoline/tetrahydropyran ligands proved superior to all other ligand classes in enabling the β-C(sp3)–H bromination. Consequently, we evaluated the effect of different substitutions on the quinoline ring (L5–L14). With ligand L5, a yield of 49% of 2a was observed. The yield was increased to 55% by installing a tert-butyl group at the 6-position (L6). A slightly lower yield was observed when a methyl group was installed at C7 (L7). For sterically encumbered ligands L8 and L9, a significant decrease in yields was observed. Other substitutions, such as aryl and amino groups, on the 6-postion (L10, L11) proved inferior to L5. The installation of a methoxy group at C8 (L12) was detrimental to the success of the reaction, presumably due to the ability of the methoxy group to occupy a vacant site of Pd(II), effectively rendering L12 a bidentate ligand. Since L6 had been identified as the optimal ligand, we pursued modification of L6 in an effort to increase the yield of the reaction; however, neither the presence of electron-withdrawing nor electron-donating substituents at C7 gave better yields (L13, L14, respectively). Using L6, we were able to dramatically improve the yield to 73% by reducing the molar equivalents of NBS and using AcOH (1 equiv) as an additive.
Table 1.

|
Conditions: 0.1 mmol of 1a, 2.0 equiv of NBS, 10 mol% of Pd(OAc)2, 10 mol% of ligand, 1.5 equiv of PhI(OAc)2, 1.0 mL of DCE, 80 °C, under air, 20 h.
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
Optimal conditions: 0.1 mmol of 1a, 1.75 equiv of NBS, 10 mol% of Pd(OAc)2, 10 mol% of ligand L6, 1.5 equiv of PhI(OAc)2, 5.0 equiv of AcOH, 1.0 mL of DCE, 70 °C, under air, 20 h.
After optimizing the conditions for C(sp3)–H bromination, we attempted C(sp3)–H iodination by simply switching NBS to NIS. However, the yield for the iodinated product was very low. We turned our attention to another iodinating source inspired by our early studies, iodoacetate (IOAc) which can be generated in situ by the reaction of I2 and PhI(OAc)2.6a,6b Fortunately, with the optimal ligand L6 for C(sp3)–H bromination and slight changes in conditions, the iodinated product was afforded in 42% yield (L6). It was found this iodination reaction was also exclusively driven by quinoline ligands. Screening of the same scaffold (L6, L15, L14, L9, L8) of ligands gave comparable results to that of the bromination. Noting that the tricyclic pyridine ligand L16 slightly enhanced the yield of 3a, we explored ligands containing fused C2/C3 quinoline/tetrahydrofuran motifs. We observed that L17 was superior to ligand L6. Acridine-type ligands (L4, L18) were inferior to L17 and when we saturated one or both of the phenyl rings of the acridine type ligands, the yields decreased dramatically (L19–L21). Other quinoline ligands (L22, L2) failed to increase the yield. Based on the optimal ligand L17, we were able to improve the yield to 75% after the systematic modification of reaction conditions (see supplementary information).
With the optimal ligands and reaction conditions in hand, we set out to explore the substrate scope for C(sp3)–H bromination and iodination. As shown in Table 3, a variety of α-alkyl substituted carboxylic amides were brominated in good-to-excellent yields (2a, 2c, 2d, 2e, 2f). Less hindered propionic amides could also be functionalized, albeit in moderate yield (2b). The difference in reactivity of 2a–2e highlights the profound effect large substituents and the resulting Thorpe-Ingold effect has in C(sp3)–H cyclometallation. Other moieties, such as trifluoromethyl groups as well as cyclic ethers were compatible in this reaction (2g, 2h). It is worth noting that when aromatic ring-containing substrates were used, no C(sp2)–H bromination of aromatic rings were detected; C(sp3)–H brominated products were formed exclusively (2i, 2j). Given the importance of non-natural amino acids in drug discovery, a N-nosyl-N-benzyl-protected L-alanine (1k) derivative was brominated, affording 2k in 65% yield. This product is attractive in that, through simple functional group interconversion of 2k, a wide-variety of chiral non-natural amino acids could be formed. Further, for the first time the N-nosyl-N-benzyl protecting groups, as opposed to the more cumbersome N-phthaloyl group, was also effective in C(sp3)–H activation.
Table 3.

|
Bromination conditions: 0.1 mmol of 1a, 1.75 equiv of NBS, 10 mol% of Pd(OAc)2, 10 mol% of ligand L6, 1.5 equiv of PhI(OAc)2, 5.0 equiv of AcOH, 1.0 mL of DCE, 70 °C, under air, 20 h.
Iodination conditions: 0.1 mmol of 1a, 1.5 equiv of I2, 1.5 equiv of PhI(OAc)2, 10 mol% of Pd(OAc)2, 10 mol% of ligand L17, 1.0 equiv of AcOH, 0.8 mL of 1,4-dioxane, 0.2 mL of DCE, 80 °C, under air, 20 h.
Isolated yields.
2.0 mmol scale.
Next, we sought out to explore the substrate scope of C(sp3)–H iodination. Different α-alkyl substituted carboxylic amides were iodinated in high yields (3a, 3c). This reaction could also be applied to conformationally less favored propionic amide substrates with moderate efficiency (3b). The trifluoromethyl substituted substrate (1d) was successfully iodinated in 75% yield (3d). This iodination method was compatible with β-amino acid derivatives (3e). Esters were also tolerated, though in modest yield (3f). Moreover, this is the first time that α-fluoro-substrates have been tolerated in β-C(sp3)–H activation, giving the iodinated product in good yield (3g).
In order to demonstrate the versatility of the halogenated products afforded by the aforementioned reactions, brominated product 2b was successfully converted to a range of other functionalities through nucleophilic substitutions. A diverse array of chemical bonds, such as C–C, C–N, C–O and C–S bonds, were easily forged (Scheme 2). Surprisingly, in attempting to displace the bromide in 2b with NaCN in THF to afford the corresponding nitrile, we observed the formation of the structurally interesting THF-substituted product 4g in 70% yield. A series of control experiments were conducted (see SI for details) which suggest the following mechanism: (A) the bromide in 2b is eliminated by the action of NaCN as base, generating the corresponding acrylamide; (B) the α-radical of THF adds to the acrylamide and then (C) the corresponding radical adduct abstracts an α-hydrogen from another THF to form a new α-radical of THF and continue the radical chain reaction. Importantly, the brominated N-nosyl-N-benzyl-protected L-alanine (2k) could be aminated smoothly with morpholine. These products are otherwise difficult to access through C(sp3)–H functionalizations.
Scheme 2.

Diversifiations of Brominated Productsa
a Conditions: (a) NaCN (2.0 equiv), THP (tetrahydropyran), 80 °C, 24 h; (b) NaN3 (4.0 equiv), MeOH, 40 °C, 24 h; (c) pyrrolidine (2.0 equiv), K2CO3 (1.5 equiv), THF, r.t., 1 h; (d) Bn2NH (2.0 equiv), K2CO3 (1.5 equiv), THF, 80 °C, 24 h; (e) AgNO3 (2.0 equiv), EtOAc, 80 °C, 24 h; (f) PhSH (2.0 equiv), NaOH (2.0 equiv), EtOH, 40 °C, 1 h; (g) NaCN (2.0 equiv), THF, 80 °C, 24 h; (h) morpholine (2.0 equiv), K2CO3 (1.5 equiv), THF, r.t., 1 h.
The long-term goal of our laboratory is to allow for innate functionality (acids, esters, amides, amines, etc.) to direct C(sp3)–H activation, therefore evading the necessity of foreign directing group-installation and removal. Presently, acid-directed C(sp3)–H activation is limited to arylation of α-quaternay carboxylic acids.9 Using ligand L6, C(sp3)–H bromination of pivalic acid proceeded to give the mono-brominated product in 40% yield. Further yield enhancement will likely be achieved by ligand modification.
In summary, we report a ligand-enabled C(sp3)–H bromination and iodination of various carboxylic acids and amino acids. The ligand effect is crucial for this C(sp3)–H halogenation. The diversifications of the product showcase the versatility of this method. Further, the first example of a free carboxylic acid directed C(sp3)–H bromination is demonstrated. Future work will focus on developing ligands for asymmetric C(sp3)–H halogenation, methylene C(sp3)–H halogenation and the free carboxylic acid directed C(sp3)–H halogenation.
Supplementary Material
Figure 1.

C(sp3)–H Bromination of Pivalic Acid.
Table 2.

|
Conditions: 0.1 mmol of 1a, 1.5 equiv of I2, 1.5 equiv of PhI(OAc)2, 10 mol% of Pd(OAc)2, 10 mol% of ligand, 1.0 mL of 1,4-dioxane, 80 °C, under air, 20 h.
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
Optimal conditions: 0.1 mmol of 1a, 1.5 equiv of I2, 1.5 equiv of PhI(OAc)2, 10 mol% of Pd(OAc)2, 10 mol% of ligand L17, 1.0 equiv of AcOH, 0.8 mL of 1,4-dioxane, 0.2 mL of DCE, 80 °C, under air, 20 h.
Acknowledgments
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, 2R01GM084019) for financial support. R.-Y. Zhu is supported by the Boehringer Ingelheim Fellowship. T. G. Saint-Denis is supported by the NSF GRFP and the TSRI Dean’s Fellowship.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected reviews: Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem. Rev. 2006;106:3364. doi: 10.1021/cr050313i.Saikia I, Borah AJ, Phukan P. Chem. Rev. 2016;116:6837. doi: 10.1021/acs.chemrev.5b00400.Petrone DA, Ye J, Lautens M. Chem. Rev. 2016;116:8003. doi: 10.1021/acs.chemrev.6b00089.
- 2.For selected reviews: Netherton MR, Fu GC. Adv. Synth. Catal. 2004;346:1525.Silverman GS, Rakita PE, editors. Handbook of Grignard Reagents. Dekker; New York: 1996. Kambe N, Iwasaki T, Terao J. Chem. Soc. Rev. 2011;40:4937. doi: 10.1039/c1cs15129k.
- 3.For selected examples of radical halogenation reactions: Schreiner PR, Lauenstein O, Kolomitsyn IV, Nadi S, Fokin AA. Angew. Chem. Int. Ed. 1998;37:1895.Liu W, Groves JT. J. Am. Chem. Soc. 2010;132:12847. doi: 10.1021/ja105548x.Schmidt VA, Quinn RK, Brusoe AT, Alexanian EJ. J. Am. Chem. Soc. 2014;136:14389. doi: 10.1021/ja508469u.Liu T, Myers MC, Yu JQ. Angew. Chem. Int. Ed. 2017;56:306. doi: 10.1002/anie.201608210.
- 4.For selected examples of C(sp2)—H Halogenation: Fahey DR. J. Chem. Soc. D. 1970:417.Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300. doi: 10.1021/ja031543m.Kalyani D, Dick AR, Anani WQ, Sanford MS. Tetrahedron. 2006;62:11483. doi: 10.1021/ol060747f.Wan X, Ma Z, Li B, Zhang K, Cao S, Zhang S, Shi Z. J. Am. Chem. Soc. 2006;128:7416. doi: 10.1021/ja060232j.Mei TS, Giri R, Maugel N, Yu JQ. Angew. Chem. Int. Ed. 2008;47:5215. doi: 10.1002/anie.200705613.Kakiuchi F, Kochi T, Mutsutani H, Kobayashi N, Urano S, Sato M, Nishiyama S, Tanabe T. J. Am. Chem. Soc. 2009;131:11310. doi: 10.1021/ja9049228.Schröder N, Wencel-Delord J, Glorius F. J. Am. Chem. Soc. 2012;134:8298. doi: 10.1021/ja302631j.Sun X, Shan G, Sun Y, Rao Y. Angew. Chem. Int. Ed. 2013;52:4440. doi: 10.1002/anie.201300176.Wang XC, Hu Y, Bonacoris S, Hong Y, Burrell R, Yu JQ. J. Am. Chem. Soc. 2013;135:10326. doi: 10.1021/ja4055492.
- 5.For selected reviews on C—H functionalizations: Chen X, Engle KM, Wang DH, Yu JQ. Angew. Chem. Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273.Daugulis O, Do HQ, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058.Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem. Soc. Rev. 2009;38:3242. doi: 10.1039/b816707a.Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e.Colby DA, Tsai AS, Bergman RG, Ellman JA. Acc. Chem. Res. 2012;45:814. doi: 10.1021/ar200190g.Zhu RY, Farmer ME, Chen YQ, Yu JQ. Angew. Chem. Int. Ed. 2016;55:10578. doi: 10.1002/anie.201600791.
- 6.Examples of Pd(II)-catalyzed C(sp3)—H Halogenation Giri R, Chen X, Yu J-Q. Angew. Chem. Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884.Giri R, Wasa M, Brazzano SP, Yu JQ. Org. Lett. 2006;8:5685. doi: 10.1021/ol0618858.(c) Two substrates have been reported. Wasa M, Yu JQ. J. Am. Chem. Soc. 2008;130:14058. doi: 10.1021/ja807129e.(d) One substrate has been reported. Stowers KJ, Kubota A, Sanford MS. Chem. Sci. 2012;3:3192. doi: 10.1039/C2SC20800H.Rit RK, Yadav M, Ghosh RK, Shankar M, Sahoo AK. Org. Lett. 2014;16:5258. doi: 10.1021/ol502337b.Yang X, Sun Y, Sun TY, Rao Y. Chem. Commun. 2016;52:6423. doi: 10.1039/c6cc00234j.
- 7.For selected examples: Wasa M, Chan KSL, Zhang XG, He J, Miura M, Yu JQ. J. Am. Chem. Soc. 2012;134:18570. doi: 10.1021/ja309325e.He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu JQ. Science. 2014;343:1216. doi: 10.1126/science.1249198.Li S, Chen G, Feng CG, Gong W, Yu JQ. J. Am. Chem. Soc. 2014;136:5267. doi: 10.1021/ja501689j.Zhu RY, He J, Wang XC, Yu JQ. J. Am. Chem. Soc. 2014;136:13194. doi: 10.1021/ja508165a.Zhu RY, Tanaka K, Li GC, He J, Fu HY, Li SH, Yu JQ. J. Am. Chem. Soc. 2015;137:7067. doi: 10.1021/jacs.5b04088.
- 8.Chen G, Gong W, Zhuang Z, Andrä MS, Chen YQ, Hong X, Yang YF, Liu T, Houk KN, Yu JQ. Science. 2016;353:1023. doi: 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; (b) Chen G, Zhuang Z, Li GC, Saint-Denis TG, Hsiao Y, Joe CL, Yu JQ. Angew. Chem. Int. Ed. 2017;56:1506. doi: 10.1002/anie.201610580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.