

Abstract
Complete genome sequences of 19 strains of monkey B virus (Macacine alphaherpesvirus 1; BV) isolated from several macaque species were determined. A low level of sequence variation was present among BV isolates from rhesus macaques. Most variation among BV strains isolated from rhesus macaques was located in regions of repetitive or quasi-repetitive sequence. Variation in coding sequences (polypeptides and miRNAs) was minor compared to regions of non-coding sequences. Non-coding sequences in the long and short repeat regions of the genome did however exhibit islands of conserved sequence. Oral and genital isolates from a single monkey were identical in sequence and varied only in the number of iterations of repeat units in several areas of repeats. Sequence variation between BV isolates from different macaque species (different BV genotypes) was much greater and was spread across the entire genome, confirming the existence of different genotypes of BV in different macaque species.
Keywords: Macacine alphaherpesvirus 1, monkey B virus, genome sequence, genotype, macaque
INTRODUCTION
The genus Macaca is the most ecologically successful and widely distributed nonhuman primate (NHP) taxon, reaching its zenith in Asia where 18 of the 19 species of macaques are found. Like other NHP, macaques are naturally infected with their own complement of herpesviruses analogous to their human counterparts. (Estep et al., 2010) The macaque alpha-herpesvirus (Macacine alphaherpesvirus 1; monkey B virus, BV) is closely related to the human herpes simplex viruses HSV1 and HSV2. In macaques BV infection can be oral and/or genital, is usually asymptomatic, the virus establishes latency in sensory ganglia, and various forms of stress can result in reactivation of BV from latency and shedding of infectious virus in bodily secretions. (Huff et al., 2003; Huff and Barry, 2003; Keeble, 1960; Weigler, 1992) A few studies have shown that free-ranging macaques in habitat countries are seropositive for BV, but the genetic diversity of BV in naturally occurring macaque populations has not been investigated. (Engel et al., 2002; Engel et al., 2008; Jones-Engel et al., 2006; Lee et al., 2015)
While BV rarely causes serious disease in healthy adult macaques, when transmitted to humans or other primate species BV can be neuropathogenic. Only about 50 cases of zoonotic BV infection, all of which have occurred in the context of exposure to laboratory or captive animals, have been documented since the virus was first recognized in 1932, but most (~75%) have been lethal. (Davidson and Hummeler, 1960; Huff and Barry, 2003; Palmer, 1987; Weigler, 1992) The high mortality associated with zoonotic BV infection makes this virus the single most serious zoonotic concern for animal care and research personnel working with or around macaque monkeys.
In the 1960s a strain of BV isolated from a laboratory rhesus macaque (M. mulatta) was adapted to replicate in primary rabbit kidney cells and used to prepare a formalin-inactivated vaccine. (Hull, 1971; Hull and Nash, 1960; Hull et al., 1962) However, given the high pathogenicity of BV in humans and the low incidence of zoonotic BV infections, this vaccine has not been used beyond initial clinical testing. This BV strain (designated E2490) has since served as the reference strain of BV and its genome sequence has been determined. (Ohsawa et al., 2002a; Ohsawa et al., 2003; Perelygina et al., 2003)
The high lethality associated with zoonotic BV infections in the laboratory led to BV being considered extremely neuropathogenic in humans. Consistent with this assumption, asymptomatic zoonotic BV infections have not been reported, although several cases are suspected to be the result of reactivation of latent BV in patients lacking a history of overt BV infection. (Fierer et al., 1973; Palmer, 1987; Weigler, 1992) Inconsistent with this assumption of extreme neuropathogenicity are the hundreds of thousands of exposure incidents that occur outside of the laboratory setting. Millions of free-ranging, urban, pet and temple macaques are distributed throughout the natural range of these highly commensal animals and contact with humans is ubiquitous and often results in bites, scratches and mucosal splashes. (Engel et al., 2002; Fuentes, 2006) However, there has never been a documented case of pathogenic zoonotic BV transmission following exposure to macaques in Asia or Gibraltar. (Craig et al., 2015; Engel et al., 2002; Jones-Engel et al., 2006; Jones-Engel et al., 2008) Obviously, factors such BV strain variation the amount of virus transmitted likely play a part in determining whether a clinically apparent zoonotic infection results.
For decades rhesus macaques have been the most widely used macaque species in biomedical research. As far as is known all zoonotic BV patients had contact with rhesus monkeys, although some had contact with other macaque species as well. Based on DNA sequence of very small regions of the BV genome (~1.3 kbp) it has been suggested that perhaps BV isolates from rhesus macaques were somehow different from BV of other macaque species, having greater pathogenicity to humans. Several studies have examined BV isolates from different macaque species housed in zoos or research centers in the US and found that BV isolates can be classified into distinct ‘genotypes’ based on DNA sequence differences. (Ohsawa et al., 2002b; Smith et al., 1998; Thompson et al., 2000) Since the number of macaque species examined is small and all BV isolates were from captive bred as opposed to wild monkeys, the veracity of BV genotypes remains to be seen. If distinct BV genotypes related to the macaque species do exist, the question of their relative neurovirulence or zoonotic potential is obviously of importance. Aside from one study in mice, comparative neurovirulence of different BV strains or genotypes has not been reported. The one study that tested several rhesus BV (BVrh) isolates in mice using i.m. inoculation did observe wide differences in neurovirulence, with the 50% infectious dose ranging from 101.5 to >106 PFU. (Ritchey et al., 2005)
Aside from the reference BVrh strain E2490, the only other published BV genome sequence is that of an isolate (E90-136) from a young cynomolgus macaque (M. fascicularis) that died of a generalized infection. (Ohsawa et al., 2014; Simon et al., 1993) Since E2490 and the cynomolgus BV isolate viruses are different BV genotypes (BVrh & BVcy, resp.), comparison of these genome sequences provides little insight regarding the degree of sequence variation within or between BV genotypes. Here we report the sequence of several BVrh isolates as well as some isolates of different BV genotypes to begin to address the extent of genetic variation among BV isolates among captive macaques.
MATERIALS AND METHODS
Viruses
BV strains sequenced and what is known regarding their origin are listed in Table 1. The reference strain of BV (E2490) was isolated from a rhesus macaque (M. mulatta; BVrh) in the early 1950s when most monkeys used in research were still imported from the wild, so this isolate may have been from a wild-caught monkey. Since there are a number of differences between published E2490 sequences from different laboratories (Ohsawa et al., 2002a; Ohsawa et al., 2003; Perelygina et al., 2003), the E2490 genome was re-sequenced. Twelve strains were isolated from captive bred rhesus macaques, two isolates from pigtail macaques (M. nemistrina; BVpt), one isolate from a lion-tailed macaque (M. Silenus; BVlt), one isolate from a Japanese macaque (M. fuscata; BVfu), and one isolate made from primary kidney cells. One additional BV strain was isolated in 2005 from necropsy tissues of a 23 year old bonnet macaque (M. radiata; BVbn) that developed typical herpetic oral lesions shortly before being humanely euthanized for cardiopulmonary complications due to congestive heart failure. (Scharf et al., 2008)
Table 1.
Summary of Characteristics of BV Isolates
| BV | Isolated | Common | Genotype | Year | Sourcec | LD50 | |||
|---|---|---|---|---|---|---|---|---|---|
| CNSD50 | ID50 Accession Refd | Unique Characteristics | |||||||
| Straina Number |
From | Name | Presumed Confirmed | Isolatedb | |||||
| E2490 | M. mulatta | Rhesus | BVrh | BVrh | 1950s | Lilly Res Labs | 104.0 | 104.0 | 104.0 |
| 24105-G | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 104.8 | 104.8 | 104.8 |
| 32425-G | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 103.8 | 103.8 | 103.8 |
| 26896-O | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 104.1 | 104.1 | 104.1 |
| iterations in repeat regions RL2, RL5 & RS1 | |||||||||
| 32157-G | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 104.0 | 104.0 | 104.0 |
| 28443-O | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 104.0 | 104.0 | 104.0 |
| 31612-G | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 103.2 | 103.2 | 103.2 |
| 31618-G | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 104.0 | 104.0 | 104.0 |
| 32188-O | M. mulatta | Rhesus | BVrh | BVrh | 2002 | CaNPRC | 104.5 | 104.5 | 104.5 |
| 20620 | M. mulatta | Rhesus | BVrh | BVrh | Unkn | SFBR | 104.1 | 104.1 | 104.1 |
| 12930 | M. mulatta | Rhesus | BVrh | BVrh | Unkn | SFBR | 105.4 | 103.6 | 103.6 |
| 16293 | M. mulatta | Rhesus | BVrh | BVrh | Unkn | SFBR | 103.8 | 103.8 | 103.6 |
| 9400371 | M. fascicularis | Cynomolgus | BVcy or BVrhe | Unkn | VRL (primary cell culture) | 103.8 | 103.8 | 103.8 | |
| 7709642 | M. fuscata | Japanese | BVfu | BVrh | Unkn | VRL (S Texas Ranch) | NDf | ND | ND |
| M12-O | M. radiata | Bonnet | BVbn | BVrh | 2005 | Downstate Med Sch, NY | 104.1 | 104.1 | 104.1 |
| E90-136 | M. fascicularis | Cynomolgus | BVcy | BVcy | 1993 | NENPRC | 104.3 | 104.3 | 104.3 |
| 1504-11 | M. nemistrina | Pigtail | BVpt | BVpt | Unkn | SFBR | >107 | 104.3 | 104.1 |
| KQ | M. nemistrina | Pigtail | BVpt | BVpt | Unkn | VRL | >107 | 105.3 | 105.3 |
| 8100812 | M. silenus | Lion-tailed | BVlt | BVlt | 1981 | VRL | >107 | 104.5 | 104.0 |
Appended O or G indicates whether the isolate was made from orophyrangeal or genital swabs.
The exact date of isolation for isolates listed as “Unkn” is not known although all were isolated prior to 1990.
Ca (California) and NE (New England) National Primate Research Center; SFBR, Southwest Foundation for Biomedical Research, San Antonio TX; VRL, diagnostic virology lab in San Antonio TX.
1: Hull, 1960; 2: Hilliard et al., 1987; 3: Huff et al., 2003; 4: Smith et al., 1998; 5: Scharf et al., 2008; 6: Thompson et al., 2000; 7: Hirano et al., 2002
Submitting institution and testing laboratory disagreed on species identification of tissue donor monkey.
ND, not done.
Viral genome sequencing
Viral DNA was purified from infected Vero cells on NaI gradients and used for both PCR and MiSeq genomic sequencing (600 cycle paired-end runs) as described. (d’Offay et al., 2013; Fulton et al., 2013) For all sequencing runs total read counts were 2.9 – 18.2 × 106 (avg. 8.2 × 106), average read length was 165 – 284 bp (avg. 250 bp), and average depth of coverage was 4.3 – 31.1 × 103 (avg. 13.1 × 103). Where very poor MiSeq results were obtained, new DNA preparations were made and sequenced. Analysis of sequence data, assembly of sequences, alignments and phylogenetic analyses were all conducted using the CLC Main Workbench 7.0.3 programs.
Sequence data of all strains were initially assembled both de novo and by alignment to a reference sequence. Coding sequences were readily assembled by both methods, but some intergenic and much of the RL and RS regions required manual extension of assembled sequences by searching unassembled reads. The initial two BVrh isolates sequenced (24105-G & 32425-G) were each assembled de novo and using both the published sequence for BVrh E2490 (Genbank NC_004812.1) and BVcy E90-136 sequence (Genbank KJ566591) as a scaffold/reference. Since there were a number of sequence discrepancies between published E2490 sequences and between E2490 and that of 24105-G, the 24105-G sequence was used as a reference sequence for assembly of all other BV isolates except those from pigtail or lion-tailed macaques. In addition to de novo assembly, both the 24105-G and E90-136 sequences were used as references to assemble one BVpt isolate (1504-11). Once the 1504-11 sequence was completed and confirmed (PCR/sequencing of all questionable areas), it was used (in addition to de novo assembly) as a reference to assemble the remaining BVpt and BVlt sequences.
Once Illumina generated sequences were assembled, sequence assemblies and sequence data of each isolate were manually reviewed to identify areas of low coverage, sequence discrepancies, manually extended areas of concern, or other areas where there was any question regarding the veracity of the draft sequence assembly. The sequence of all such areas in each isolate were determined/confirmed by PCR amplification and dideoxy sequencing of the products. Deep vent polymerase (New England BioLabs, Ipswich, MA) and 32% betaine (Sigma Chemical Co, St Louis, MO) were used in PCR reactions. Primers having Tms of 70–74°C were used to amplify and sequence areas of repetitive sequence. When completed, genome sequences were aligned and all conflicts resolved with additional PCR/sequencing. All genome sequences have been deposited in GenBank and their accession numbers are listed in Table 1.
Neurovirulence testing
Neurovirulence of BV isolates was assessed using the mouse skin scarification model as described previously. (Black et al., 2014; Brush et al., 2014; Rogers, 2006) Briefly, the left flank of 10–12 gm female Balb/c mice was shaved, lightly scarified with an 18 Gg needle, and 10 ul of virus applied to the scarified area. Ten-fold dilutions of virus ranging from 106–103 PFU/10 ul were tested. Mice were observed twice daily for signs of neurological involvement and scored on a scale of 0 – 5. (Brush et al., 2014) Any mice showing signs of brain involvement (ataxia, tremors) were humanely euthanized. Blood was collected when surviving mice were euthanized at 14 days PI (DPI). Whether or not survivors were infected was determined by testing serum for anti-BV IgG. For each isolate the dose required to induce neurological symptoms (CNSD50), death/euthanasia (LD50) or infection (ID50; based on clinical signs plus positive serology in survivors) in 50% of inoculated mice was determined as described. (Black et al., 2014; Ritchey et al., 2005)
RESULTS
Sequencing of the BV reference E2490 strain genome revealed a number of differences from published sequences. (Perelygina et al., 2003, Ohsawa et al., 2002b, 2003) Most were single nucleotide differences or small indels, the majority being located in noncoding regions of the genome. In one published E2490 sequence UL1 lacked a conventional ATG start codon and the UL region extended ~1000 bp 3′ of the UL56 initiation codon. WSith all these differences in the E2490 genome, we re-sequenced the E2490 genome. This identified an additional 719 bp that included a conventional UL1 start codon. This additional sequence resulted in ~600 bp of what was previously UL (past the UL56 ORF) now being part of the RL region. There was overall a 658 bp difference in size of the previously reported E2490 genome and the re-sequenced E2490 genome. The vast majority of this size difference was due to the shifting of sequence from UL into RL and differences in the number of repeat unit iterations within repeat regions in the E2490 genome.
Since the number of iterations present in the repeat region at the RL/UL junction was not determined for any of the isolates in this study, the precise length of all BV genomes was not determined. Although the number of repeat units comprising the RL-UL repeats was not determined, the sequence of the repeat units was determined and all isolates had ≥3 copies of the repeat unit, so in final sequences three iterations of these repeats appear in all isolates. With this limitation, the genome of all BV isolates varied between 154,958 and 157,447 bp. However, BVrh isolates varied less (154,958 – 156,131 bp), with much of the size variation being due to differences in the number of iterations in areas of repeated sequence blocks. The two BVpt genomes were 156,887 and 157,120 bp long, and the BVlt isolate being 157,447 bp. Since repeats were found in all these BV genomes but some were not identified in the BVcy E90-136 genome. (Ohsawa et al., 2014). We therefore re-examined these areas of the E90-136 genome by PCR using Deep Vent pol/betaine and high Tm primers. Repeats were found in all positions where they are in the E2490 genome, resulting in the E90-136 BVcy genome being 155,157 bp rather than the 153,891 bp originally reported.
Despite the variation in genome size, there was no variation in the overall genetic arrangement of any BV isolates or different BV genotypes. With only two exceptions all ORFs, splice junctions and pre-miRNAs were conserved among all BV isolates. One exception was the termination point of the UL50 ORF in one isolate (9400371) where a single base insertion in a homopolymeric C tract near the 3′ end of the ORF resulted in extension of the ORF by 287 bp (96 amino acids) to the next termination codon. However, the UL50 polyA signal is located only 24 bp 3′ of the original termination codon, so it is entirely possible that the UL50 mRNA of this isolate is polyadenylated and terminates before the next downstream termination codon is reached, resulting in a UL50 protein that would not bet much larger than that of other BV isolates. The second exception was the UL45 ORF in the lion-tailed macaque isolate (8100812). A predicted amino acid sequence with 90% identity to the UL45 protein of the BVpt isolates was clearly evident in isolate 8100812, but there was not a conventional ATG start codon anywhere near the initiation point of other BV UL45 ORFs; presumably if the 8100812 UL45 ORF is expressed, an unconventional ATC initiation codon is used. Other than these few exceptions, the coding regions of BVrh isolates were very conserved. Blocks of repetitive sequences were however present in the UL36, UL46, RL1, RS1, US4, US7 and US11 ORFs, with the length of repeat units and the number of repeat unit iterations varying among isolates.
The long (RL) and short (RS) repeat regions flanking the unique long (UL) and short (US) regions were more variable in sequence than were the unique regions, but again had a consistent genetic arrangement among all BV isolates. This included not only the RL1 (ICP0), RS1 (ICP4) and pre-miRNA coding sequences and the RL1, RS1 and LAT polyA signals, but the location of areas of repeated sequence blocks as well (Fig 1). The number of repeat unit iterations in all areas of reptitive sequence were determined except the repeats at the RL/UL junction. Within RL there are six areas of repeats: RLR6 abutting the UL region, RLR5 located between the B14RC/B22 pre-miRNAs (Amen and Griffiths, 2011; Besecker et al., 2009) and Exon 3 of RL2, RLR4 in the RL2 1–2 intron adjacent to exon 1, and RLR1, 2 & 3 all located between RL2 and the B19/B26/B20 pre-miRNAs (Fig 1). There were two areas of repeats with the RS region, RSR2 abutting the US region and RSR1, a large area of repeats/quasi-repeats located between the RS1 ORF and the OriS DNA replication origin palindromes. Despite the conserved location of all 8 repeat regions in all BV isolates, the sequence of the repeat units and the number of repeat unit iterations varied considerably among strains and genotypes.
Fig 1.
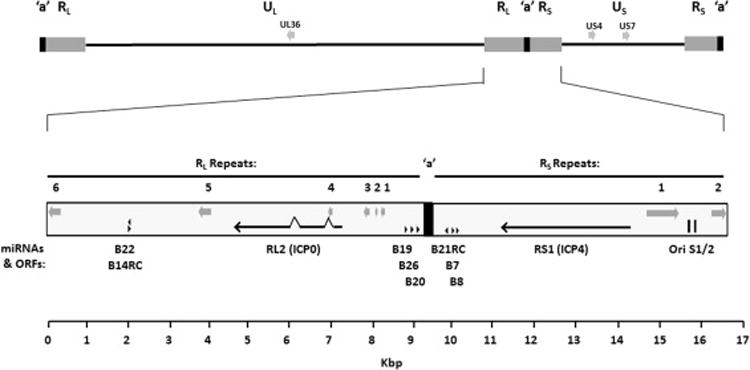
Location of conserved repeat islands in the BV genome. The location of eight areas of repeat sequence present in the RL (RLR1-6) and RS (RSR1-2) regions of all BV isolates are indicated by gray arrows located below the corresponding repeat number. The position of the RL1 (ICP0) and RS1 (ICP4) ORFS is shown by arrows, and the position/orientation of Pre-miRNAs is indicated by black arrowheads.
The BVlt and BVpt isolates varied somewhat from the BVrh and BVcy isolates. Repeat region RSR1 (between RS1 and OriS) in these isolates is comprised of two separate regions of repeats separated by some unique sequence. In both BVpt isolates (but not the BVlt isolate) the RS region also extended 30 bp into the US1 and US12 ORFs such that the N-terminal 10 amino acids of the US1 and US12 proteins are identical.
Strain variation among BVrh isolates
Differences in the overall size of BVrh genomes were due primarily to differences in the number of repeat unit iterations comprising various areas of repeat sequences. These include the RSR1 region of repeats/quasi-repeats located between the RS1 (ICP4) ORF and OriS and the RSR2 repeats at the RS/US junction. In addition to the repeats in RL and RS, there were also variable repeat sequences in some isolates within the UL36, UL46, RL2, RS1, US4, US7 and US11 ORFs. There was very little variation in the size of ORFs or pre-miRNAs among the 12 BVrh isolates (excluding areas of repetitive sequence). Even at the whole genome level, sequence identity among aligned BVrh isolates varied only slightly from 97.2 to 99.9% (Table 2).
Table 2.
Percent DNA Sequence Identity of Complete BV Genomes and Coding Sequences % Sequence Identity of Coding Sequences
| BVrha | BVrh | BVrh | BVrh | BVrh | BVrh | BVrh | BVrh | BVrh | BVrh | BVrh | BVrh | BVrh | Bvcy | BVbn | Bvfu | Bvcy | BVpt | BVpt | BVIt | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||||||
| E2490 | 12930 | 16293 | 20620 | 24105-G | 26896-G | 26896-O | 28443-O | 31612-G | 31618-G | 32157-G | 32188-O | 32425-G | 9400371 | M12-O | 7709642 | E90-136 | KQ | 1504-11 | 8100812 | |||
| % DNA Sequence Identity of Complete Genome | BVrha | E2490 | 99.7 | 99.6 | 99.2 | 99.2 | 99.0 | 99.0 | 98.9 | 99.2 | 99.5 | 99.1 | 98.9 | 99.5 | 99.0 | 99.3 | 99.39 | 95.0 | 89.3 | 89.4 | 89.78 | |
| BVrh | 12930 | 98.8 | 99.4 | 99.1 | 99.1 | 99.2 | 99.2 | 99.1 | 99.1 | 99.4 | 99.2 | 99.1 | 99.4 | 99.1 | 99.5 | 949 | 94.9 | 89.4 | 89.5 | 9.78 | ||
| BVrh | 16293 | 98.2 | 98.5 | 99.2 | 99.2 | 99.0 | 99.0 | 98.9 | 99.2 | 99.6 | 99.1 | 98.9 | 99.5 | 99.0 | 99.3 | 949 | 95.0 | 89.3 | 89.4 | 978 | ||
| BVrh | 20620 | 97.7 | 98.0 | 98.1 | 99.9 | 99.8 | 99.8 | 99.7 | 99.9 | 99.3 | 99.9 | 99.7 | 99.4 | 99.3 | 99.1 | 91 | 95.1 | 89.4 | 89.5 | 98 | ||
| BVrh | 24105-G | 97.7 | 98.1 | 98.4 | 99.4 | 99.7 | 99.71 | 99.6 | 99.91 | 99.3 | 99.8 | 99.6 | 99.4 | 99.2 | 99.1 | 9919 | 95.1 | 89.4 | 89.5 | 8988 | ||
| BVrh | 26896-G | 97.3 | 98.0 | 97.9 | 99.3 | 99.1 | 00.0 | 99.9 | 00.0 | 99.1 | 99.9 | 99.8 | 99.2 | 99.3 | 99.2 | 91 | 95.0 | 89.5 | 89.6 | 98 | ||
| BVrh | 26896-O | 97.4 | 98.0 | 97.8 | 99.2 | 99.1 | 99.9 | 99.9 | 99.9 | 99.1 | 99.9 | 99.8 | 99.2 | 99.3 | 99.2 | 991 | 95.1 | 89.5 | 89.6 | 898 | ||
| BVrh | 28443-O | 97.1 | 97.9 | 97.6 | 99.0 | 98.8 | 99.2 | 99.3 | 99.8 | 99.0 | 99.8 | 99.8 | 99.1 | 99.3 | 99.1 | 9909 | 95.0 | 89.5 | 89.6 | 8988 | ||
| BVrh | 31612-G | 97.5 | 97.9 | 97.9 | 99.3 | 99.2 | 99.5 | 99.6 | 99.0 | 99.3 | 99.8 | 99.7 | 99.3 | 99.3 | 99.1 | 919 | 95.2 | 89.4 | 89.5 | 998 | ||
| BVrh | 31618-G | 97.4 | 98.5 | 98.9 | 98.3 | 98.7 | 97.9 | 97.9 | 97.6 | 98.0 | 99.2 | 99.0 | 99.9 | 99.1 | 99.4 | 949 | 95.1 | 89.3 | 89.5 | 988 | ||
| BVrh | 32157-G | 97.4 | 97.9 | 97.8 | 99.3 | 99.0 | 99.7 | 99.7 | 99.1 | 99.6 | 97.9 | 99.8 | 99.3 | 99.3 | 99.2 | 92 | 95.0 | 89.4 | 89.5 | 98 | ||
| BVrh | 32188-O | 97.2 | 97.9 | 98.0 | 99.3 | 99.3 | 99.3 | 99.2 | 99.0 | 99.1 | 98.0 | 99.1 | 99.1 | 99.1 | 99.1 | 9909 | 94.9 | 89.4 | 89.5 | 8968 | ||
| BVrh | 32425-G | 98.5 | 98.5 | 98.8 | 98.4 | 98.6 | 97.9 | 97.9 | 97.7 | 98.0 | 99.6 | 97.9 | 98.1 | 99.1 | 99.3 | 93 | 95.0 | 89.3 | 89.4 | 98 | ||
| Bvcy | 9400371 | 97.6 | 98.2 | 98.3 | 98.6 | 98.7 | 98.4 | 98.4 | 98.2 | 98.3 | 98.4 | 98.3 | 98.6 | 98.4 | 99.1 | 991 | 95.1 | 89.4 | 89.6 | 897 | ||
| BVbn | M12-O | 98.2 | 98.6 | 98.4 | 98.1 | 98.1 | 98.1 | 98.1 | 97.8 | 97.9 | 98.6 | 97.9 | 98.0 | 98.5 | 98.2 | 994 | 95.0 | 89.4 | 89.6 | 897 | ||
| Bvfu | 7709642 | 97.7 | 98.3 | 98.3 | 98.1 | 98.1 | 97.9 | 97.9 | 97.8 | 97.8 | 98.3 | 97.8 | 97.9 | 98.2 | 98.3 | 98.2 | 95.0 | 89.4 | 89.5 | 897 | ||
| BVcy | E90136 | 90.3 | 90.5 | 90.7 | 90.8 | 90.9 | 90.8 | 90.8 | 90.5 | 90.9 | 90.8 | 90.8 | 90.6 | 90.7 | 91.0 | 90.7 | 9068 | 89.1 | 89.2 | 8949 | ||
| BVpt | KQ | 82.3 | 82.6 | 82.5 | 82.7 | 82.7 | 82.8 | 82.8 | 82.7 | 82.6 | 82.5 | 82.7 | 82.7 | 82.5 | 82.7 | 82.6 | 258 | 81.4 | 99.2 | 209 | ||
| BVpt | 150411 | 82.5 | 82.7 | 82.7 | 82.9 | 82.9 | 82.9 | 82.9 | 82.9 | 82.8 | 82.7 | 82.8 | 82.9 | 82.7 | 82.9 | 82.8 | 27 | 81.6 | 98.2 | 20 | ||
| BVlt | 8100812 | 82.7 | 83.0 | 82.8 | 82.9 | 83.0 | 83.1 | 83.1 | 82.9 | 83.1 | 82.9 | 83.1 | 82.8 | 82.8 | 83.0 | 82.9 | 829 | 81.9 | 83.8 | 83.9 | ||
The presumed genotype of each isolate (based on the macaque species from which they were isolated) is indicated above/to the left of each isolate number.
A low level of sequence variation among HSV2 isolates outside of discrete regions of high sequence variability (primarily unsequenced areas of repeats) has been reported. (Newman et al., 2015) A similarly low level of sequence variation in these same areas was evident among BVrh strains, with coding sequences (exclusive of areas of repeats) being highly conserved (98.87 – 99.98% DNA sequence identity; Table 2). When coding sequences of all 12 BVrh were aligned (114,176 bp; only one copy of RL2 and RS1) there were a total of 810 single nucleotide substitutions. Of these, 88 (10.9%) were isolate-specific, of which 67 (76.1%) were present in only one isolate (16293). In contrast, while there were a total of 27 indels in the aligned coding sequences, only one was isolate-specific (to 16293). Of the 27 indels, 25 were associated with variation in the copy number of repeated sequences. Variation in amino acid sequence identity of BVrh proteins ranged from 0% to a maximum of 3.9%. As shown in Figure 2, the level of BVrh protein sequence variation is markedly less than the level of variation in proteins of its phylogenetically closest relative virus of baboons, HVP2. (Black et al., 2014) Two BVrh strains isolated from the same monkey (26896-O & -G, oral and genital isolates, resp.) had identical genome sequences except for the number of repeat unit iterations in UL36, RLR2, RLR5 and RSR1.
Fig 2.

Comparison of BVrh vs. HVP2 protein sequence variation. Translated sequences of each ORF were aligned and given a score calculated by assigning nucleotide substitutions that resulted in no amino acid change a value of 0, a conservative amino acid change a value of 1, a non-conservative amino acid change a value of 2, and an insertion/deletion a value of 2 with the total sum value being divided by the ORF size (number of amino acids) and multiplied by 100.
Variation among BV genotypes
Genomes of non-rhesus BV isolates were all orthologous with the BVrh genome. The genomes of the 7 BV isolates from non-rhesus macaque species fell into two very distinct groups based on sequence (Table 2) and phylogenetic analyses (Fig 3): those that were very similar to BVrh (9400371, 7709642 & M12-O) and those that were substantially different from BVrh isolates [E90-136 (BVcy), 8100812 (BVlt), KQ & 1504-11 (BVpt)]. Regarding the latter four isolates that are substantially different from BVrh, comparison of complete genome sequences confirmed that genotype-specific differences are present in both coding and non-coding sequences (Table 2). Since the number of non-rhesus BV isolates was small, strain variation within genotypes could not be accurately assessed. However, the two BVpt isolates were 98.2% identical to each other, a level comparable to that among BVrh isolates (97.2 – 99.9%). The BVcy isolate E90-136 varied from BVrh isolates by 9.1 – 9.7% while BVpt and BVlt isolates varied from BVrh isolates by 16.9 – 17.7%. Phylogenetic trees based on aligned complete genome sequences and coding sequences only (Fig 3) were completely consistent with trees generated using smaller sequence data sets (Ohsawa et al., 2002b; Smith et al., 1998; Thompson et al., 2000) and their topology reflected the phylogeny of the host macaque host species. (Fan et al., 2017)
Fig 3.

Phylogenetic relationship of BV isolates.BV genome sequences (A) or concatenated coding sequences (B) were aligned and Neighbor Joining trees constructed using Jukes-Cantor distance and 100 bootstrap replicates. Trees were rooted using HVP2 as an outgroup.
Three BV isolates from non-rhesus macaques were found to be very closely related to BVrh isolates (isolates 7709642, 9400371 and M12-O). The percent sequence identity at the complete genome level among these three isolates was 98.2%, and sequence identity between these isolates and BVrh isolates was 97.6 – 98.7%, well within the variation occurring among BVrh isolates (97.2 – 99.9%). This raised the question of whether or not these three BV isolates were truly different genotypes or if they were possibly the result of cross-species infection of non-rhesus macaques with BVrh from a rhesus macaque. When the DNA coding sequences of these three isolates were added to the alignment with other BVrh isolates, the number of total single nucleotide substitutions increased from 810 to 1259 and indels increased from 26 to 34. These three isolates accounted for 449/537 (83.6%) of the total isolate-specific single nucleotide substitutions and 6/7 (85.7%) of isolate-specific indels. Thus, while these three isolates all appear to be BVrh genotype viruses, they are somewhat variant from other BVrh strains isolated from rhesus monkeys.
To further assess isolate 7709642 (presumed to be BVfu), limited regions of the conserved UL19 (major capsid protein; 929 nucleotides) and UL27 (glycoprotein gB; 311 nucleotides) ORFs amplified from ganglia of three wild-caught Japanese macaques resident in Japan (Ohsawa et al., 2002b) were aligned with the corresponding BVrh and 7709642 sequences. As summarized in Table 3 variation among sequences from the three wild Japanese macaques varied by 0.6% (UL19) or 0% (UL27). In contrast, the UL19 and UL27 sequences of both BVrh and 7709642 isolates varied from the Japanese monkey sequences by 3.2% and 3.3%, respectively. Also, at all points where substitutions were present in the alignment, the 7709642 and BVrh sequences were identical to one another. Thus, isolate 7709642 does appear to be a BVrh virus rather than a strain of BVfu. Similar analysis was performed for isolate 9400371. This virus was isolated from primary macaque kidney cell cultures, but the submitting and testing institutions differed in their species identification of the donor monkey (rhesus vs. cynomologus). Using two areas of sequence spanning coding and non-coding sequences (US5–US7; 2652 bp and US8–US10; 2142 bp) of a BVcy isolate from England and homologous sequences from BVrh E2490 and 9400371, it was again clear that isolate 9400371 is a BVrh genotype virus rather than BVcy (Table 3). Furthermore, alignment of complete genome sequences of 9400371 with BVrh and BVcy isolates did not reveal any areas of the 9400371 genome that were more closely related to BVcy than to BVrh, thus providing no evidence of BVrh-BVcy recombination within the 9400371 genome. No sequence data from another bonnet macaque BVbn isolate were available to compare the M12-O sequence with, but given the results above for the other two “outliers” and the similar relatedness of M12-O to all other BVrh isolates (Table 2), it is likely that M12-O is also a BVrh strain.
Table 3.
Sequence-based genotype classification of two outlying BV isolates
| Isolate 7709642 (Presumed BVfu) |
% Sequence Identitya | Isolate 9400371 (Presumed BVcy) |
% Sequence Identityb |
|---|---|---|---|
| BVrh:BVrh (E2490:24105-G) |
99.8 | BVrh:BVrh (E2490:24105-G) |
98.3 |
| BVfu:BVfu (mfu#4,23) |
99.5 | BVcy:BVcy (E90-136:SHBV) |
97.7 |
| BVrh:BVfu E2490:mfu#4,23 |
96.5 – 96.7 | BVrh:BVcy (E2490:E90-136) |
87.9 – 88.9 |
| 7709642:E2490 | 99.8 – 100 | 9400371:BVrh | 98.7 – 99.1 |
| 7709642:mfu#4,23 | 96.5 – 96.7 | 9400371:BVcy | 88.1 – 89.2 |
Based on 1707 nt sequence from UL19 and UL27 ORFs (Genbank AB087736-AB087741 for BVfu and homologous sequences from E2490 and 24105-G).
Comparative neurovirulence of BV isolates
The neurovirulence of BV isolates was assessed using a mouse skin scarification model. (Black et al., 2014; Brush et al., 2014) This model relies on the virus undergoing sufficient replication in the skin to reach titers high enough to invade sensory neurons via unmyelinated nerve endings in the epidermis with subsequent ascension to the dorsal root ganglia and from there into the CNS. As summarized in Table 1, LD50 values for BVrh isolates ranged from 103.8 – 105.4 PFU, with an average value of 104.1 PFU. Despite having originally been adapted to replicate in primary rabbit kidney cells, the E2490 reference strain was not attenuated in this model, having an LD50 of 104.0 PFU. With the exception of one BVrh isolate (12930), all BVrh isolates were within 1 standard deviation from the mean LD50. With the exception of only two isolates (12930 & 16293), the CNSD50 and ID50 were the same as the LD50; every mouse that was infected developed clinical signs of neurological involvement and the infection was fatal (progressed to the point that euthanasia was necessary). A single mouse inoculated with 104 PFU of 16293 survived and was seropositive at 14 DPI despite not having developed any clinical signs of infection. Isolate 12930 produced clinical signs of neurological involvement in all mice that were seropositive at 14 DPI, but in several mice the infection did not progress to the point that euthanasia was necessary. Consistent with their possible classification as BVrh isolates, two of the outlying isolates (9400371 & M12-O; 7709642 was not tested) had LD50 values that were consistent with those of BVrh isolates.
As previously reported (Black et al., 2014) the neurovirulence of BVcy E90-136 was comparable to that of BVrh isolates (104.3 PFU). In contrast, both BVpt isolates and the BVlt isolate had LD50 values of >107 PFU. Despite the development of neurological signs of infection by all three isolates, the infection never progressed to the point that euthanasia was indicated. The CSD50 and ID50 for the BVpt isolates were however similar to those of BVrh isolates indicating that these isolates were able to infect mice and invade the CNS.
DISCUSSION
The genus Macaca is the most ecologically successful and widely distributed NHP taxon. Biogeographical barriers have led to species divergence and different levels of isolation, particularly among species that have evolved on islands such as M. fuscata in Japan, M. cyclopis on Taiwan, M. sinica in Sri Lanka, and the seven species of macaques on Sulawesi. On mainland Asia, inter-species hybridization may occur in areas where the taxa are sympatric, raising the possibility of cross-species transmission of their viruses. Humans have also introduced this highly adaptable Macaca genus to regions well outside their historical natural range, including the Americas and Mauritius. Three species of macaques (rhesus, cynomolgus and pigtail) have been widely used for decades as important animal models in biomedical research.
Prior to the 1950’s, NHPs were wild caught in habitat countries and shipped to the USA for use in research. In the late-1950’s, concerns over the lack of macaques available to import for biomedical research as well as a desire to explore the utility of other NHP species for research led to the development through the National Institutes of Health of seven National Primate Research Centers (NPRC) in the US. These Centers, established in 1960–1965, maintained breeding colonies of 45 different NHP species. At this time, husbandry concerns surrounding potential cross-species transmission of infectious agents in these heterogeneous NHP colonies were largely underappreciated as the focus was on biomedical research on human diseases. (Anonymous, 1971; Gibson, 1994) Over the next five decades tens of thousands of NHP of diverse taxa were imported, bred and transferred between facilities. Consequently, many macaques, particularly Indian-origin rhesus used in research today, have been bred for generations in captivity. When the founder animals of current captive breeding colonies were originally imported, it was common practice among capture teams to co-house monkeys of different species, and infectious disease outbreaks often caused significant losses of newly captured monkeys. Thus, it is possible that some BV isolates from captive macaques could in fact reflect viruses that are not natural to the host or even chimeric/recombinant viruses resulting from the practice of co-housing multiple taxa of NHP in captive environments. All these factors need to be borne in mind when assessing genetic diversity among viral isolates.
The results of this study reveal several interesting observations regarding BV strains isolated from captive macaques. Regarding different BV genotypes, the identification of various BV genotypes in different macaque species was originally based on comparison of only 1.3 kbp of sequence (3′ end of US4 – US5 - 5′ end of US4 ORF). (Smith et al., 1998) Despite the small number of non-rhesus BV isolates sequenced in this study, alignment of complete genome sequences confirmed that similar genotypic sequence differences occur throughout the BV genome. While BV isolates from only 5 of the 20 macaque species have been analyzed to date, the phylogenetic relatedness of various BV isolates consistently parallels that of the host macaque species, whether isolates are compared at the complete genome level, coding sequences only, or even just the 1.3 kbp of sequence originally used. These genotypic sequence differences can be useful in classifying BV isolates, but this same sequence divergence needs to be considered when using highly specific tests for detection of BV. For example, diagnostic PCR tests need to use primers located in conserved regions such that BV from different macaque species will be detected.
Differences between genotypes were also evident at the biological level in that all BVrh and the single BVcy isolate were uniformly lethal in mice while BVpt and BVlt isolates did not produce lethal infections. Whether or not this is true when these BV genotypes infect human or other primate species remains unknown, although a number of DeBrazza’s monkeys (Cercopithecus neglectus) did die of (and some survived) BVlt infection. (Thompson et al., 2000) The variation between coding sequences of BVrh/BVcy vs. BVpt/BVlt isolates was only ~10%, but differences were greater at the whole genome level and especially in non-coding regions. Sequence differences in intergenic non-coding sequences were largely unremarkable, and the location of regions of repeat sequences was the same in all BV isolates. Two things did however stand out between BVrh/BVcy and BVpt/BVlt isolates. First was the nature of the repeat region between the RS1 ORF and OriS. In BVrh and BVcy, this repeat region was comprised of multiple iterations of a mixture of repetitions of one sequence plus a partial fraction of it a ‘quasi-repeat region’). In contrast this region of both BVpt isolates consisted of 15 copies of a 7 bp repeat followed by three imperfect (quasi) copies of a 68 bp repeat. This region of the BVlt was also quite different from that of BVrh isolates, consisting of 12 copies of a 13 bp repeat and 4 copies of a 17 bp repeat, with 5 quasi-copies of an ~35 bp sequence interspersed before, between and following the other two repeat regions.
The second noteworthy difference between the BVrh/BVcy and BVpt/BVlt isolates was the UL39 ORF. In the closely related virus HVP2, the UL39 ORF determines neurovirulence of the virus in mice. (Black et al., 2014; unpublished) Of all the amino acid differences between the UL39 ORFs of lethal vs. non-lethal HVP2 subtypes, 59.0% of subtype-specific sequence differences are located in the N-terminal 20% of the protein with the C-terminal 80% being much more conserved (~2.0% amino acid sequence divergence). Comparing the aligned UL39 ORFs of BVrh/BVcy vs. BVpt/BVlt clades, 52.3% of all clade-specific amino acid differences are similarly confined to the N-terminal region and the C-terminal 80% of UL39 being highly conserved (~5.0% sequence divergence). While the UL39 gene is a neurovirulence factor in HSV, (Jacobson et al., 1989) the role of the N-terminal region of the HSV UL39 protein in neurovirulence is not known, nor has the role of the N-terminal region in neurovirulence been tested. Whether or not BV clade differences in UL39 influence neurovirulence in the mouse model remains to be determined.
The identification of three non-rhesus BV isolates as BVrh genotypes was unexpected since BV isolates to date have separated phylogenetically into distinct genotypes (Ohsawa et al., 2002b; Smith et al., 1998; Thompson et al., 2000) that phylogenetically parallel that of different macaque species. (Fan et al., 2017) Based on comparative sequence analysis it seems certain that two of these atypical BV isolates (7709642 & 9400371) are actually BVrh viruses. The identification of 9400371 as BVrh is not so surprising since there was a question of whether it was derived from a rhesus or cynomologus macaque. However, the identification of 7709642 (and M12-O) as BVrh implies that the macaques these isolates came from were at some point infected with a BVrh virus. Years ago it was not at all uncommon for different macaque species to be housed very near one another, and in some cases different NHP species were not even separated during capture/importation. Certainly such direct contact between NHP species would present an opportunity for cross-species BV infections, and cross-species BV infections where non-macaques were housed in close proximity to macaques have been documented. (Coulibaly et al., 2004; Thompson et al., 2000; Wilson et al., 1990)
While detailed records are no longer available, it is known that isolate 7709642 was obtained from a diagnostic specimen from a Japanese macaque in “the south Texas colony”. This is probably the “Arashiyama A troop” that was imported from Arashiyama, Japan in 1968 and released onto a ranch in south Texas. (Fedigan and Asquith, 1992) These Arashiyama monkeys would be expected to carry a BVfu virus since they were originally from Japan, a country with only one endemic taxa of nonhuman primate (M. fuscata). However, historical data indicate that rhesus macaques were inadvertently introduced into Japan in the 1960’s following their escape from a private tourist facility on the main Japanese island of Honshu. (https://www.nies.go.jp/biodiversity/invasive/DB/detail/10390e.html) The ecologically and behaviorally adaptive rhesus macaques proceeded to establish feral populations, successfully interbreeding and hybridizing with the native M. fuscata on Honshu. (Kawamoto et al., 2004; 2007; http://www.japantimes.co.jp/news/2017/02/21/national/japan-zoo-culls-monkeys-carrying-invasive-genes/#.WO7wB39SLV8) Given our findings that isolate 7709642 was consistent with BVrh and the documentation of rhesus macaques in Japan as early as the 1960’s, it is likely that this isolate represents an inadvertent cross-species transmission of BV in the wild. Despite being members of the same monophyletic species group, (Delson, 1980) Japanese macaques have been geographically isolated from mainland rhesus macaques for millennia; contact only occurs through human agency. Identifying introgression, both genetic and viral, following contact among species that would otherwise remain separate has implications for reconstructing evolutionary relationships and may be key for interpreting the inexplicable variability in apparent BV pathogenicity observed in captive versus free-ranging macaques (i.e., the lack of fatla zoonotic BV infections in Asia where exposure incidents are common).
The third outlier virus that typed as BVrh (M12-O) was isolated in 2005 from oral lesions of a 23 year old female bonnet macaque that had been born in a closed colony. (Scharf et al., 2008) One possible explanation for this is that the BV genotype indigenous to bonnet macaques is phylogenetically the same as BVrh. However, the topology of phylogenetic trees of all BV isolates to date parallel that of their host macaques species. Based on the phylogeny of macaque species, (Fan et al., 2017) it would be expected that BV indigenous to bonnet macaques would fall in a separate clade distinct from the other two major BV clades (BVrh/BVcy/BVfu and BVpt/BVlt). Since this is not the case, it seems likely that isolate M12-O represents a cross-species infection of the bonnet macaque with a BVrh virus. While the individual M12 was from a closed colony without contact with any other macaque species, the colony was at one time housed adjacent to pigtail macaques. While this could not explain the presence of BVrh in this monkey (BVpt would be expected), it is possible that long ago founders of the bonnet macaque colony acquired BVrh at some time when housed near rhesus or even as a result of shared support facilities, the virus since continuing to be transmitted within the colony.
The low level of genetic diversity among BVrh isolates is also of interest. While a similar low level of sequence diversity has been reported for HSV2, (Newman et al., 2015) the more closely related baboon virus HVP2 shows a much higher level of sequence diversity among strains. (Black et al., 2014) One possible explanation for this apparent discrepancy is that while the baboons from which HVP2 isolates were made were imported from various geographical locations over many years, this may not be true for the macaques from which BV isolates were made. Also, all 14 of the 15 BVrh strains sequenced here were isolated from rhesus monkeys bred at one of the US NPRCs, and nine came from the CaNPRC rhesus colony. (Huff et al., 2003) This particular rhesus colony was established in 1965 and subsequent to the ban on exportation of macaques from India in the 1970s all rhesus monkeys were captive bred, leading to concerns regarding the genetic diversity of captive macaque breeding colonies. (Kanthaswamy et al., 2012) Phylogenetic analyses grouped the nine CaNPRC isolates with other BVrh isolates. Five of these CaNPRC isolates (24105-G, 26896-G & -O, 31612-G, 32188-O and 32425-G) came from female macaques (2–3 years old plus one 14 year old) that had a history of having been in the same corral, and these isolates did constitute a phylogenetically very closely related subgroup of isolates together with one other BVrh strain (20620), so it is possible that some of the lack of genetic diversity among BVrh isolates may be due to a single virus strain circulating among animals in this one corral.
A lack of genetic diversity of BV isolates could reflect a general lack of genetic diversity of captive rhesus in the US. Rhesus macaques are one of the most geographically widespread monkey species, ranging from Afghanistan south into India, east into China and throughout Southeast Asia. (Burton and Eaton, 1995) As far as can be determined, all BVrh isolates used in this study were from Indian origin rhesus. Wild rhesus macaques have not been imported to the US from India since the late 1970’s, most recent imports being of Chinese origin. Rhesus from India and China are known to have a number of genetic differences, some of which affect the immune response. (Kanthaswamy et al., 2014) Given many years of captive breeding in the US without much external genetic import from wild animals, it is possible that captive rhesus do not represent anywhere near the level of genetic diversity present among the six subspecies of wild rhesus macaques. Comparing BV isolates from captive Indian origin rhesus with BV isolates obtained from free ranging rhesus across their natural geographical distribution will be necessary to address the true level of sequence diversity of BVrh isolates.
It was interesting that the two viruses that came from non-rhesus macaques (M12-O and 7709642) accounted for so many isolate-specific indels and nucleotide substitutions in alignments of all BVrh coding or whole genome sequences. One possible explanation for this is that if the non-rhesus host monkeys (M. fuscata and M. radiata) were infected with a BVrh virus, mutations within the genome of the virus could be expected to accumulate more rapidly than in the natural host as the virus adapts to the different macaque species. This would however not explain why isolates 16293 and especially 9400371 (both also BVrh viruses) had so many isolate-specific substitutions and indels. While isolate 9400371 could conceivably have been isolated from a cynomolgus monkey infected with a BVrh virus, isolate 16293 was isolated from a rhesus monkey. One possibility is that these four isolates (16293, 7709642, 9400371 & M12-O) that together account for >90% of all isolate-specific substitutions and indels among all BVrh isolates plus the remaining group of BVrh isolates represent five “lineages” of BVrh present in captive macaques, these lineages possibly being derived from BVrh isolates of monkeys captured in distinct geographical areas or from different sub-species of M. mulatta.
CONCLUSIONS
Complete genome sequencing confirmed the separation of BV isolates into distinct genotypes. Some isolates from non-rhesus macaques were found to be of the rhesus genotype, possibly resulting from cross-species infections. While substantial sequence variation exists among BV isolates of different genotypes, sequence variation is much less among isolates within a given genotype. Whether this low level of sequence variation is characteristic of BV or is possibly due to the restricted nature of captive macaques in terms of their bio-geographical origins remains to be determined. A clear understanding the genetic diversity (or lack of) among BV isolates will require analysis of many more virus strains isolated from free-ranging macaques, from macaques inhabiting varied geographical locations, and from a number of different macaque species.
Highlights.
Genome sequences of 19 B virus (Macacine alphaherpesvirus 1) isolates were determined
Differences between isolates from various macaque species were consistent with previously identified BV genotypes, and differences occurred throughout the viral genome
Coding regions of the BV genome were strongly conserved, showing a low level of sequence variation
Regions of repeated sequence occurred in specific areas of coding and non-coding regions, but the number of iterations of repeat units varied widely among isolates
Several BV isolates from were identified as BVrh genotypes despite having been isolated from non-rhesus macaques, indicating the occurrence of cross-species BV infections
Acknowledgments
The services of the OUHSC DNA Sequencing/Genomics Core Facility which provided Illumina genomic sequencing service and the OSU Genomics and Proteomics Center for Sanger sequencing are also acknowledged. This study was supported in part by US Public Health Service grants P40 OD010988, P20 GM103648 and R24 OD022013.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amen MA, Griffiths A. Identification and expression analysis of herpes B virus-encoded small RNAs. J Virol. 2011;85:7296–7311. doi: 10.1128/JVI.00505-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. NIH Primate Research Centers: A Major Scientific Resource, in: National Institutes of Health. U.S. Government Printing Office; 1971. [Google Scholar]
- Bennett AM, Harrington L, Kelly DC. Nucleotide sequence analysis of genes encoding glycoproteins D and J in simian herpes B virus. J Gen Virol. 1992;73:2963–2967. doi: 10.1099/0022-1317-73-11-2963. [DOI] [PubMed] [Google Scholar]
- Besecker MI, Harden ME, Li G, Wang XJ, Griffiths A. Discovery of herpes B virus-encoded microRNAs. J Virol. 2009;83:3413–3416. doi: 10.1128/JVI.02419-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D, Ohsawa K, Tyler S, Maxwell L, Eberle R. A single viral gene determines lethal cross-species neurovirulence of baboon herpesvirus HVP2. Virol. 2014:452–453. doi: 10.1016/j.virol.2013.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush LA, Black DH, Mccormack KA, Maxwell LK, Wright G, Ritchey JW, Payton ME, Eberle R. Papiine herpesvirus 2 as a predictive model for drug sensitivity of Macacine herpesvirus 1 (monkey B virus) Compar Med. 2014;64:386–93. [PMC free article] [PubMed] [Google Scholar]
- Burton F, Eaton M. The Multimedia Guide to the Non-Human Primates. Prentice Hall; Canada, Scarborough, Ontario: 1995. [Google Scholar]
- Coulibaly C, Hack R, Seidl J, Chudy M, Itter G, Plesker R. A natural asymptomatic herpes B virus infection in a colony of laboratory brown capuchin monkeys (Cebus apella) Lab Anim. 2004;38:432–438. doi: 10.1258/0023677041958891. [DOI] [PubMed] [Google Scholar]
- Craig KL, Hasan MK, Jackson DL, Engel GA, Soliven K, Feeroz MM, Wang X, Jones-Engel L, Linial ML. A seminomadic population in Bangladesh with extensive exposure to macaques does not exhibit high levels of zoonotic simian foamy virus infection. J Virol. 2015;89:7414–7416. doi: 10.1128/JVI.01065-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Offay JM, Fulton RW, Eberle R. Complete genome sequence of the NVSL BoHV-1.1 Cooper reference strain. Arch Virol. 2013;158:1109–1113. doi: 10.1007/s00705-012-1574-6. [DOI] [PubMed] [Google Scholar]
- Davidson WL, Hummeler K. B virus infection in man. Ann N Y Acad Sci. 1960;85:970–979. doi: 10.1111/j.1749-6632.1960.tb50017.x. [DOI] [PubMed] [Google Scholar]
- Delson E. Fossil macaques phyletic relationships and a scenario of development. In: Lindburg DG, editor. The macaques: Studies in Ecology Behavior and Evolution. Van Nostrand Reinhold Co; New York: 1980. pp. 10–30. [Google Scholar]
- Engel GA, Jones-Engel L, Schillaci MA, Suaryana KG, Putra A, Fuentes A, Henkel R. Human exposure to herpesvirus B-seropositive macaques, Bali, Indonesia. Emerg Infect Dis. 2002;8:789–795. doi: 10.3201/eid0808.010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel GA, Pizarro M, Shaw E, Cortes J, Fuentes A, Barry P, Lerche N, Grant R, Cohn D, Jones-Engel L. Unique pattern of enzootic primate viruses in Gibraltar macaques. Emerg Infect Dis. 2008;14:1112–1115. doi: 10.3201/eid1407.071643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estep RD, Messaoudi I, Wong SW. Simian herpesviruses and their risk to humans. Vacc. 2010;28(Suppl 2):B78–84. doi: 10.1016/j.vaccine.2009.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, Liu Y, Zhang Z, Zhao C, Li C, Liu W, Liu Z, Li M. Phylogenetic position of the white-cheeked macaque (Macaca leucogenys), a newly described primate from southeastern Tibet. Molec Phylog Evol. 2017;107:80–89. doi: 10.1016/j.ympev.2016.10.012. [DOI] [PubMed] [Google Scholar]
- Fedigan LM, Asquith PJ. Arashiyama Research as a Microcosm of Larger Trends in Primatoology. In: Itoigawa N, Sugiyama Y, Sackett GP, Thompson KR, editors. Topics in Primatology. Vol. 2. University of Tokyo Press; 1992. pp. 67–77. [Google Scholar]
- Fierer J, Bazely P, Braude AI. Herpes B virus encephalomyelitis presenting as ophthalmic zoster. A possible latent infection reactivated. Annal Intern Med. 1973;79:225–228. doi: 10.7326/0003-4819-79-2-225. [DOI] [PubMed] [Google Scholar]
- Fuentes A. Human culture and monkey behavior: Assessing the contexts of potential pathogen transmission between macaques and humans. Am J Primatol. 2006;68:880–896. doi: 10.1002/ajp.20295. [DOI] [PubMed] [Google Scholar]
- Fulton RW, d’Offay JM, Eberle R. Bovine herpesvirus-1: comparison and differentiation of vaccine and field strains based on genomic sequence variation. Vacc. 2013;31:1471–1479. doi: 10.1016/j.vaccine.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Gibson DC. Communication and collaboration in primatology. Am J Primatol. 1994;34:97–100. doi: 10.1002/ajp.1350340115. [DOI] [PubMed] [Google Scholar]
- Huff JE, Eberle R, Capitano J, Zhou SS, Barry PA. Differential detection of B virus and rhesus cytomegalovirus in rhesus macaques. J Gen Virol. 2003;84:83–92. doi: 10.1099/vir.0.18808-0. [DOI] [PubMed] [Google Scholar]
- Huff JL, Barry PA. B-virus (Cercopithecine herpesvirus 1) infection in humans and macaques: potential for zoonotic disease. Emerg Infect Dis. 2003;9:246–250. doi: 10.3201/eid0902.020272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull RN. B virus vaccine. Lab Anim Sci. 1971;21:1068–1071. [PubMed] [Google Scholar]
- Hull RN, Nash JC. Immunization against B virus infection. I. Preparation of an experimental vaccine. Am J Hygiene. 1960;71:15–28. doi: 10.1093/oxfordjournals.aje.a120086. [DOI] [PubMed] [Google Scholar]
- Hull RN, Peck FB, Jr, Ward TG, Nash JC. Immunization against B virus infection. II. Further laboratory and clinical studies with an experimental vaccine. Am J Hygiene. 1962;76:239–251. [PubMed] [Google Scholar]
- Jacobson JG, Leib DA, Goldstein DJ, Bogard CL, Schaffer PA, Weller SK, Coen DM. A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virol. 1989;173:276–283. doi: 10.1016/0042-6822(89)90244-4. [DOI] [PubMed] [Google Scholar]
- Jones-Engel L, Engel GA, Heidrich J, Chalise M, Poudel N, Viscidi R, Barry PA, Allan JS, Grant R, Kyes R. Temple monkeys and health implications of commensalism, Kathmandu, Nepal. Emerg Infect Dis. 2006;12:900–906. doi: 10.3201/eid1206.060030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Engel L, May CC, Engel GA, Steinkraus KA, Schillaci MA, Fuentes A, Rompis A, Chalise MK, Aggimarangsee N, Feeroz MM, Grant R, Allan JS, Putra A, Wandia IN, Watanabe R, Kuller L, Thongsawat S, Chaiwarith R, Kyes RC, Linial ML. Diverse contexts of zoonotic transmission of simian foamy viruses in Asia. Emerg Infect Dis. 2008;14:1200–1208. doi: 10.3201/eid1408.071430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthaswamy S, Johnson Z, Trask JS, Smith DG, Ramakrishnan R, Bahk J, Ng J, Wiseman R, Kubisch HM, Vallender EJ, Rogers J, Ferguson B. Development and validation of a SNP-based assay for inferring the genetic ancestry of rhesus macaques (Macaca mulatta) Am J Primatol. 2014;76:1105–1113. doi: 10.1002/ajp.22290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthaswamy S, Trask JS, Ross CT, Kou A, Houghton P, Smith DG, Lerche N. A large-scale SNP-based genomic admixture analysis of the captive rhesus macaque colony at the California National Primate Research Center. Am J Primatol. 2012;74:747–757. doi: 10.1002/ajp.22025. [DOI] [PubMed] [Google Scholar]
- Kawamoto Y, Kawamoto S, Kawai S, Sirai K, Toshida J, Ogiwara H, Shiratori D, Naoi Y. Status report of hybridization in an introduced population of rhesus macaques (Macaca mulatta) with native Japanese macaques (M. fuscata) in the Boso Peninsula, Chiba, Japan. Primate Res. 2007;23:81–89. [Google Scholar]
- Kwawmoto Y, Hagihara K, Aizawa K. Finding of hybrid individuals between native Japanese macaques and introduced rhesus macaques in the Bousou peninsula, Chiba, Japan. Primate Res. 2004;20:89–95. [Google Scholar]
- Keeble SA. B virus infection in monkeys. Ann NY Acad Science. 1960;85:960–969. doi: 10.1111/j.1749-6632.1960.tb50016.x. [DOI] [PubMed] [Google Scholar]
- Killeen AM, Harrington L, Wall LVM, Kelly DC. Nucleotide sequence analysis of a homologue of herpes simplex virus type 1 gene US9 found in the genome of simian herpes B virus. J Gen Virol. 1992;73:195–199. doi: 10.1099/0022-1317-73-1-195. [DOI] [PubMed] [Google Scholar]
- Lee MH, Rostal MK, Hughes T, Sitam F, Lee CY, Japning J, Harden ME, Griffiths A, Basir M, Wolfe ND, Epstein JH, Daszak P. Macacine herpesvirus 1 in long-tailed macaques, Malaysia, 2009–2011. Emerg Infect Dis. 2015;21:1107–1113. doi: 10.3201/eid2107.140162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RM, Lamers SL, Weiner B, Ray SC, Colgrove RC, Diaz F, Jing L, Wang K, Saif S, Young S, Henn M, Laeyendecker O, Tobian AA, Cohen JI, Koelle DM, Quinn TC, Knipe DM. Genome sequencing and analysis of geographically diverse clinical isolates of herpes simplex virus 2. J Virol. 2015;89:8219–8232. doi: 10.1128/JVI.01303-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa K, Black D, Ohsawa M, Eberle R. Genome sequence of a pathogenic isolate of monkey B virus (species Macacine herpesvirus 1) Arch Virol. 2014;159:2819–2821. doi: 10.1007/s00705-014-2130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa K, Black DH, Sato H, Eberle R. Sequence and genetic arrangement of the unique short region of monkey B virus (Cercopithecine herpesvirus 1) genome and its comparison with other primate herpesviruses. J Virol. 2002a;76:1516–1520. doi: 10.1128/JVI.76.3.1516-1520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa K, Black DH, Sato H, Rogers K, Eberle R. Sequence and genetic arrangement of the UL region of the monkey B virus (Cercopithecine herpesvirus 1) genome and comparison with the UL region of other primate herpesviruses. Arch Virol. 2003;148:989–997. doi: 10.1007/s00705-003-0011-2. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Black DH, Torii R, Sato H, Eberle R. Detection of a unique genotype of monkey B virus (Cercopithecine herpesvirus 1) indigenous to native Japanese macaques (Macaca fuscata) Compar Med. 2002b;52:546–550. [PubMed] [Google Scholar]
- Palmer AE. Herpesvirus simiae: historical perspective. J Med Primatol. 1987;16:99–130. [PubMed] [Google Scholar]
- Perelygina L, Zhu L, Zurkuhlen H, Mills R, Borodovsky M, Hilliard JK. Complete sequence and comparative analysis of the genome of herpes B virus (Cercopithecine herpesvirus 1) from a rhesus monkey. J Virol. 2003;77:6167–6177. doi: 10.1128/JVI.77.11.6167-6177.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchey JW, Payton ME, Eberle R. Clinicopathological characterization of monkey B virus (Cercopithecine herpesvirus 1) infection in mice. J Compar Pathol. 2005;132:202–217. doi: 10.1016/j.jcpa.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Rogers KM, Ritchey JW, Payton M, Black DH, Eberle R. Neuropathogenesis of Herpesvirus papio 2 in mice parallels infection with Cercopithecine herpesvirus 1 (B virus) in humans. J Gen Virol. 2006;87:267–76. doi: 10.1099/vir.0.81476-0. [DOI] [PubMed] [Google Scholar]
- Scharf BA, Wan CH, Bluth M, Eberle R, Videan EN, Smith E, Coplan J. Lethargy, ulcers, bronchopneumonia and death in two aged female bonnet macaques presumed to be caused by Cercopithicine herpesvirus I. J Med Primatol. 2008;37(Suppl 1):60–64. doi: 10.1111/j.1600-0684.2007.00264.x. [DOI] [PubMed] [Google Scholar]
- Simon MA, Daniel MD, Lee-Parritz D, King NW, Ringler DJ. Disseminated B virus infection in a cynomolgus monkey. Lab Anim Sci. 1993;43:545–550. [PubMed] [Google Scholar]
- Smith AL, Black D, Eberle R. Molecular evidence for distinct genotypes of monkey B virus (Herpesvirus simiae) which are related to the host macaque species. J Virol. 1998;72:9224–9232. doi: 10.1128/jvi.72.11.9224-9232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, Hilliard JK, Kittel D, Lipper S, Giddens WE, Black DH, Eberle R. Retrospective analysis of an outbreak of B virus in a colony of DeBrazza’s monkeys (Cercopithecus neglectus) Compar Med. 2000;50:649–657. [PubMed] [Google Scholar]
- Weigler BJ. Biology of B virus in macaque and human hosts: a review. Clin Infect Dis. 1992;14:555–567. doi: 10.1093/clinids/14.2.555. [DOI] [PubMed] [Google Scholar]
- Wilson RB, Holscher MA, Chang T, Hodges JR. Fatal Herpesvirus simiae (B virus) infection in a patas monkey (Erythrocebus patas) J Vet Diagn Invest. 1990;2:242–244. doi: 10.1177/104063879000200321. [DOI] [PubMed] [Google Scholar]