

Abstract
Adoptive cellular therapy (ACT) is now considered a bona fide treatment modality within the evolving field of anticancer immunotherapy. Great advances have enabled the adoptive transfer of tumour-selective lymphocytes for the treatment of a variety of malignancies. Unfortunately, this selectivity has led to the emergence of antigen-loss variants. New strategies need to be employed to minimize the incidence of this phenomenon, enabling the full potential of ACT to be realized.
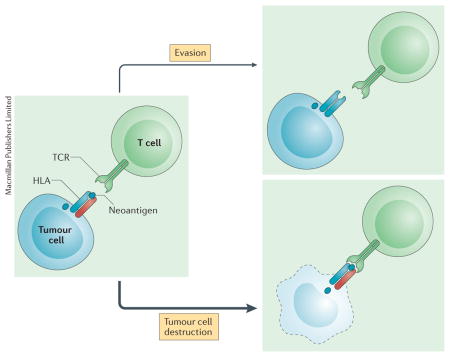
Over the past decade, adoptive cellular therapy (ACT) has moved from the fringes to the mainstream of experimental immunotherapeutic research. Clinical successes have been reported with each of the three main pillars of this treatment modality: ex vivo expanded tumour-infiltrating T lymphocytes (TILs); genetically engineered T cells expressing tumour-reactive T-cell receptors (TCRs); and tumour-recognizing chimeric antigen receptors (CARs). ACT is based on the principle that bulk delivery of tumour-recognizing T cells will create an immune response that overwhelms malignant lesions, resulting in complete and durable clinical remission. In the case of TCR-based ACTs, transferred lymphocytes are capable of recognizing an antigen that is either exclusive to, or over-expressed in tumour cells in the form of a peptide in the context of a human leukocyte antigen (HLA). By using CAR T-cell-based ACT, these tumour-associated antigens can be detected independently of an HLA and must simply be expressed on tumour surfaces. In a recent report by Tran and colleagues1, ex vivo expanded TILs recognizing a KRAS-derived neoantigen in the context of an HLA were used to treat a patient with metastatic colorectal cancer. This work highlights the far-reaching clinical implications of such an approach, and also sheds light on tumour immune escape as an unfortunate and limiting consequence of such a targeted T-cell-based approach.
Mutant KRAS is an oncogenic driver found in a multitude of human cancers and is critical for tumour development, progression and survival. A mutant peptide derived from KRAS G12D can be displayed as a neoepitope in the context of HLA–C*08:02, a class I major histocompatibility complex (MHC) molecule. The authors report on the isolation of a poly-clonal population of CD8+ TILs capable of recognizing this mutant KRAS neoepitope in the corresponding HLA molecule. Following isolation and ex vivo expansion, these clones were infused back into the patient, resulting in successful long-term regression of most metastatic lesions, except one. This single unresponsive lesion was noted to have undergone a copy-neutral loss of heterozygosity at chromosome 6, a location encoding HLA–C*08:02, leading to the loss of the HLA molecule capable of presenting the mutant KRAS neoepitope to T cells.
Tran and colleagues1 demonstrate the feasibility and potential of targeting tumour cells via their HLA-restricted display of neoepitopes derived from driver oncogenic mutations, which are present in both primary tumours and subsequent metastases. The appeal of such a strategy is clear, as a tumour is highly unlikely to lose a driver mutation and hence will most likely continuously express fragments of such a mutant oncogene, making it susceptible to elimination by T cells with cognate TCRs. However, as reported by Tran et al.1, the introduction of such selective pressures led to the outgrowth of a single metastatic lesion that had lost the chromosomal region encoding the HLA–C*08:02, in essence rendering the tumour cells undetectable to adoptively transferred T cells.
In the past 5 years, the level of interest in ACT has grown substantially, primarily in the area of CAR T cells, with most current clinical experience relating to the targeting of CD19 on B cells in patients with haematological malignancies. In this space, a loss of tumour containment has occurred through a number of rather interesting mechanisms: the emergence of alternatively spliced CD19 isoforms ablating the previously recognizable epitope in patients with B-cell-acute lymphoblastic leukaemia (B-ALL)2; differentiation of a mature B-cell malignancy to a plasmablastic lymphoma lacking CD19 expression3; and lineage switching of B-ALL to a clonally related CD19-negative myeloid leukaemia4,5. Of interest, the latter escape mechanism has also been documented with the use of the bidirectional T-cell engager, blinatumomab6, which functions by crosslinking CD19-positive targets with the CD3 molecule on endogenous T cells, thereby leading to the destruction of the former. Interestingly, despite these troubling occurrences, preclinical researchers continue to focus on strategies that prolong the persistence of CAR T cells in vivo, and in essence prolong the exposure of tumours to selective pressures, with less emphasis on methods of minimizing the escape and subsequent proliferation of antigen-loss variants.
Most of the evasion mechanisms described above occur as a result of the highly selective pressures created by targeting a single antigen. For the true potential of ACT to be realized, investigators need to research ways to minimize the risk of immune escape by broadening the selectivity of our interventions. One such approach is currently being tested in the setting of a clinical trial assessing the use of ipilimumab (an inhibitory antibody targeting cytotoxic T-lymphocyte-associated protein 4) before, and in parallel with treatment with ex vivo expanded melanoma-derived TILs7. A rationale for such a combination, as it relates to minimizing immune escape, is provided by the observation that ipilimumab substantially broadens the T-cell repertoire of patients with metastatic cancer8. Such a diversification would enable more-comprehensive coverage of the neoantigens presented by multiple HLA-class molecules, and hence minimize or even eliminate the risk of antigen-negative outgrowth that results from the introduction of a singular selective pressure. In the case of CAR T-cell-based ACT, simultaneously targeting two tumour antigens has been proposed as a way to prevent immune escape, with data from preclinical models successfully demonstrating the feasibility of such an intervention9. One final strategy that might synergize with all forms of ACT is the combined use with checkpoint inhibitors targeting the programmed cell death protein 1 (PD-1)/programmed cell death protein 1 ligand 1 (PDL-1) axis. Whether or not enhanced tumour clearance actually minimizes the risk of antigen-escape variants emerging remains unclear, although such an approach is worthy of clinical assessment.
The future of ACT is bright and very promising. As with any other antineoplastic therapy, we are rapidly learning that cancers are able to adopt sophisticated contingency measures to evade complete eradication by cellular immunotherapies. As we uncover such escape strategies, we are able to further augment our interventions with the hopes of minimizing the possibility of such escapes. Immunotherapy is shaping up to be an arms race, one that we can and will win.
Acknowledgments
The authors thank the following for financial support: The Leukemia & Lymphoma society; The U.S. National Institutes of Health Grants (R01CA138738-05, PO1CA059350, PO1CA190174-01); The Annual Terry Fox Run for Cancer Research (New York, NY) organized by the Canada Club of New York; Kate’s Team; Carson Family Charitable Trust; William Lawrence and Blanche Hughes Foundation; Emerald Foundation; the Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center (Innovations in the structures, functions and targets of monoclonal antibody-based drugs for cancer).
Footnotes
Competing interests statement
R.J.B. is a scientific co-founder of, reports receiving a commercial research grant from, has ownership interest (including patents) in, and is a consultant/advisory board member of JUNO Therapeutics. A.F.D. declares no competing interests.
References
- 1.Tran E, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sotillo E, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans AG, et al. Evolution to plasmablastic lymphoma evades CD19-directed chimeric antigen receptor T cells. Br J Haematol. 2015;171:205–209. doi: 10.1111/bjh.13562. [DOI] [PubMed] [Google Scholar]
- 4.Gardner R, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacoby E, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7:12320. doi: 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rayes A, McMasters RL, O’Brien MM. Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer. 2016;63:1113–1115. doi: 10.1002/pbc.25953. [DOI] [PubMed] [Google Scholar]
- 7.US National Library of Medicine. ClinicalTrials.gov. 2016 https://clinicaltrials.gov/ct2/show/NCT01701674.
- 8.Cha E, et al. Improved survival with T cell clonotype stability sfter anti-CTLA-4 treatment in cancer patients. Sci Transl Med. 2014;6:238ra70. doi: 10.1126/scitranslmed.3008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruella M, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126:3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]