

Abstract
Rationale
Mitochondrial oxidative stress (mitoOS) has been shown to be increased in various cell types in human atherosclerosis and with aging. However, the role of cell type-specific mitoOS in atherosclerosis in the setting of advanced age and the molecular mechanisms remain to be determined in vivo.
Objective
The aim of this study was to examine the role of myeloid-cell mitoOS in atherosclerosis in aged mice.
Approach and Results
Lethally irradiated Ldl receptor-deficient mice (Ldlr−/−) were reconstituted with bone marrow from either wild type or mitochondrial catalase (mCAT) mice. mCAT transgenic mice contain ectopically expressed human catalase gene in mitochondria, which reduces mitoOS. Starting at the age of 36 weeks, mice were fed the Western-type diet (WD) for 16 weeks. We found that mitoOS in lesional myeloid cells was suppressed in aged mCAT→Ldlr−/− chimeric mice compared to aged controls, and this led to a significant reduction in aortic root atherosclerotic lesion area despite higher plasma cholesterol levels. Neutrophil extracellular traps (NETs), a pro-inflammatory extracellular structure that contributes to atherosclerosis progression, were significantly increased in the lesions of aged mice compared with lesions of younger mice. Aged mCAT→Ldlr−/− mice had less lesional neutrophils and decreased NETs compared with age-matched WT→Ldlr−/− mice, while young mCAT→ and WT→ Ldlr−/− mice had comparable numbers of neutrophils and similar low levels of lesional NETs. Using cultured neutrophils, we showed that suppression of mitoOS reduced 7-ketocholesterol-induced NET release from neutrophils of aged but not younger mice.
Conclusions
MitoOS in lesional myeloid cells enhanced atherosclerosis development in aged mice, and this enhancement was associated with increased lesional NETs. Thus, mitoOS-induced NET formation is a potentially new therapeutic target to prevent atherosclerosis progression during aging.
Keywords: atherosclerosis, neutrophil extracellular trap, mitochondria, reactive oxygen species
Graphical Abstract
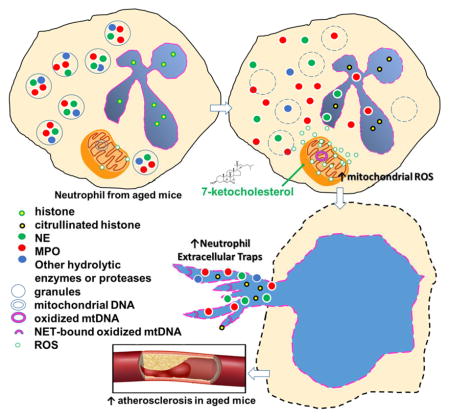
Introduction
Excessive mitochondrial oxidative stress (mitoOS) results in increased oxidative damage to mitochondrial DNA (mtDNA), proteins, and lipids. This process has been implicated in several human diseases including atherosclerosis and aging1–3. Studies from humans and animal models have confirmed that atherosclerosis and mtDNA damage increase exponentially with aging4–6, and mitoOS level correlates with atherosclerotic lesion progression during aging5, 7. We previously showed that excessive mitoOS in atherosclerotic lesional myeloid cells (macrophages) accelerated lesion development via enhanced inflammation and increased recruitment of monocytes in young Ldlr−/− mice8. However, roles of myeloid cell mitoOS in atherosclerosis during aging and cell-specific mechanisms of mitoOS require further investigation.
Neutrophils are the most abundant type of white blood cells and form an essential part of innate immunity. They contribute to acute inflammation by a number of different mechanisms including phagocytosis and degranulation or release of neutrophil extracellular traps (NETs).9–11 NETs are networks of extracellular fibrous material composed of neutrophil derived DNA and granule proteins. Neutrophils use these fibrous structures to trap and kill extracellular pathogens12. NET formation has also been implicated in the regulation of innate and acquired immune responses in chronic inflammatory and autoimmune diseases9, 13. The association of NETs with human and murine atherosclerotic lesions has been recently identified14. Further studies have revealed that cholesterol-crystal-induced NET formation in atherosclerotic lesions primes macrophages for cytokine release, thus amplifying immune cell recruitment and promoting diseases progression15. Mitochondrial ROS was also necessary for spontaneous NET release in granulocytes in individuals with systemic lupus erythematous, and released NET bound to oxidized form of mitochondrial mtDNA contributed to lupus-like disease13. However, the molecular mechanisms underlying NET formation in atherosclerosis are poorly understood.
Human NETs induced by certain stimuli (e.g., IL-8 or LPS) have been reported to be impaired in advanced aging16. Neutrophils from aged mice generated significantly fewer NETs when challenged with Staphylococcus Aureus17. These observations provided one of the mechanisms underlying declined innate immune functions in response to bacterial infection in aging. However, NETs induced by sterile inflammation or atherosclerotic-relevant stimuli in the setting of hyperlipidemia in advanced aging remains elusive. Given that mitoOS has been shown to increase with aging18, 19, we hypothesized that increased myeloid-cell mitoOS during aging could accelerate atherosclerosis progression by promoting NET formation in mice.
To address this question, we utilized the mitochondrial catalase (mCAT) transgenic mouse. Normally, catalase is an endogenous enzyme exclusively located in peroxisome. Catalase catalyzes the reduction of H2O2 and prevents its conversion into the most detrimental hydroxyl nitrites. The mCAT transgenic mouse ectopically expresses human catalase in mitochondria through a mitochondrial matrix–targeting motif, which quenches mitoOS in vivo and protects against mitoOS-induced damage3. To focus on myeloid cell in atherosclerosis, we performed transplantation of mCAT transgenic bone marrow cells into aged athero-prone Ldlr−/−mice. We showed that aged mCAT→Ldlr−/−mice had less lesional neutrophils and decreased NETs compared with age-matched WT→Ldlr−/− mice. Using cultured neutrophils, we showed that suppression of mitoOS reduced 7-ketocholesterol-induced NET release from neutrophils of aged but not younger mice.
Materials and Methods
We used male Ldlr−/− mice on C57BL/6 background. Materials and Methods are available in the online-only Data Supplement.
Results
Suppression of myeloid-cell mitoOS protects against atherosclerosis and reduces myeloid-cell accumulation in aged mice
Bone marrow from mCAT transgenic3 or littermate wild-type (WT) mice was transplanted into 8-week-old male Ldlr−/− recipient mice. Beginning at 36 weeks of age, mice were placed on a western-type diet (WD) for an additional 16 weeks. Oxidative damage to nuclear and mitochondrial DNA can be assessed by immunostaining for nuclear and non-nuclear 8-hydroxydeoxyguanosine (8OHDG) respectively20, 21. We utilized non-nuclear 8OHDG as the marker of mitoOS in the lesion8, 20. Because mitoOS occurs in various cell types in the lesions, including blood derived cells, vascular endothelial cells and smooth muscle cells8, sections were immunostained with the anti-Ly6G (neutrophil) 22, 23, anti-Mac3 (macrophage)24, the nuclear marker DAPI, and anti-8OHDG, or the respective isotype-matched IgGs as negative controls (Figure 1, Supplementary Figure I and IIA) to quantify the relative distribution of mitoOS in specific types of myeloid cells. We defined cells that contain Ly6G or Mac3 immunofluorescence signals surrounding a DAPI positive nuclei as authentic neutrophils or macrophages. For quantification, lesional sections from multiple mice were viewed and visualized by confocal fluorescence microscopy to look for punctate 8OHDG staining that was either cytoplasmic (localization of 8OHDG signal in juxtaposed with DAPI) or nuclear (co-localization of 8OHDG with DAPI). In lesions of mCAT mice, 8OHDG staining was reduced in both lesional neutrophils (Ly6G+)(Figure 1) and macrophages (Mac3+) (Supplementary Figure IIA). Moreover, in comparison with WT mice, aortic root lesion area was significantly reduced in the mCAT→Ldlr−/− chimeric mice (Figure IIA), whereas necrotic areas or necrotic core areas were comparable between the two groups (Supplementary Figure III). These findings suggest that myeloid-cell mitoOS regulates lesion size, but does not change necrotic core area in aged animals. Immunofluorescence staining showed that mCAT-positive lesions had >60% reduction of Ly6G+ cell number or cell density (cell number normalized to lesion area), as well as ~40% reduction of Mac3+ areas (Figure IIB–C, Supplementary Figure IIB) compared with control lesions. The protective effect mCAT was observed despite a ~30% increase in plasma cholesterol levels in the mCAT cohort (Supplementary Figure IV A) but comparable levels of weight gain, blood glucose and triglyceride between the two cohorts (Supplementary Figure IV B–D). Taken together, these data show that suppression of myeloid-cell mitoOS protects against atherosclerosis and reduces myeloid-cell accumulation despite elevated plasma cholesterol levels in aged mCAT→Ldlr−/− mice.
Figure 1. Suppression of myeloid cell mitochondrial oxidative stress in aged mCAT→Ldlr−/− mice.

36-week-old mCAT→Ldlr−/− and WT →Ldlr−/− (littermate control) chimeric mice were fed the Western-type diet for another 16 weeks and aortic root lesions were analyzed by confocal fluorescence microscopy. (A) Representative immunofluorescence staining of Ly6G (the marker of neutrophil, red), and non-nuclear 8OHDG staining (a marker of DNA oxidative damage, green) in the aortic root lesions from two groups of mice, with the intima outlined by the dotted line. In the merged images, the green 8OHDG signal locates either within the blue nuclei (arrowheads) or in juxtaposed with the blue nuclei (arrows). When mitochondrial DNA oxidative damage is suppressed, the green 8OHDG signal was exclusively restricted to nuclei region. The non-nuclei 8OHDG signal was abrogated and myeloid cells give red signals (arrowheads). The images in the 2nd or 4th rows are of higher magnifications of the boxed areas in the images above. Scale bar, 20μm (1st and 3rd rows) and 5μm (2nd and 4th rows). A, adventitia; I, intima; L,lumen. (B) Data were quantified as the percentage of non-nuclear 8OHDG+ cell among all Ly6G+ cells in intima per section, the absolute number of 8OHDG+Ly6G+ cells in intima per section, and number of 8OHDG+Ly6G+ cell normalized to lesion area. n = 9 WT vs. 5 mCAT mice; *P<0.05 using student’s t test.
Suppression of myeloid-cell mitoOS reduces NET formation in atherosclerotic lesions of aged mice
Given that mCAT lesions contained reduced number of Ly6G+ neutrophils in aged mice, we next examined whether this could lead to a decreased content of lesional NETs. Aortic root lesions from two groups of mice were stained with antibodies against citrullinated histone 3 (Cit-H3), myeloperoxidase (MPO), the key enzyme required for NET formation, and counter-stained with DAPI for nuclei (Figure 3). NET formation was then quantified as the areas that were covered by extracellular Cit-H3+MPO+structures (Figure 3A)15. We found that NETs were formed in the necrotic core areas in the intima, extending into a region of damaged medial elastic fibers throughout tunica media and reaching the adventitia in the control lesions (1st column in Figure 3A and Supplemental Figure V). These extracellular structures co-localized with neutrophil marker Ly6G and the oxidized mtDNA marker 8OHDG (Figure 3C). Such NET enriched with oxidized DNA are positive for mitochondrial inner membrane protein Tom20, and has been reported to activate type-I interferon gamma signaling and contribute to lupus like phenotypes in mice13. NETs also formed in non-necrotic areas, particularly in lesions adjacent to the opening of the coronary artery in aged WT lesions (Figure 3, 2nd). Most importantly, such NETs structures were seen much less frequently in the lesions of the mCAT group (Figure 3A, 3rd and 4th columns), with a ~65% reduction in average NET-covered areas and a ~60% reduction in the percentage of NET-covered areas normalized to total lesion size (Figure 3B). In addition, we observed that media erosion occurred in 4 out of 9 WT→Ldlr−/− lesions but in none of the 8 mCAT→Ldlr−/− lesions (Fisher Exact Test, P = 0.082, n = 9 WT and 8 mCAT) (Supplementary Figure V). These findings indicated a potential link between suppressed NET release and a strong trend toward reduced media degradation in mCAT lesions. The reduction in NET formation was associated with decreased number of cells positive for MPO, an enzyme required for NET formation (Supplementary Figure VI, 3rd and 4th plots).
Figure 3. The formation of neutrophil extracellular traps (NET) in atherosclerotic lesions was increased during aging, and mCAT expression limited NET formation only in the aged mice.

(A) Aortic root lesions from 36-week-old (aged) and 14-week-old (young) mice were placed on WD for additional 16 weeks were stained for citrullinated histone H3 (Cit-H3, a NET marker, red), myeloperoxidase (MPO, green) and nuclei (DAPI, blue), with intima and tunica media outlined. The bottom row were the H&E of neighboring sections with intima outlined in red dotted lines and necrotic core area outlined in blue lines (the 5th row). NET formed in both necrotic areas (the 1st and 3rd columns) and non-necrotic areas (the 2nd and 4th columns). White arrows depicted media erosion and white arrowheads depicted NET that extended to adventitia (the 1st column). Red stars depicted the emergence of the coronary artery in the bottom row. The images in the 4th row are of higher magnifications of the boxed areas in the images above. Scale bars, 40μm for rows #3 and 5; and 20μm for row #4. I, intima; NC, necrotic core; M, tunica media; A, adventitia. (B) NET formation was quantified as the areas that were covered by extracellular Cit-H3+ structures. Shown are the quantification of NET-covered areas per section and % of NET-covered areas per section normalized to lesion area. n = 9 WT vs. 5 mCAT aged mice; 6 WT vs. 7 mCAT young mice; * P < 0.05 using one-way ANOVA followed by Bonferroni post hoc test. (C) Aortic root lesions from two groups of aged mice were stained for Cit-H3 (magenta), Ly6G (red) and 8OHDG (green) and nuclei (blue). Shown are the co-localization of extracellular Cit-H3+structures with Ly6G and 8OHDG immunofluorescence signals in the arotic root lesions, which are depicted by white arrows. When mitoOS was suppressed, the extracellular Cit-H3 positive DNA structures was diminished, and thus did not overlap with 8OHDG signals. The images in the 2nd and 4th rows are of higher magnifications of the boxed areas in the 1st and 3rd rows respectively. Scale bars, 20 μm for rows #1 and 3, 10 μm for rows #2 and 4.
Suppression of myeloid-cell mitoOS does not reduce NET formation in atherosclerotic lesions of the younger mice
In view of the striking reduction of NET formation in aged mCAT lesions, we wondered if this was also the case in younger mice, who were placed on a WD for additional 16 weeks beginning at 14 weeks of age. We first compared the total lesion sizes between young and aged animals, and found that young animals have significant smaller lesion size than the aged cohorts (young vs. old WT→Ldlr−/− mice: 332504±139889 vs. 562548±79320 μm2; mCAT→Ldlr−/− mice: 229901±78137 vs. 491615±87921 μm2)8(Figure 2A). In contrast to the older WT mice, no medial erosion was observed in two groups of young mice (data not shown). The numbers of Ly6G+ were comparable between young WT and mCAT mice, and total numbers of Ly6G+ cells in the young mice were significantly less than the older cohorts (left graph of Figure 2C and Supplementary Figure VII-A). This was not due to the enhanced migratory capacity of neutrophils from aged mice (Supplementary Figure VIII). However, the Ly6G+ cell density (the number of Ly6G+ cells normalized to lesion size) was similar between aged and younger groups (right graph of Figure 2C and Supplementary Figure VII-B). Despite similar Ly6G+ cell density, there was similarly less lesional NETs area (Figure 3B upper graph) and NETs density (NETs area normalized to lesion area) in the young WT→ Ldlr−/− and mCAT→ Ldlr−/− mice (Figure 3B lower graph). Taken together, NET formation was increased in atherosclerosis during aging. Suppression of myeloid mitoOS decreased NETs in atherosclerotic lesions of aged mice but not in younger mice.
Figure 2. Suppression of myeloid cell mitochondrial oxidative stress protects against atherosclerosis and reduces myeloid-cell accumulation in aged mice.

(A) Shown are hematoxylin and eosin–stained aortic root lesions, with the intima marked by dotted lines, and total lesion area quantification (n= 9 WT vs. 8 mCAT; *P<0.05 using student’s t test). Scale bar, 40 μm. (B) Representative images of Ly6G staining (green) and counter-stained nuclei DAPI (blue) in the lesions from two groups of mice, with the intima outlined by the dotted line. The images in the 2nd or 4th rows are of higher magnifications of the boxed areas in the images above. Scale bar, 20 μm (1st and 3rd rows) and 10 μm (2nd and 4th rows). I, intima; A, adventitia. (C) Data were quantified and presented as No. of Ly6G+ cell in the intimal area per section or No. of Ly6G+ cell normalized to the intimal area. n= 9 WT vs. 5 mCAT; *P<0.05 using student’s t test.
Suppression of mitoOS abrogates 7KC-triggered NET formation in neutrophils from aged mice in vitro
Finally, we questioned whether the findings in vivo were related to a neutrophil cell-intrinsic effect that could be observed in neutrophils from aged mice in vitro. We first tested which stimuli could induce NET in neutrophils in vitro by testing several athero-relevant stimuli (oxLDL and 7-ketocholestero [7KC], the most abundant oxysterol in human oxLDL25 and atheroma26) and bacterial infection-relevant stimuli (LPS primed with TNF-α)16, 27. Neutrophils isolated from peripheral blood of three 58-week-old mice were treated with oxLDL (100μg/ml), 7KC (50μg/ml), or LPS (100ng/ml) with TNF-α priming (10ng/ml pretreatment) for 4 hr. PMA (200ng/ml), a potent chemical NET inducer, was used as the positive control. NETs were seen as string-like structure extending from the cell body that were positive for DNA and histone, which we quantified as extracellular material that was double-positive for DNA (DAPI+) and citrullinated H3 (Cit-H3+)28. We found that PMA and 7KC triggered NET formation in cultured neutrophils (Supplementary Figure IX). Although oxLDL or LPS with TNF-α priming was reported to induce NET formation in human neutrophils, a similar dose and period of stimulation only minimally induced NET formation in cultured mouse neutrophils (Supplementary Figure IX). We therefore focused on 7KC-induced NET as the in-vitro model for further investigation.
To quantify mitoOS in vitro, we used MitoSOX to selectively detect superoxide in the mitochondria of the live cells29. Confocal microscope images showed that MitoSOX signals exclusively localized to mitochondria marker Mitotracker Green (MTG), confirming the usefulness of MitoSOX to quantify ROS located in mitochondria (Supplementary Figure X). We isolated neutrophils from three 58-week-old mice and three 16-week-old gender-matched C57/BJ mice, and then incubated with 7KC. 7KC led to a more pronounced increase in the mean fluorescence intensity (MFI) of MitoSOX in neutrophils from both mice by flow cytometry, but the increment was greater in neutrophils from the aged mice (Figure 4A, the middle two bars). This increment was not related to changes in neutrophil mitochondrial mass, as quantified by the MFI of MTG using flow cytometry (Supplementary Figure XI). Pretreatment with the mitochondrial antioxidant mitoTEMPOL(10 μM) reduced the MFI of MitoSOX (Figure 4A, the right two bars) without reducing the MFI of the cytosolic ROS indicator, CellROX (Supplementary Figure XII), indicating that 10μM mitoTEMPOL suppressed mitochondrial ROS without affecting cytosolic ROS30. As another control, neutrophils treated with NADPH oxidase activator PMA also increases CellROX MFI. Such enhancement of CellROX MFI could be suppressed by 20μM NADPH oxidase inhibitor DPI31 but not by 10μM mitoOS inhibitor mitoTEMPOL (Supplementary Figure XIII). These observations further confirmed the specific role of mitoTEMPOL in quenching mitochondrial ROS but not cytosolic ROS. Previous work has shown that NETs-bound 8OHDG positive structures co-localized with the mitochondrial marker13, we therefore utilized 8OHDG as the marker for oxidized mitochondrial DNA (mtDNA). As shown in Figure 4B, 7KC led to NET formation in ~40% of the neutrophils from aged mice. Most importantly, suppression of mitochondrial ROS with mitoTEMPOL, which was verified by a decrease in mitoSOX MFI (Figure 4A), suppressed NETs release, decreased extracellular DAPI+ Cit-H3+ area (Figure 4B) and reduced oxidized mtDNA-bound NET formation (depicted by arrows in Figure 4B and Supplementary Figure XIV). In contrast, NETs were formed in only ~20% of neutrophils from the younger mice, and mitoTEMPOL did not decrease total NETs or oxidized mtDNA-bound NETs in these cells (Figure 4B and Supplementary Figure XIV). The impact of 7KC and mitoTEMPOL on NET formation in both mice (Figure 4B) well paralleled that on MitoSOX (Figure 4A).
Figure 4. Inhibition of mitoOS by mitoTEMPOL reduced NET formation in neutrophils from aged mice.

(A) Neutrophils isolated from 58-week-old (old) and 16-week-old (young) WT mice were stained with 5μM mitochondrial superoxide indicator MitoSOX. Cells were then incubated with vehicle or mitochondria-targeted antioxidant mitoTEMPOL (10μM) for 30 min followed by incubation with vehicle or 7KC for additional 4 hr. Mitochondrial ROS production was quantified as the mean fluorescence intensity (MFI) of MitoSOX by flow cytometry. (B) Isolated neutrophils were pre-incubated with vehicle or 10μM mitoTEMPOL for 30 min followed by incubation with 7KC for 4 hr. Cells were then fixed and stained with antibody against Cit-H3 (red), 8OHDG (reflective of mitochondrial DNA oxidative damage, green) and DAPI (blue), and visualized by fluorescence microscopy. Shown were the representative images. The images in the 5th row are of higher magnifications of the boxed areas in the 4th row. Scale bars, 5μm for the 1st – 4th rows, 1.25μm for the 5th row. Arrows depicted oxidized mitochondrial DNA-bound NETs where antibodies against 8OHDG co-localized with Cit-H3 positive chromosome DNA. Data were quantified in the graphs below, as the percentage of neutrophils that release NETs (the upper graph) and the area of NET+ structure normalized to the number of NET+ cells (the lower graph). Each dot represented the average of 16 randomly picked-up areas in each well, quadruplets for each animal. n = 3 young and 3 old mice. * P <0.05 using one-way ANOVA followed by Bonferroni post hoc test. N.S., not significant.
Discussion
In view of the presence of mitoOS in aging and human atherosclerotic vascular diseases, the current study provides in vivo evidence for the role of endogenous neutrophil mitoOS in atherosclerosis during aging and demonstrated that one of the mechanisms might involve the formation of NETs. Given that NETs promoted atherosclerosis progression15, 32, we think that suppression of NET formation in aged mCAT→Ldlr−/− mice could be at least partly responsible for the decrease in lesion area and cellularity. Interestingly, suppression of mitoOS reduced NETs in aged mice but not in younger mice. These observations suggest that neutrophil mitoOS contributes to atherosclerosis lesion progression specifically during aging. In agreement with these in-vivo observations, we found that younger Ldlr−/− mice did not display significant NET formation, paralleling their lower level of mitoOS, which may explain previous observations that mitochondrial membrane potential uncouplers, FCCP and dinitrophenol, suppressed mitoOS but did not inhibit PMA-induced NET formation in cultured neutrophils isolated from healthy young adults33. Increased ROS production by mitochondria, increased 8OHDG content in the mtDNA and mitochondrial oxidative damage have been consistently reported to be associated with aging18, 34. We showed that 7KC, an oxysterol found in human atheroma26, led to a more profound mitoOS production in aged neutrophils compared to the young neutrophils. The mechanism may have been related to decreased antioxidant enzyme activity, cumulative damage to mitochondria, and mitochondrial DNA (mtDNA) during aging35, 36. Further, the increase in mitoOS production correlated with the enhanced NET formation in neutrophils from aged mice, and both were suppressed by mitoTEMPOL. These findings are consistent with our hypothesis that increased mitoOS contributes to neutrophil NET formation during aging via a cell-intrinsic manner. However, these findings does not rule out the possibilities that reduced content of 7KC37 or other NET inducers, such as damage-associated molecular pattern (DAMP) released or ROS leaking from other types of cells38, also contributed to decreased neutrophil mitoOS and NET formation in aged mCAT lesion in vivo, especially with the observation that total macrophage contents and macrophage mitoOS also decreased in mCAT lesions (Supplementary Figure II).
Several stimuli have been reported to stimulate NET in human neutrophils in vitro, such as LPS plus TNF-α priming as pathogen associated molecular pattern (PAMP), oxLDL and cholesterol crystal as athero-prone stimuli, High Mobility Group Box 1(HMGB1) and histones as DAMP38. However, a similar dose and incubation period with LPS plus TNF-α priming did not induce NET formation in mouse neutrophils in vitro (Supplementary Figure IX). OxLDL seems to induce neutrophil apoptosis rather than NET formation, as indicated by the shrinkage of nuclei stained by DAPI (Supplementary Figure IX). Such a discrepancy between human and mouse neutrophils could be due to the differences in TLR properties and signaling between man and mouse, particularly the stronger effect of LPS in human than mouse immune cells39. From the translational perspective, it would be interesting to investigate the existence of 7KC-mitoOS-NET pathway and related pathological roles in human neutrophils from aged subjects.
In this study, we first reported that 7KC could trigger NETs in mouse neutrophils partly through mitoOS (Supplementary Figure IX and Figure 4), although the molecular mechanisms remain to be elucidated. Studies have shown that 7KC triggers multiple types of stress and signaling pathways, including NADPH oxidative-mediated oxidative stress40, mitochondrial oxidative stress8, ER stress and protein kinase R (PKR) activation40, autophagy and even caspase-mediated cell apoptosis41. Therefore, there may be other mechanisms involved in 7KC-induced NETs such as NADPH oxidase-induced oxidative burst27, peptidylarginine deiminase (PAD) enzyme activation42, neutrophil elastase translocation to the nucleus15, as well as paracrine effects including HMGB1 release from macrophage 38 or NET enriched in oxidized mtDNA from neighboring neutrophils13.
Several recently identified NET inducers, including cholesterol crystals and components of oxidized LDL15, 27, are also capable of generating mitoOS and generating oxidized mtDNA8. In this regard, previous work has shown that binding of oxidized mtDNA components to released chromatin activates monocytes and macrophages, triggers IL-1, IL-17, and interferon signaling reactions, promotes inflammation in the spleen and kidney, and amplifies sustained inflammation in lupus-like diseases13. The current study identified a novel athero-relevant NETs inducer 7KC. Importantly, 7KC also resulted in the structure of oxidized mtDNA-bound NETs, which could be quenched by mitoTEMPOL in neutrophils from aged animals (Figure 3B and Supplemental Figure VII). We therefore propose that mitoOS promotes atherosclerosis in aged animals by promoting NET formation and by producing highly pro-inflammatory oxidized mtDNA-bound NETs.
Finally, it is important to note that lesions of the two groups of aged mice had comparable necrotic (core) areas, suggesting a dispensable role for myeloid-mitoOS in regulating plaque necrosis during aging. Interestingly, young mCAT chimeric mice had significantly reduced total lesion area and less Mac3+ macrophages in the intima compared with young WT chimeric mice8, while neutrophil infiltration and NET formation did not differ between the two groups of younger mice. However, we observed that NETs localized to necrotic areas and regions with medial degradation suggesting a potential unidirectional link between medial degradation and NETs (Figure 3 and Supplementary Figure V). Observations in human lesion by Inota, et al. also showed that high neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions43. Due to the limited sample size in our study, we could not firmly conclude that suppression of mitoOS in myeloid cells were associated with decreased medial degradation in age mice (Supplementary Figure V). Future studies will be needed to establish causation and elucidate potential mechanisms linking plaque necrosis to NET formation. If plaque necrosis is indeed trigger NET formation, that process would be yet another pathologic consequence of necrotic cores in vulnerable plaques, i.e., in addition to the role of necrotic cores in inflammation, plaque disruption, and thrombosis44, 45.
Supplementary Material
Highlights.
This article provides causative in vivo evidence that quelling mitoOS in myeloid cells suppresses atherosclerosis in aged mice.
Suppression of mitoOS in myeloid is associated with reduced formation of neutrophil extracellular traps (NETs) in atherosclerotic lesions of aged mice.
7-ketocholesterol, the most abundant oxysterol in human atheroma, induces NET release from neutrophils in vitro.
Suppression of mitoOS abrogates 7-ketocholesterol-triggered NET formation in neutrophils from aged mice but not from the younger mice in vitro.
Acknowledgments
Acknowledgements: We thank George Kuriakose for technical contributions related to the atherosclerosis assays; Dr. Gary Wang for editing the manuscript; Dr. Peter Rabinovitch and Jeanne Fredrickson for help with the mCAT-expressing mice; Dr. Carrie Welch for the help with atherosclerosis histology analysis.
Source of Funding: This study was supported by an American Heart Association predoctoral training grant and the American Philosophical Society Daland Fellowship in Clinical Investigation to Y.W, by National Institutes of Health (NIH) grants to I.T. (R01HL075662, R01HL106019), to A.T. (R01HL107653) and to N.W.(R01HL118567).
Disclosures: none.
Abbreviations
- mCAT
mitochondrial catalase
- ROS
reactive oxygen species
- mitoOS
mitochondrial oxidative stress
- mtDNA
mitochondrial DNA
- NETs
neutrophil extracellular traps
- 8OHDG
8-hydroxydeoxyguanosine
- WD
western-type diet
- Cit-H3
citrullinated histone 3
- MPO
myeloperoxidase
- 7KC
7-ketocholesterol
References
- 1.Lee HC, Wei YH. Mitochondria and aging. Advances in experimental medicine and biology. 2012;942:311–327. doi: 10.1007/978-94-007-2869-1_14. [DOI] [PubMed] [Google Scholar]
- 2.Madamanchi NR, Zhou RH, Vendrov AE, Niu XL, Runge MS. Does oxidative DNA damage cause atherosclerosis and metabolic syndrome?: New insights into which came first: The chicken or the egg. Circulation research. 2010;107:940–942. doi: 10.1161/CIRCRESAHA.110.230904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 4.Simons LA. Epidemiologic considerations in cardiovascular diseases in the elderly: International comparisons and trends. The American journal of cardiology. 1989;63:5H–8H. doi: 10.1016/0002-9149(89)90108-2. [DOI] [PubMed] [Google Scholar]
- 5.Dorighello GG, Paim BA, Leite AC, Vercesi AE, Oliveira HC. Spontaneous experimental atherosclerosis in hypercholesterolemic mice advances with ageing and correlates with mitochondrial reactive oxygen species. Experimental gerontology. 2017 doi: 10.1016/j.exger.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Sobenin IA, Zhelankin AV, Sinyov VV, Bobryshev YV, Orekhov AN. Mitochondrial aging: Focus on mitochondrial DNA damage in atherosclerosis - a mini-review. Gerontology. 2015;61:343–349. doi: 10.1159/000368923. [DOI] [PubMed] [Google Scholar]
- 7.Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, Runge MS, Madamanchi NR. Nox4 nadph oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxidants & redox signaling. 2015;23:1389–1409. doi: 10.1089/ars.2014.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappab-mediated inflammation in macrophages. Circulation research. 2014;114:421–433. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 10.Nathan C. Neutrophils and immunity: Challenges and opportunities. Nature reviews. Immunology. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 11.Segal AW. How neutrophils kill microbes. Annual review of immunology. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nature immunology. 2014;15:1017–1025. doi: 10.1038/ni.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nature medicine. 2016;22:146–153. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Megens RT, Vijayan S, Lievens D, Doring Y, van Zandvoort MA, Grommes J, Weber C, Soehnlein O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thrombosis and haemostasis. 2012;107:597–598. doi: 10.1160/TH11-09-0650. [DOI] [PubMed] [Google Scholar]
- 15.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–320. doi: 10.1126/science.aaa8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hazeldine J, Harris P, Chapple IL, Grant M, Greenwood H, Livesey A, Sapey E, Lord JM. Impaired neutrophil extracellular trap formation: A novel defect in the innate immune system of aged individuals. Aging cell. 2014;13:690–698. doi: 10.1111/acel.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tseng CW, Kyme PA, Arruda A, Ramanujan VK, Tawackoli W, Liu GY. Innate immune dysfunctions in aged mice facilitate the systemic dissemination of methicillin-resistant s. Aureus. PloS one. 2012;7:e41454. doi: 10.1371/journal.pone.0041454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:4533–4537. doi: 10.1073/pnas.87.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawada M, Carlson JC. Changes in superoxide radical and lipid peroxide formation in the brain, heart and liver during the lifetime of the rat. Mechanisms of ageing and development. 1987;41:125–137. doi: 10.1016/0047-6374(87)90057-1. [DOI] [PubMed] [Google Scholar]
- 20.Wu LL, Chiou CC, Chang PY, Wu JT. Urinary 8OHDG: A marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clinica chimica acta; international journal of clinical chemistry. 2004;339:1–9. doi: 10.1016/j.cccn.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rotzius P, Thams S, Soehnlein O, Kenne E, Tseng CN, Bjorkstrom NK, Malmberg KJ, Lindbom L, Eriksson EE. Distinct infiltration of neutrophils in lesion shoulders in apoe−/− mice. The American journal of pathology. 2010;177:493–500. doi: 10.2353/ajpath.2010.090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. The Journal of experimental medicine. 2013;210:1283–1299. doi: 10.1084/jem.20122220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watt V, Chamberlain J, Steiner T, Francis S, Crossman D. Trail attenuates the development of atherosclerosis in apolipoprotein e deficient mice. Atherosclerosis. 2011;215:348–354. doi: 10.1016/j.atherosclerosis.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terasaka N, Wang N, Yvan-Charvet L, Tall AR. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via abcg1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15093–15098. doi: 10.1073/pnas.0704602104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;142:1–28. doi: 10.1016/s0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 27.Awasthi D, Nagarkoti S, Kumar A, Dubey M, Singh AK, Pathak P, Chandra T, Barthwal MK, Dikshit M. Oxidized ldl induced extracellular trap formation in human neutrophils via tlr-pkc-irak-mapk and nadph-oxidase activation. Free radical biology & medicine. 2016;93:190–203. doi: 10.1016/j.freeradbiomed.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Hirose T, Hamaguchi S, Matsumoto N, Irisawa T, Seki M, Tasaki O, Hosotsubo H, Yamamoto N, Yamamoto K, Akeda Y, Oishi K, Tomono K, Shimazu T. Presence of neutrophil extracellular traps and citrullinated histone h3 in the bloodstream of critically ill patients. PloS one. 2014;9:e111755. doi: 10.1371/journal.pone.0111755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clark RA. Activation of the neutrophil respiratory burst oxidase. The Journal of infectious diseases. 1999;179(Suppl 2):S309–317. doi: 10.1086/513849. [DOI] [PubMed] [Google Scholar]
- 31.O’Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil nadph oxidase. The Biochemical journal. 1993;290( Pt 1):41–49. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT, Kaplan MJ. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circulation research. 2014;114:947–956. doi: 10.1161/CIRCRESAHA.114.303312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirchner T, Moller S, Klinger M, Solbach W, Laskay T, Behnen M. The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediators of inflammation. 2012;2012:849136. doi: 10.1155/2012/849136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Capel F, Rimbert V, Lioger D, Diot A, Rousset P, Mirand PP, Boirie Y, Morio B, Mosoni L. Due to reverse electron transfer, mitochondrial h2o2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mechanisms of ageing and development. 2005;126:505–511. doi: 10.1016/j.mad.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. Journal of signal transduction. 2012;2012:646354. doi: 10.1155/2012/646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnheim N, Cortopassi G. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutation research. 1992;275:157–167. doi: 10.1016/0921-8734(92)90020-p. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez IR, Clark ME, Lee JW, Curcio CA. 7-ketocholesterol accumulates in ocular tissues as a consequence of aging and is present in high levels in drusen. Experimental eye research. 2014;128:151–155. doi: 10.1016/j.exer.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, Tsung A. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62:600–614. doi: 10.1002/hep.27841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Critical reviews in immunology. 2014;34:433–454. [PubMed] [Google Scholar]
- 40.Li G, Scull C, Ozcan L, Tabas I. Nadph oxidase links endoplasmic reticulum stress, oxidative stress, and pkr activation to induce apoptosis. The Journal of cell biology. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell metabolism. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo netosis, which impairs wound healing. Nature medicine. 2015;21:815–819. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ionita MG, van den Borne P, Catanzariti LM, Moll FL, de Vries JP, Pasterkamp G, Vink A, de Kleijn DP. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1842–1848. doi: 10.1161/ATVBAHA.110.209296. [DOI] [PubMed] [Google Scholar]
- 44.Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circulation research. 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721. [DOI] [PubMed] [Google Scholar]
- 45.Tabas I. Pulling down the plug on atherosclerosis: Finding the culprit in your heart. Nature medicine. 2011;17:791–793. doi: 10.1038/nm0711-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.