

Abstract
Structure-based vaccine design depends on extensive structural analyses of antigen-antibody complexes. Single-particle electron cryomicroscopy (cryoEM) can circumvent some of the problems of x-ray crystallography as a pipeline for obtaining the required structures. We have examined the potential of single-particle cryoEM for determining the structure of influenza-virus hemagglutinin (HA):single-chain Fv (scFv) complexes, by studying a complex we failed to crystallize in pursuing an extended project of the human immune response to influenza vaccines. The result shows that a combination of cryoEM and molecular modeling can yield details of the antigen:antibody interface, although small variation in the twist of the rod-like HA trimer limited the overall resolution to about 4.5Å. Comparison of principal 3D classes suggests ways to modify the HA trimer to overcome this limitation. A closely related antibody from the same donor did yield crystals when bound with the same HA, giving us an independent validation of the cryoEM results The two structures also augment our understanding of receptor-binding site recognition by antibodies that neutralize a wide range of influenza-virus variants.
Graphical Abstract
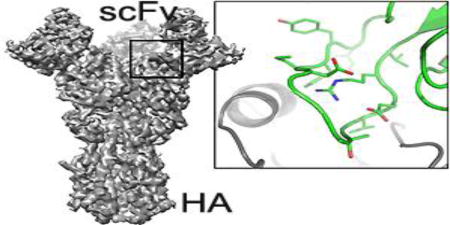
Introduction
Structure-based vaccine design, made possible by new technologies for generating pathogen-specific human monoclonal antibodies, requires large-scale structural analysis of antigen-antibody complexes [1]. The outlines of the problem have become particularly evident from work on HIV and influenza virus [2]. Single-particle electron cryomicroscopy (cryoEM), which can circumvent some of the problems of x-ray crystallography, has contributed substantially to the study of antibody complexes with the HIV envelope glycoprotein (Env) [3, 4]. It has helped to define epitopes not restricted to the core of the receptor-binding gp120 fragment and to establish the relationship of all identifiable epitopes with respect to the trimeric Env ectodomain.
The trimeric influenza-virus hemagglutinin (HA) ectodomain [5], in association with one antigen-binding fragment (Fab) or single-chain variable-domain fragment (scFv) per subunit, generates a complex with a total mass of 250–300 kDa, well within the range of contemporary, high-resolution cryoEM methods, but nonetheless substantially smaller than any comparable HIV gp140:Fab complex. We have examined, in the work described here, the potential of single-particle cryoEM for determining the structure of influenza-virus HA:scFv complexes. In particular, we describe the structure of a complex we had failed to crystallize, part of a broad study of antibodies from individuals who received the 2007–2008 trivalent inactivated vaccine (TIV) [6]. We compare the result with the crystal structure of the Fab of a closely related antibody from the same donor, bound with the same HA. These antibodies bind HA from nearly all members of a large panel of historical H1 influenza isolates, and a further goal of the studies was to visualize the HA:antibody interface.
The cryoEM structure shows that a combination of cryoEM and molecular modeling can yield details of the antigen:antibody interface. Comparison of principal 3D classes suggests ways to modify the HA trimer to achieve an overall resolution higher than the nominal 4.8 Å of our current reconstruction. The related crystal structure, an independent validation of the cryoEM results, pointed to a local adjustment in the model for the third heavy-chain complementarity region (CDRH3) that improved its agreement with the cryoEM density map. The two structures augment our understanding of receptor-binding site recognition by antibodies that neutralize a wide range of influenza-virus variants.
Results
B-cell repertoire of donor TIV24
The HA-directed antibodies we have studied came from a vaccinee who received the 2007–2008 TIV [6]. Paired-chain sequencing of heavy- and light-chain cDNA, from individual B cells obtained seven days post vaccination, allowed expression of recombinant antibodies and in some cases derivation of lineages when more than one sequence derived from the same germline rearrangement were available. Two antibodies, K1912 and K1915 had the same heavy chain gene rearrangement and very closely related third complementarity determining regions (CDRH3s), but paired with a different light chain (Fig. S1). K1912 is one of four antibodies defining a lineage, designated clone 2897 (Fig. S1). Because its light chain is distinct from that of the 2897 clone, K1915 is technically an "orphan" (no other lineage members), but its heavy chain is almost certainly a fifth descendent of the same V(D)J recombination event that led to the clone. The two cDNA sequences are consistent with receptor "revision" after an initial light-chain gene rearrangement [7, 8].
Reconstructions from rosettes
We began by considering "rosettes" of intact HA trimers [9], as a way of generating higher image contrast than isolated HAs and aiding particle alignment. HA, solubilized from virions with mild detergent (e.g., β-octyl glucoside) and then dialyzed against detergent-free buffer, associates through its transmembrane segments into small aggregates with the ectodomains projecting radially outwards. We obtained initial, low resolution results by this approach, but it soon became apparent that new instrumentation and software (in particular, direct detectors, dose fractionation, movie processing, and maximum-likelihood 3D classification) would allow us to reconstruct individual HA ectodomain trimers with bound Fabs. Although we could therefore avoid working with rosettes, the rosette method, modified as described here, helped us obtain an objective starting point for the single-particle analysis of individual Fab:HA complexes. Besides aiding the alignment of small proteins and complexes, this type of approach may also be useful when particles show strongly preferred orientations. By forming rosettes, a broader range of orientations is imposed to allow reconstruction with more isotropic resolution.
Instead of relying on hydrophobic association of transmembrane segments to generate rosettes, we used His-tagged, trimeric HA ectodomain and Ni-NTA-coated gold clusters as nucleation sites. The resulting rosettes are readily recognized in cryoEM images (Fig. 1). The gold clusters allow accurate localization of the rosette centers and help identify side views of individual HA trimers for further processing as single particles. We could usually select two to three single trimers from good clusters (see Methods). Starting with about 5000 HA trimers thus selected, we used a simple cylinder model as an initial reference (Fig. 1) to perform iterative refinement by projection matching (see Methods and Fig. S2). Knowledge of the approximate in-plane alignments of the trimers, based on their mean radial direction from the center of the cluster, substantially facilitated projection matching. The prior knowledge of approximate orientation of the threefold axis allowed us to exclude particles that deviated significantly from their initial alignments and to calculate a reconstruction at a resolution between 15 and 20 Å (Fig. 1) from a subset of about 3000 trimers. This reconstruction served as an initial model for alignment of HA trimers bound to antibody fragments.
Fig. 1.

Use of gold-bead HA rosettes to derive starting model. Upper row: (left) a field from a cryomicrograph with two rosettes in red circles, scale bar = 500 Å; (center) magnified images of those two rosettes, with arrows indicating the HA trimers shown boxed on the right. An arbitrary, cylindrical model (one example shown) led to the low-resolution HA trimer model, used for subsequent work.
Single-particle reconstruction of HA bound with K1915 antibody fragments
Preliminary studies with the Fab suggested that the constant module, which projected radially, might have enough variability in orientation to compromise refinement of particle parameters. We therefore carried out subsequent studies with the single-chain Fv (scFv). We collected a large dataset of scFv:HA complexes using a K2 Summit counting detector (Gatan, Inc.) mounted on an FEI Titan Krios electron microscope, operated at 300 kV and 80° K (see Methods). We initially selected about 250,000 particles from 10,280 "movies" and carried out 2D classification in RELION [10] to detect and remove distorted particles as well as any that had lost bound scFv (Fig. 2). We aligned the 142,314 particles that passed this filter and calculated a 3D reconstruction with Frealign [11]. Inspection of the resulting map showed that density in the region of the HA stem and head was better defined than the density corresponding to the scFv. We therefore carried out 3D classification, with four classes, again using Frealign (Fig. 3). Comparing superposed maps from each of the classes revealed that a twist of the complex around its threefold axis related one class to another (Figs. 4 and S3). The source of the twisting is not evident, but it could be related to particle distortion during sample freezing or simply to alternative fee-energy minima in the trimer, due to the absence of constraints at either end (such as crystal-lattice contacts or membrane-embedded transmembrane segments). The overall resolution of the reconstructed densities ranged from 4.8 to 6.0 Å (Fig. S4A). Three of the four classes showed high-resolution features with side-chain density in the best-resolved regions (e.g., the central helices of HA). The interface between the scFv and the HA was not as well defined, however, and parts of the scFv distal to the HA interface were even less so, suggesting that four classes may not have captured all conformational states. More classes would have reduced the overall definition of the reconstructions owing to the smaller numbers of particles in each class, and we attempted instead to improve the map by local averaging of densities (see next section).
Fig. 2.

Examples of results at different data processing stages for HA:scFv complex. (a) Field from cryomicrograph. (b) Selected particles. (c) 2D class averages showing views both along and normal to the threefold axis. The selected particles in (b) belong to the classes in the corresponding positions in (c) Scale bars in (a), (b), (c) represent 250 Å.
Fig. 3.

Surface representations of 3D class averages for HA:scFv complex. The labels include the number of particles in each class and the corresponding percentage of the total particles included in the computation. Beside each map is a detail from the long central helix of HA2, to illustrate resolution differences among the classes. The resolutions given are from the half-map FSC for each class (see Fig. S4, which also includes the FSC for each map with its corresponding refined model).
Fig. 4.

Superposition of class 2 (blue) and class 4 (cyan) models, illustrating difference in twist around the threefold axis. The models were fit to the final reconstructions by real-space refinement and inspection shows that they accurately represent the key differences between the two maps. The two models are aligned at the base of HA2, so that a relative circumferential displacement of the upper surface of HA1 illustrates the overall twist of the molecule. Both views include only the HA part of the model. The top view (right) shows about a 10° rotation of one head with respect to the other.
HA:antibody interface of K1915
We averaged density, after applying rotational and translational alignments to superpose the densities of the three best classes near the HA:Fv interface, in order to enhance the definition of features in the density map at the antigen-antibody contact. We docked atomic models of HA (from x-ray crystallography) and Fab (from a near homolog) into the density, obtained optimal fits using UCSF Chimera [12], and calculated rigid-body transformations of HA-head:scFv substructures to derive the density-averaging operators. This procedure made the density at the interface somewhat more interpretable (Fig. 5). We assembled a model as described in the next paragraph and adjusted it to fit the map for each of the four classes, using real-space refinement as implemented in Phenix [13] (Table S1).
Fig. 5.

Density from classes 1,2, and 4 for an HA "head" and bound scFv and average of the three superposed on the region shown in the red ovals. There was a slight enhancement of density at the interface, helpful for building the CDRH3 loop.
The model building used the known structure of the H1 A/Solomon Islands/03/2006 HA [14], which we re-refined against reprocessed data (revised PDB deposition 5UGY), and a good homology model for the scFv, from known structures of heavy- and light-chain variable domains with similar sequences (PDB 4K8R). The one segment that required fully de novo modeling was the unusually long, heavy-chain third complementarity determining region (CDRH3), which inserts into the HA receptor-binding pocket. Features for several large side chains helped establish an unambiguous sequence register (Fig. 6). The loop folds into the receptor-binding pocket, with some of the features of sialic-acid mimicry we have seen in other receptor-binding site (RBS) directed antibodies. In particular, the model showed contacts of Val102-Gly103 with Gly135-Ser136 and Trp153 on HA. The valine position and likely non-bonded contacts were like those of the sialic-acid acetamido group, and the glycine carbonyl appeared to occupy the same position as one of the sialic-acid carboxylate oxygens.
Fig. 6.

Conformation of K1915 CDRH3 in the HA receptor-binding site. (a) Backbone trace, with density break at Gly-Gly sequence (asterisks in figure and in sequence below it) and side-chain density for tyrosine (arrow). (b) As in (a), but with side chains.
Crystal structure of Fab K1912 bound with an HA head domain
We crystallized the Fab of K1912 bound with the Solomon Islands HA head (see Methods); the crystals yielded a structure of the complex at 3.4 Å resolution (Fig. 7 and Table S2). Because the CDRH3 sequences of K1915 and K1912 are nearly the same (Fig. S1), we could compare the two structures directly (Fig. S5). Superposition of the EM and x-ray models suggested that a discrepancy in the main-chain fit of the Arg100-Glu101 dipeptide was due to an incorrect modeling of that segment into the EM map. Reconfiguration of that dipeptide, with further small adjustments to reset the fit of CDRH3 to the EM map and subsequent real-space refinement, yielded good agreement with the EM density and an alpha-carbon rmsd of 1.7 Å for 24 residues spanning CDRH3. The largest difference is at the glycine-rich tip of the loop; eliminating just those three residues reduces the rmsd to 1.2 Å. A comparison of structures before and after these adjustments is shown, together with the x-ray structure, in Fig. S5.
Fig. 7.

Receptor-binding site specificity of K1915 and K1912 antibodies. (a) Structures of HA with scFv from antibody K1915 (top), determined by cryoEM, and of K1912 with Fab from antibody K1912 (bottom), determined by x-ray crystallography. HA, in gray; K1915 scFv, in green; K1912 Fab, in blue. See further comparison in Fig. S5. (b) Contacts in the HA sialic-acid binding site. The side chain of Val102 is in the conserved, non-polar pocket bounded by HA residues Trp153, Thr155, and Leu194, into which would insert the methyl group of the receptor acetamido group, and a hydrogen bond from the main-chain NH of Gly103 in CDRH3 to the main-chain carbonyl of HA residue 135 resembles a similar bond from the acetamido NH. The carbonyl of Gly103 and the carboxylate of Glu101 have, respectively, polar interactions similar to those of the sialic-acid receptor carboxylate with Ser136 Oγ and Ala137 main-chain NH. Non-polar contacts between the Phe110 side chain and HA Leu194 and between the Gly103 main chain and HA Trp153 resemble contacts from the non-polar surface of the sialic acid pyranose ring.
Receptor-binding site recognition
The principal contacts with HA are from the heavy chains of the K1912 and K1915 antibodies, as might be expected from their likely ontogeny (Figs. 7, S1 and S5). In addition to insertion of CDRH3 into the RBS, there are a set of contacts, both polar and non-polar, between two residues in CDRH2 and residues 144–145 in HA and a single van der Waals contact from CDRH1. Light-chain contacts involve just two residues in CDRL1 that are present in the light chains of both antibodies (Tyr32 and Tyr33).
Receptor mimicry by CDRH3 is similar to examples we have described previously [14–16], although the loop itself is substantially longer. Fig. 7B illustrates the sialic acid-like contacts; Fig. S6 shows an explicit comparison of interactions in the K1912 complex with those in a receptor complex. Arg100 (one of the residues adjusted after examining the crystal structure) fixes the conformation of the CDRH3 loop through a network of polar hydrogen bonds with main-chain carbonyls; thus positioned, the arginine side chain salt-bridges to the carboxylate of Asp190, a conserved residue at the Nterminus of the HA "190s helix".
Residue 226 is Gln in most human H1 isolates, but passage of vaccine strains in chicken eggs can select for a mutation to Arg, because of its preference for the avian receptor [16]. In the vaccine-strain Solomon Islands HA we used for crystallization, residue 226 is indeed Arg, which bridges between HA S136 and T105 at the flexible tip of the antibody CDRH3; Gln could do the same (Fig. 7B; Fig. S6). Affinities of K1912 for a set of H1 HAs (Fig. S1B) show that binding is essentially indifferent to the substitution. Moreover, both K1912 and K1915 have similar polar contacts with HA residues 226–227, despite their local backbone conformational differences.
Discussion
Receptor mimicry
The long CDRH3 loops of antibodies that recognize the RBS of influenza HA all exhibit some degree of sialic-acid mimicry. The essential HA-receptor contacts are with the sialic-acid carboxylate and acetamido groups and secondarily with the glycerol moiety [5, 17]. In the various structures of RBS-directed antibodies bound with HA, groups near the apex of CDRH3 recapitulate most or all of these contacts. The same is true of K1912 and K1915, but with some variations not previously seen. The hydrogen bond between the main-chain amide of Gly103 and the main-chain carbonyl of HA residue 135 and the non-polar contacts of Val 102 with Trp153, Thr 155, and Leu 194 are like the replicas of sialic-acid acetamido group contacts seen in other antibody complexes. Instead of a single acidic residue, however, the side chain of Glu 101 and the main-chain carbonyl of Gly103 share the interactions made in the receptor complex by the sialic-acid carboxylate. The contacts of the antibody with non-conserved residues around the rim of the RBS ignore almost completely the 190s helix, the site of much variation among HAs of influenza isolates [18], except for the salt bridge between Arg 100 and Asp 190; the latter residue is conserved in nearly all H1 influenza HAs in the sequence database. Many of the mutations that lead to escape from neutralization by other RBS-directed antibodies would probably have little effect on K1912 or K1915.
A cryoEM "pipeline" for high-resolution epitope mapping?
Our results illustrate the problems that require solutions before we can depend on cryoEM instead of x-ray crystallography for structures of HA:Fab complexes. The principal limitations appear to be: (1) variation of twist around the long axis of the HA trimer; (2) incomplete scFv occupancy; (3) preferential particle orientation.
HA twist may be a continuous rather than a discrete variable, but a substantially greater number of particles would permit a great number of classes, subdividing the limited range of twists finely enough to achieve higher resolution. The increased speed of image analysis software since completing the work reported here and improved automatic particle-picking routines (e.g., [19]) will facilitate an efficient pipeline. More particles and hence more classes might also allow relaxation of threefold symmetry at an intermediate stage, to eliminate or sub-classify particles with less than three bound antibody fragments.
Preferential orientation is a general problem, for which our rosette method is one of many ad hoc solutions. Because the preference is rarely absolute, the problem resembles that of anisotropy in x-ray crystallography, once a reasonable molecular model has been built or fit. Constrained refinement of a model to fit a map of anisotropic resolution can yield a robust result, because stereochemical constraints correlate all three spatial directions.
Materials and Methods
Preparation of influenza hemagglutinin, K1915 antibody single-chain variable fragment (scFv), and K1912 Fab
Recombinant HA (rHA) H1 Solomon Islands/03/2006 (GenBank ABU50586.1) was expressed in Trichoplusia ni (Hi-5 cells) using recombinant baculovirus and purified as previously described [14]. The supernatant was harvested and clarified by centrifugation 72 h post infection. The rHA was purified by immobilized metal affinity chromatography using TALON resin (Clontech). For crystallography, the C-terminal foldon and 6xHis tag were removed using thrombin-conjugated agarose resin (Sigma Aldrich) followed by separation on a Superdex 200 column (GE Healthcare). The resulting rHA was HA0, with the fusion peptide uncleaved. For cryoEM, when rosettes were not used (see below), the purified rHA was treated for several days at 4°C with trypsin, resulting in removal of both the foldon and the 6xHis tag and cleavage between HA1 and HA2.
The K1915 single chain Fv (scFv) and Fab were codon-optimized for mammalian cell expression as previously described [15]. The VL (residues 1–109) and VH (residues 1–129) domains were joined using a (GGGS)3 linker and included a non-cleavable, C-terminal 6xHis tag. For expression, 293T cells were transiently transfected using Lipofectamine 2000 (Thermofisher Scientific). Supernatants were harvested 5 days post transfection and clarified isolated on TALON resin (Clontech) followed by purification over a Superdex 200 column (GE Healthcare). For the Fab, the VL and VH were fused to the human CL (kappa) and CH domains, respectively with a non-cleavable C-terminal 6xHis tag on the CH domain. Equimolar amounts of the heavy- and light-chain plasmids were transiently transfected into 293T cells and purified as described for the scFv.
Preparation of hemagglutinin rosettes
His-tagged HA solution at a concentration of 0.1 mg/ml was mixed with 10 nM solution of 5 nm Ni-NTA functionalized Nanogold (Nanoprobes). The HA:Nanogold ratio was experimentally optimized to produce rosettes that had about five HA trimers bound per Nanogold particle.
Electron microscopy
Rosette samples were prepared for cryoEM by applying 3 µL of rosette solution to glow-discharged R 1.2/1.3 holey carbon 200-mesh copper grids (Quantifoil Micro Tools), plunge-frozen with a Vitrobot Mark I (FEI). We recorded 412 images, with a one second exposure of 20 electrons/Å2, on a US4000 CCD camera (Gatan, Inc.) and an F30 electron microscope (FEI) operated at 300 kV and a nominal magnification of 50,000×, resulting in a calibrated pixel size of 2.2 Å at the specimen level.
HA:scFv cryo samples were prepared in the same way as the rosette samples, after addition of β-octylglucoside to a final concentration of 0.07% wt/vol. The detergent prevented a strongly preferred orientation; without it, particles presented views almost exclusively along the symmetry axis. We recorded 10,281 movies on a K2 Summit detector (Gatan, Inc.) and a Titan Krios (FEI) operated at 300 kV and a nominal magnification of 18,000× and controlled by SerialEM (http://bio3d.colorado.edu/SerialEM); the calibrated physical pixel size was 1.64 Å. We recorded 38 frames/movie in super-resolution mode using an exposure rate of 8 electrons/physical pixel/second, and a total exposure of 40 electrons/Å2.
Image processing
We selected 9544 HA rosettes manually using e2boxer.py [20], taking special care to center each rosette on the gold clusters. We then picked 5201 suitable HA trimers, again carefully centering on each trimer to obtain rough in-plane orientation angles by connecting the selected locations with the rosette centers. Further processing steps were carried out with the Spider processing software [21]. We boxed and masked each trimer with a rectangular mask aligned with the trimer axis (Fig. 1). We used a cylinder as an initial reference to adjust alignment of each particle by projection matching and calculated a reconstruction (Fig. S2), iterating seven times. In each iteration, shifts were limited to about 10 Å relative to the original picked positions. Trimers that changed their in-plane rotations by more than 18° or that had a correlation coefficient with their reference projection of less than 0.3 were rejected, leaving 3302 particles in the final reconstruction (Fig. 1) with an estimated resolution of 15 to 20 Å, based on visual inspection of recognizable features.
Frame alignment of movies of the HA:Fv complex was performed with Unblur [22]; frames were summed without exposure filtering. We selected 252,130 particles semi-automatically using e2boxer.py [20]. Particles were boxed and downsampled to 3.28 Å/pixel to accelerate processing. A subset of particles was rejected based on 2D classification using RELION [10], leaving 229,237 particles for 3D analysis. Particles were aligned against the HA trimer reconstruction obtained from the rosettes using projection matching implemented in IMAGIC [23] with a step size if 7.5° and assuming C3 symmetry. Particle alignments were further refined using 10 cycles of Mode 1 in Frealign [11], followed by a few cycles of Mode 2 to align any remaining, incorrectly aligned particles. Frealign 3D classification (two classes) yielded a final dataset of 142,314 particles. Particles in the rejected class appeared to suffer from missing antibody fragments, distortions and other imperfections.
At this point, processing switched to 1.64 Å/pixel downsampled data and particles were refined further in 60 cycles of Frealign Mode 1, then split into four classes and refined in another 60 cycles of Mode 1. The resolution limit during processing in Frealign never exceeded 10 Å, and the estimated resolutions of the final four classes were between 4.8 Å and 6.0 Å (FSC = 0.143 criterion, Fig. S4A). The final maps were scaled in resolution zones against a density map derived from the atomic model of a previously determined structure of HA bound to a different antibody fragment [14]. The scaled maps were then filtered using a figure-of-merit filter [24] based on the estimated FSC curve output by Frealign, adjusted for the volume occupied by the particle (Part_FSC). Finally, the maps were sharpened by a B-factor of −100 A2.
Model building and refinement for K1915 scFv:HA(A/Solomon Islands/03/2006) complex
We placed the Solomon Islands HA trimer structure (PDB 5UGY) into the EM maps with the program O [25]. For the structure of the K1915 scFv we used Modeller [26], starting with a structural template from PDB 4K8R (chain D residues 1–130 and chain C residues 2–109, corresponding to heavy and light chain sequence, respectively). We identified 4K8R as a good template structure for Modeller by aligning the K1915 sequence to the sequences of all non-redundant structures of the PDB. The heavy and light chains of Fab1 in 4K8R (chains D and C) have 78 and 71% identity with aligned sequences of the corresponding chains in K1915. Using Modeller’s automodel class, we generated 10 K1915 models. Superposition of the 10 models showed that they were essentially identical except at the N terminus, the scFv linker and the CDRH3 loop. Since the conformation of these residues could not be modeled accurately, we removed them before placing the model with the highest molpdf score into the EM map. We then manually built the CDRH3 loop in O, guided by the class 4 EM density, and added N-linked glycans with phenix.carbo_load where supported by density.
We refined coordinates and B factors with phenix.real_space_refine (version 1.11.1-2575, protocol: rigid_body, minimization_global, adp) [13]. In addition to standard geometry and B-factor restraints, we applied Ramachandran, rotamer and secondary structure restraints throughout the refinement. We also used non-crystallographic symmetry (NCS) torsion angle restraints, thereby essentially imposing three-fold symmetry on the model, corresponding to the symmetry imposed on the EM reconstruction. The FSCs between the final maps of the four classes and the refined models are shown in Fig. S4B. The estimated resolutions from these analyses (5.1 – 7.1 Å, FSC = 0.5 criterion) agree with the values reported above obtained from the half-map analyses (Fig. S4A). We analyzed the final model with MolProbity [27]; statistics are in Table S1.
Crystallography of K1912 Fab:HA(A/Solomon Islands/03/2006) complex
For crystallization of the K1912 Fab:HA complex, we incubated Fab K1912 with H1 Solomon Islands HA ectodomain at a 1.3:1 molar ratio. Complexes were separated from excess K1912 by gel filtration and concentrated to ~22 mg/mL. Hanging-drop vapor diffusion from a 1:1 mixture of the concentrate with reservoir solution containing 10% (w/v) PEG 8000 and 100 mM HEPES gave crystals of the complex in 3 days at 18 °C. The crystals were cryo-protected by soaking for 5 seconds in reservoir solution augmented with 15% (v/v) MPD, harvested into loops, and flashed cooled by plunging into liquid N2.
Diffraction data were collected at 100 K on NE-CAT beamline 24-ID-C at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). Diffraction images were indexed, integrated and scaled with XDS [28]. Models of Solomon Islands HA (PDB 5UGY) and Fab (PDB ID 4K8R) were used as probes for molecular replacement with PHASER [29]. Density modification was performed with DM [30], and model rebuilding was completed manually with COOT [31]. Refinement used BUSTER (Global Phasing Ltd). Statistics are in Table S2. Figures were generated with PyMOL (Schrödinger LLC).
Supplementary Material
Fig. S1. History of affinity maturation in antibody lineage 2897. (a) Phylogenetic tree of antibody lineage 2897 [6], inferred with Clonalyst [32, 33], and alignment of heavy-chain variable domain sequences for the unmutated common ancestor (UCA) of the lineage, antibody K1912, and antibody K1915. We have assigned the K1915 heavy chain to the same UCA, based on nearly identical nnucleotide sequences (and n-nucleotide encoded residues, shown here), and inferred that the antibody acquired a different light chain by receptor revision. (b) Binding (of Fab with monomeric HA head) of the lineage 2897 UCA and Fab K1912 for selected H1 HAs from 1977 to 2009, i.e., from the reappearance of circulating H1 to the early twenty-first century "new pandemic H1". The donor was born in 1986; we infer that strains circulating at that time (e.g., strains similar to A/Kawasaki//1986 or A/Massachusetts//1990) probably elicited the initial response (UCA) [34]. Affinity maturation in response to subsequent exposures (infection or vaccination) gave rise to antibodies with high affinity for strains from 1977 through 2008. Symbols represent the following approximate equilibrium dissociation constants (µM) for Fab and HA head: ++++, < 1; +++, 1–10; ++, 10–50; +, 50–100; −, >100.
Fig. S2. Orientation determination for an HA trimer from a rosette. The model was positioned in the cube as shown. Projection images from this volume result in the rod shaped reference with the "head" directed away from the center. The in-plane rotation angle from 12 o'clock for the reference can be calculated as arctan{tan(ϕ)*cos(θ)} (ϕ and θ are the values from the Spider VO EA command). Two constraints from the rosette can be used in the image processing. (1) During alignment, the boxed trimer is allowed to rotate freely, but shift can vary only within a small radius. For the example shown, the image in the middle can either rotate about 0° to align with the reference on the left or rotate about 180° to align with the reference on the right; it cannot, however, rotate about 0° and align with the reference on the right, due the limitation of the shift. Thus, the polarity of the molecule is preserved. (2) When the trimers are picked from the rosette, the angle of the trimer from 12 o'clock is determined. The additional in-plane rotation angle is also known after the alignment. The sum of these two values should approximately equal the in-plane rotation angle of the reference. If the difference is larger than a chosen threshold, the particle will be rejected from 3D reconstruction.
Fig. S3. Successive sections through aligned class 2 and class 4 maps. The two reconstructions were aligned by superposing the helix-bundle density in the HA stem. The two maps were sectioned and displayed alternately, to show the difference in twist between them. The scFv region can be seen in sections 8–25; the HA head, in sections 31–45.
{kind=link}
Fig. S4. Fourier shell correlations. (a) Between the two half maps for each of the four classes. (b) Between map and model for each of the four classes.
Fig. S5. K1915 and K1912 comparison: CDRH3 conformation in the RBS. Left: initial fit of the CDRH3 polypeptide chain in the cryoEM map; center, adjusted cryoEM model, based on the configuration of Arg100 in the x-ray map of K1912 bound with HA A/Solomon Islands//2006 (right).
Fig. S6. Comparison of receptor contacts (left) with contacts in the K1912:HA complex (right). The receptor shown is a human receptor analog (LSTc), bound with the HA from the 1918 H1N1 influenza virus (PDB 2WRG). Hydrogen bonds are shown as dashed, yellow lines. The most important conserved van der Waals contacts are in the pocket formed by Trp153, Thr155 and Leu 194 (asterisk), which accommodates the acetamido methyl of sialic acid and the Val102 side chain of the antibody CDRH3. HA and sialic-acid carbon atoms and ribbon backbone, cyan; K1912 CDRH3 carbons and ribbon backbone, dark blue-green; nitrogens and oxygens in blue and red, respectively.
Highlights.
Single-particle electron cryomicroscopy (cryoEM) of antigen-antibody complexes
Influenza virus hemagglutinin (HA) bound with single-chain Fv
cryoEM and molecular modeling yield details of the antigen:antibody interface
HA receptor-binding site recognition by broadly neutralizing antibodies
Acknowledgments
We thank Chen Xu (Brandeis University EM Facility), Zongli LI (Harvard Medical School EM Facility), Zhiheng Yu and Jason de la Cruz (HHMI Janelia Research Campus cryoEM Facility), for technical support, and the NE-CAT beamline staff, for help with x-ray data collection. NE-CAT is funded by NIH grant P41 GM-103403 and the Pilatus 6M detector on 24-ID-C by NIH-ORIP HEI grant S10-RR-029205. APS is operated for the DOE Office of Science by Argonne National Laboratory under contract DE-AC02-06CH11357. The work at Boston Children's Hospital and Harvard Medical School was supported by NIH grants P01 AI-089618 and P01 GM-62580. SCH and NG are Investigators in the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
The K1915 scFv:HA electron density maps and the corresponding models of the four classes are deposited in the Electron Microscopy Data Bank (EMDB accession numbers EMD-8561, EMD-8562, EMD-8563, EMD-8564) and Protein Data Bank (PDB accession numbers 5UJZ, 5UK0 5UK1, 5UK2). The K1912 Fab:HA crystal structure is deposited in the PDB (accession number 5UG0)
References
- 1.Rappuoli R, Bottomley MJ, D'Oro U, Finco O, De Gregorio E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J Exp Med. 2016;213:469–81. doi: 10.1084/jem.20151960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012;30:423–33. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1484–90. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Julien JP, Lee JH, Ozorowski G, Hua Y, Torrents de la Pena A, de Taeye SW, et al. Design and structure of two HIV-1 clade C SOSIP.664 trimers that increase the arsenal of native-like Env immunogens. Proc Natl Acad Sci U S A. 2015;112:11947–52. doi: 10.1073/pnas.1507793112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–69. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 6.Moody MA, Zhang R, Walter EB, Woods CW, Ginsburg GS, McClain MT, et al. H3N2 influenza infection elicits more cross-reactive and less clonally expanded anti-hemagglutinin antibodies than influenza vaccination. PLoS One. 2011;6:e25797. doi: 10.1371/journal.pone.0025797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelsoe G. V(D)J hypermutation and receptor revision: coloring outside the lines. Curr Opin Immunol. 1999;11:70–5. doi: 10.1016/s0952-7915(99)80013-2. [DOI] [PubMed] [Google Scholar]
- 8.Kouskoff V, Nemazee D. Role of receptor editing and revision in shaping the B and T lymphocyte repertoire. Life Sci. 2001;69:1105–13. doi: 10.1016/s0024-3205(01)01219-x. [DOI] [PubMed] [Google Scholar]
- 9.Bottcher C, Ludwig K, Herrmann A, van Heel M, Stark H. Structure of influenza haemagglutinin at neutral and at fusogenic pH by electron cryo-microscopy. FEBS Lett. 1999;463:255–9. doi: 10.1016/s0014-5793(99)01475-1. [DOI] [PubMed] [Google Scholar]
- 10.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–30. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyumkis D, Brilot AF, Theobald DL, Grigorieff N. Likelihood-based classification of cryo-EM images using FREALIGN. J Struct Biol. 2013;183:377–88. doi: 10.1016/j.jsb.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 13.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc Natl Acad Sci U S A. 2011;108:14216–21. doi: 10.1073/pnas.1111497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt AG, Therkelsen MD, Stewart S, Kepler TB, Liao HX, Moody MA, et al. Viral receptor-binding site antibodies with diverse germline origins. Cell. 2015;161:1026–34. doi: 10.1016/j.cell.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raymond DD, Stewart SM, Lee J, Ferdman J, Bajic G, Do KT, et al. Influenza immunization elicits antibodies specific for an egg-adapted vaccine strain. Nat Med. 2016;22:1465–9. doi: 10.1038/nm.4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weis W, Brown JH, Cusack S, Paulson JC, Skehel JJ, Wiley DC. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature. 1988;333:426–31. doi: 10.1038/333426a0. [DOI] [PubMed] [Google Scholar]
- 18.Koel BF, Burke DF, Bestebroer TM, van der Vliet S, Zondag GC, Vervaet G, et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science. 2013;342:976–9. doi: 10.1126/science.1244730. [DOI] [PubMed] [Google Scholar]
- 19.Scheres SH. Semi-automated selection of cryo-EM particles in RELION-1.3. J Struct Biol. 2015;189:114–22. doi: 10.1016/j.jsb.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Shaikh TR, Gao H, Baxter WT, Asturias FJ, Boisset N, Leith A, et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat Protoc. 2008;3:1941–74. doi: 10.1038/nprot.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. Elife. 2015;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 24.Sindelar CV, Grigorieff N. Optimal noise reduction in 3D reconstructions of single particles using a volume-normalized filter. J Struct Biol. 2012;180:26–38. doi: 10.1016/j.jsb.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 26.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Protein Sci. 2016;86:2 9 1–2 9 37. doi: 10.1002/cpps.20. [DOI] [PubMed] [Google Scholar]
- 27.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kepler TB, Munshaw S, Wiehe K, Zhang R, Yu JS, Woods CW, et al. Reconstructing a B-Cell Clonal Lineage. II. Mutation, Selection, and Affinity Maturation. Front Immunol. 2014;5:170. doi: 10.3389/fimmu.2014.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kepler TB. Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors. F1000Res. 2013;2:103. doi: 10.12688/f1000research.2-103.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt AG, Do KT, McCarthy KR, Kepler TB, Liao HX, Moody MA, et al. Immunogenic Stimulus for Germline Precursors of Antibodies that Engage the Influenza Hemagglutinin Receptor-Binding Site. Cell Rep. 2015;13:2842–50. doi: 10.1016/j.celrep.2015.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. History of affinity maturation in antibody lineage 2897. (a) Phylogenetic tree of antibody lineage 2897 [6], inferred with Clonalyst [32, 33], and alignment of heavy-chain variable domain sequences for the unmutated common ancestor (UCA) of the lineage, antibody K1912, and antibody K1915. We have assigned the K1915 heavy chain to the same UCA, based on nearly identical nnucleotide sequences (and n-nucleotide encoded residues, shown here), and inferred that the antibody acquired a different light chain by receptor revision. (b) Binding (of Fab with monomeric HA head) of the lineage 2897 UCA and Fab K1912 for selected H1 HAs from 1977 to 2009, i.e., from the reappearance of circulating H1 to the early twenty-first century "new pandemic H1". The donor was born in 1986; we infer that strains circulating at that time (e.g., strains similar to A/Kawasaki//1986 or A/Massachusetts//1990) probably elicited the initial response (UCA) [34]. Affinity maturation in response to subsequent exposures (infection or vaccination) gave rise to antibodies with high affinity for strains from 1977 through 2008. Symbols represent the following approximate equilibrium dissociation constants (µM) for Fab and HA head: ++++, < 1; +++, 1–10; ++, 10–50; +, 50–100; −, >100.
Fig. S2. Orientation determination for an HA trimer from a rosette. The model was positioned in the cube as shown. Projection images from this volume result in the rod shaped reference with the "head" directed away from the center. The in-plane rotation angle from 12 o'clock for the reference can be calculated as arctan{tan(ϕ)*cos(θ)} (ϕ and θ are the values from the Spider VO EA command). Two constraints from the rosette can be used in the image processing. (1) During alignment, the boxed trimer is allowed to rotate freely, but shift can vary only within a small radius. For the example shown, the image in the middle can either rotate about 0° to align with the reference on the left or rotate about 180° to align with the reference on the right; it cannot, however, rotate about 0° and align with the reference on the right, due the limitation of the shift. Thus, the polarity of the molecule is preserved. (2) When the trimers are picked from the rosette, the angle of the trimer from 12 o'clock is determined. The additional in-plane rotation angle is also known after the alignment. The sum of these two values should approximately equal the in-plane rotation angle of the reference. If the difference is larger than a chosen threshold, the particle will be rejected from 3D reconstruction.
Fig. S3. Successive sections through aligned class 2 and class 4 maps. The two reconstructions were aligned by superposing the helix-bundle density in the HA stem. The two maps were sectioned and displayed alternately, to show the difference in twist between them. The scFv region can be seen in sections 8–25; the HA head, in sections 31–45.
Fig. S4. Fourier shell correlations. (a) Between the two half maps for each of the four classes. (b) Between map and model for each of the four classes.
Fig. S5. K1915 and K1912 comparison: CDRH3 conformation in the RBS. Left: initial fit of the CDRH3 polypeptide chain in the cryoEM map; center, adjusted cryoEM model, based on the configuration of Arg100 in the x-ray map of K1912 bound with HA A/Solomon Islands//2006 (right).
Fig. S6. Comparison of receptor contacts (left) with contacts in the K1912:HA complex (right). The receptor shown is a human receptor analog (LSTc), bound with the HA from the 1918 H1N1 influenza virus (PDB 2WRG). Hydrogen bonds are shown as dashed, yellow lines. The most important conserved van der Waals contacts are in the pocket formed by Trp153, Thr155 and Leu 194 (asterisk), which accommodates the acetamido methyl of sialic acid and the Val102 side chain of the antibody CDRH3. HA and sialic-acid carbon atoms and ribbon backbone, cyan; K1912 CDRH3 carbons and ribbon backbone, dark blue-green; nitrogens and oxygens in blue and red, respectively.