

Abstract
Introduction
Increasing evidence links COPD pathogenesis with pulmonary capillary apoptosis. We previously demonstrated that plasma levels of circulating microparticles released from endothelial cells (EMPs) due to apoptosis are elevated in smokers with normal spirometry but low diffusion capacity, i.e., with early evidence of lung destruction. We hypothesized that pulmonary capillary apoptosis persists with development of COPD and assessed its reversibly in healthy smokers and COPD smokers following smoking cessation.
Methods
Pulmonary function and HRCT were assessed in 28 nonsmokers, 61 healthy smokers and 49 COPD smokers; 17 healthy smokers and 18 COPD smokers quit smoking for 12 months following the baseline visit. Total EMP (CD42b−CD31+), pulmonary capillary EMP (CD42b−CD31+ACE+) and apoptotic EMP (CD42b−CD62E+/CD42b−CD31+) levels were quantified by flow cytometry.
Results
Compared to nonsmokers, healthy smokers and COPD smokers had elevated levels of circulating EMPs due to active pulmonary capillary endothelial apoptosis. Levels remained elevated over 12 months in healthy smokers and COPD smokers who continued smoking but returned to nonsmoker levels in healthy smokers who quit. In contrast, levels remained significantly abnormal in COPD smokers who quit.
Conclusion
Pulmonary capillary apoptosis is reversible in healthy smokers who quit, but continues to play a role in COPD pathogenesis in smokers who progressed to airflow obstruction despite smoking cessation.
Keywords: Smoking cessation, endothelial microparticles, COPD, apoptosis
Introduction
Chronic obstructive pulmonary disease (COPD), the 3rd leading cause of mortality in the US, is defined by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) as a chronic lung disorder with airflow limitation that is not fully reversible1. There is overwhelming evidence that most cases of COPD are caused by cigarette smoking, with approximately 20% of smokers at risk for COPD if they continue to smoke2. The airflow obstruction that characterizes COPD is caused by a variable mixture of small airway disease (bronchitis) and parenchymal destruction (emphysema)1,3. Although the airway and alveolar disease were classically considered as separate entities, it is now recognized they usually coexist to variable degrees and are closely linked, with the parenchymal destruction evolving around areas of small airway disease3–7.
There is increasing evidence that the pathogenesis of COPD is linked, in part, to apoptosis of pulmonary capillaries8–11. Consistent with this concept, we recently demonstrated that smokers and, to a greater extent, smokers with early evidence of lung destruction [normal spirometry but low diffusing capacity (DLCO)] have elevated levels of circulating endothelial microparticles (EMPs)12. Importantly, a significant proportion of these EMPs are derived from pulmonary capillaries and have characteristics of apoptotic EMPs, i.e., they are derived from lung endothelial cells that have been induced to undergo apoptosis12–14.
If elevated levels of circulating apoptotic EMPs are a reflection of active smoking-related injury to lung endothelium, based on the knowledge that even those COPD smokers who stop smoking continue to have a decline in lung function that is more rapid than that of healthy non-smokers or healthy smokers who quit smoking15, we hypothesized that elevated levels of circulating apoptotic EMPs may persist in COPD smokers following smoking cessation, reflecting continuous lung endothelial injury that persists even after the stress of smoking is removed. To assess this hypothesis, we quantified the levels of circulating EMPs, and the fraction represented by apoptotic EMPs, in nonsmokers, healthy smokers, and smokers with COPD at baseline and at 3 more intervals over 1 year and then compared those levels to those obtained from a subgroup of healthy smokers and COPD smokers who successfully stopped smoking after baseline assessment. The data demonstrates that circulating EMP levels derived from apoptotic pulmonary capillary endothelial cells remain elevated over 1 year in healthy smokers and COPD smokers who continue smoking. However, while levels of total and apoptotic EMPs return to nonsmoker levels in healthy smokers who successfully quit smoking, total and apoptotic EMP levels remain elevated in COPD smokers who quit smoking persisting 12 months of smoking cessation.
Methods
Human Subjects and Clinical Phenotypes
All subjects were evaluated at the Weill Cornell NIH Clinical and Translational Science Center (CTSC) and Department of Genetic Medicine Clinical Research Facility, under Institutional Review Board approved clinical protocols. Recruitment was from the general population in New York City by posting advertisements in local newspapers and on electronic bulletin boards. All subjects provided written consent prior to enrollment, and then underwent thorough medical history, screening and pulmonary function tests. Smoking status was determined based on self-reported history and quantified levels of urine nicotine metabolites. For details and full inclusion/exclusion criteria see Supplemental Methods. A total of 138 subjects were assessed for circulating total and apoptotic EMP levels at baseline, 3, 6 and 12 months (28 nonsmokers, 61 healthy smokers and 49 COPD GOLD I/II smokers). See Supplemental Figure 1 for study design.
Characterization of Plasma EMPs
EMPs were quantified according to a standard operating procedure as previously described12 to eliminate variability in sample processing. Briefly, blood was collected, processed within 1 hr and stained for the endothelial markers PECAM (CD31) and E-selectin (CD62E) and the constitutive platelet-specific glycoprotein Ib (CD42b) to differentiate endothelium-originated microparticles from platelet-derived microparticles, which also express CD31. EMPs were defined as microparticles <1.5 μm in size, expressing CD31+ or CD62E+ but not CD42b microparticles. We have previously shown that staining with annexin V is comparable to CD42b−CD31+ staining12, but annexin V was not used because it is not specific for EMPs16. Circulating EMPs are present in low levels in plasma of healthy subjects, reflecting normal endothelial turnover17, but their levels increase in a variety of vascular-related disorders. As in our previous study12, total MEP levels above the nonsmoker total EMP mean level plus 2 standard deviations were considered abnormally elevated. To assess the presence of relative contribution of pulmonary capillary endothelium to the elevated total EMP levels1,12, EMPs were co-stained with anti-human angiotensin converting enzyme (ACE), which is abundantly expressed on pulmonary capillary endothelium (CD42b−CD31+ACE+)13. To quantify the proportion of EMPs that originated from apoptotic endothelium, we assessed the ratio of CD42b−CD62E+/CD42b−CD31+ EMPs in all groups. EMPs induced by apoptosis express the constitutive CD31 marker, whereas activation-induced EMPs express CD62E. Using this criteria, EMPs with a low CD42b−CD62E+ to CD42b−CD31+ ratio were defined as “apoptotic EMPs” and the percentage of subjects with apoptotic EMPs with CD42b−CD62E+/CD42b−CD31+ ratio below the lowest ratio in healthy nonsmokers was quantified. See Supplemental Methods for further details on the EMP analysis.
Assessment after Smoking Cessation
After the baseline levels of total and apoptotic EMPs were determined, all healthy smokers and COPD smokers were invited to stop smoking using a combination of varenicline and counseling for 3 months (see details in Supplemental Methods). A total of 17 healthy smokers and 18 COPD smokers successfully quit smoking as confirmed by urine tobacco metabolite level quantification at 3, 6 and 12 months after the baseline; subjects were considered true quitters only if there were no detectable levels of nicotine metabolites in the urine at months 3, 6 and 12. Healthy smokers and smokers with COPD were treated with exact same prescription of varenicline to prevent any effect it might have on EMP levels. All other healthy smokers and COPD smokers were considered current smokers if urine cotinine level was ≥ 104 ng/ml at each time point, a level based on our previous study of low level smoke exposure18, where 104 ng/ml was calculated as the induction half maximal level (ID50) at which the small airway epithelium, the initial site of smoking-related pathology, showed an abnormal response. See Supplemental Figure 1 for study design.
Statistical Analysis
Chi square test, with a Yates’ correction for small sample size, was used for comparing demographic parameters and the number of subjects with high total EMP and apoptotic EMP levels and Pair-wise ANOVA was used to compare total and apoptotic EMP levels between groups and within a group at different time points with no correction for multiple test as the number of tests was low (<21). In order to eliminate the effect of diseases known to be associated with elevated EMPs, including diabetes19,20 and systemic hypertension21 or drugs for COPD, including corticosteroids and bronchodilators, subjects with known disease state or drug treatment were removed from statistical analysis. Removal of those subjects did not alter the results.
Results
Study Population
Except for minor differences, the study population of nonsmokers was comparable to the healthy smokers and COPD smokers in all demographic parameters (see Table I for details). At each time point, the nonsmokers had undetectable urine nicotine (not shown) and cotinine levels (Figure 1). Both the healthy smokers and COPD smokers who continued smoking had urine cotinine levels consistent with tobacco smoking and comparable in both groups at each time point (p>0.07, all comparisons). In healthy smokers and COPD smokers who quit smoking, urine nicotine and cotinine levels were consistent with smoking at baseline and were undetectable at all time points after baseline (see Supplemental Methods for details of urine nicotine metabolite level criteria for smoking/abstinence).
Table I.
Study Population1
| Healthy smokers2
|
COPD smokers3
|
||||||
|---|---|---|---|---|---|---|---|
| Parameter | Non-smokers | All | Who continued smoking | Who quit | All | Who continued smoking | Who quit |
| n | 28 | 61 | 44 | 17 | 49 | 31 | 18 |
| Gender (M/F) | 15/13 | 47/14 | 37/7 | 10/7 | 46/3 | 30/1 | 16/2 |
| Ethnicity (B/W/O)4 | 10/7/11 | 33/9/19 | 23/7/14 | 10/2/5 | 27/12/10 | 18/6/7 | 9/6/3 |
| Age | 37 ± 11 | 44 ± 9 | 44 ± 9 | 45 ± 10 | 53 ± 8 | 53 ± 7 | 53 ± 9 |
| BMI | 27 ± 5 | 28 ± 5 | 27 ± 4 | 30 ± 4 | 27 ± 4 | 25 ± 3 | 29 ± 5 |
| Smoking history | |||||||
| Pack-yr | – | 23 ± 12 | 24 ± 12 | 20 ± 8 | 32 ± 14 | 32 ± 15 | 34 ± 12 |
| Pack per day | – | 0.8 ± 0.6 | 1.0 ± 0.6 | 0.6 ± 0.2 | 0.8 ± 0.4 | 0.8 ± 0.5 | 0.8 ±0.3 |
| Age of initiation | – | 16 ± 3 | 16 ± 3 | 16 ± 3 | 16 ± 3 | 16 ± 3 | 16 ± 3 |
| Urine cotinine (ng/ml) | – | 1693 ± 961 | 1828 ± 930 | 1323 ± 979 | 1747 ± 980 | 1953 ± 959 | 1393 ± 938 |
| Subjects with emphysema (n, %)5 | 1 (4%) | 0 (0%) | 0 (0%) | 0 (0%) | 13 (27%) | 9 (29%) | 4 (22%) |
| Pulmonary function6 | |||||||
| FEV1 | 106 ± 11 | 109 ± 11 | 109 ± 10 | 107 ± 13 | 85 ± 16 | 87 ± 16 | 82 ± 17 |
| FVC | 107 ± 11 | 111 ± 10 | 110 ± 10 | 111 ± 11 | 108 ± 16 | 109 ± 17 | 105 ± 15 |
| FEV1/FVC | 83 ± 5 | 80 ± 5 | 81 ± 4 | 79 ± 6 | 63 ± 6 | 64 ± 6 | 63 ± 7 |
| TLC | 99 ± 16 | 96 ± 12 | 95 ± 12 | 96 ± 10 | 99 ± 12 | 100 ± 13 | 99 ± 10 |
| DLCO | 91 ± 11 | 89 ± 8 | 89 ± 9 | 90 ± 6 | 71 ± 14 | 68 ± 13 | 77 ± 15 |
| GOLD stage (I/II) | – | – | – | – | 31/18 | 20/11 | 11/7 |
Data are presented as mean ± standard deviation; all parameters recorded at baseline; health/disease state based on screening and medical history and smoking status based on self-reported history and urine nicotine metabolite levels (detailed in Supplemental Methods); nonsmokers were comparable to all healthy smokers and all COPD smokers in ethnicity, BMI and all pulmonary function (p>0.1, all comparisons), except for FEV1 and DLCO that were lower in all COPD smokers (p<10−7, both comparisons) and FEV1/FVC, that was lower in all healthy smokers and, by definition, in all COPD smokers (p<0.02, both comparisons). Nonsmokers were younger than all healthy smokers and all COPD smokers (p<0.002, both comparisons) and there were less female COPD smokers than female nonsmokers (p<0.0002). There were more COPD smokers with emphysema compared to nonsmokers (p<10−4); All healthy smokers were comparable to all COPD smokers in ethnicity, age, BMI, all smoking history parameters (p>0.3, all comparisons) except for pack-yr that was lower in all healthy smokers (p<10−3). FVC and TLC were comparable (p>0.07, both comparisons), but FEV1, DLCO and, by definition, FEV1/FVC were lower in all COPD smokers (p<10−12, all comparisons). There were fewer females among all COPD smokers than among all healthy smokers (p<0.04). There were more COPD smokers with emphysema compared to healthy smokers (p<10−4).
Healthy smokers who continued smoking had urine cotinine ≥ 104 ng/ml (see Supplemental Methods for details) at baseline, 3,6, and 12 months. Healthy smokers who quit had undetectable urine nicotine and cotinine levels at 3, 6 and 12 months. The healthy smokers who continued smoking were comparable to those who quit in age, ethnicity, all smoking history (p>0.3, all comparisons), except for pack per day that was lower in those who quit (p<0.03), and comparable in all pulmonary function (p>0.1). There were more females and the BMI was higher in the healthy smokers who quit group (p<0.04, both comparisons).
Gold stage defined by GOLD criteria1; see Supplemental Methods for details for subjects on medications; several of those treated were on multiple classes of medications; COPD smokers who continued smoking had urine cotinine ≥ 104 ng/ml at baseline, 3, 6 and 12 months; COPD smokers who quit had undetectable urine nicotine and cotinine levels at 3, 6 and 12 months; The COPD smokers who continued smoking were comparable to those who quit in age, gender, ethnicity, all smoking history and all pulmonary function (p>0.3, all comparisons), except for DLCO that was lower in the COPD who continued smoking compared to those who quit (p<0.03). The BMI was lower in the COPD who continued smoking vs those who quit (p<0.002). There was no difference in the number of subjects with emphysema between the COPD smokers who quit smoking and those who continued smoking (p>0.6).
B=Black, W=White, O= Other.
Chest high resolution computed tomography (HRCT); % emphysema at −950 Hounsfield Units (HU); Emphysema defined as >5% lung volume, see Supplemental Methods for details.
Pulmonary function testing parameters are given as % of predicted value with the exception of FEV1/FVC, which is reported as % observed; FVC - forced vital capacity; FEV1 - forced expiratory volume in 1 sec; TLC - total lung capacity; DLCO - diffusing capacity.
Figure 1.
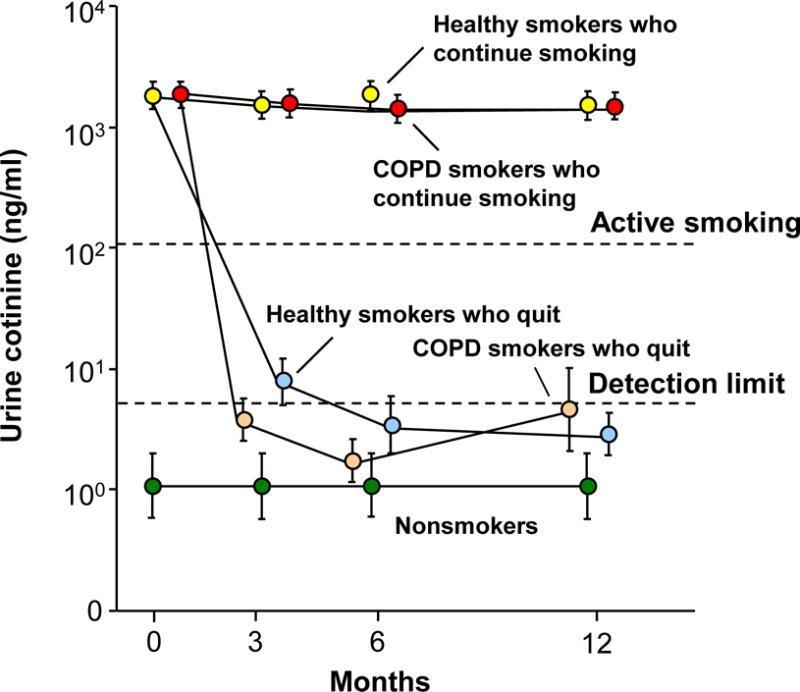
Urine cotinine levels (ng/ml) as a measure of smoking status at baseline and at 3, 6 and 12 months in nonsmokers, healthy smokers and smokers with COPD (COPD smokers). Shown is data for nonsmokers (n=28, green circles), healthy smokers who continue to smoke (n=44, yellow circles), healthy smokers who quit smoking following baseline (n=17, light blue circles), COPD smokers who continue to smoke (n=31, red circles) and COPD smokers who quit smoking following baseline (n=18, tan circles). Data represent mean ± standard error. Dashed lines indicate urine cotinine detection level of ≤ 5 ng/ml and urine cotinine level of ≥ 104 ng/ml for active smoking (see Supplemental Methods).
Total EMP Levels
Consistent with our prior study with a different cohort12, total EMP levels were higher in healthy smokers compared to nonsmokers (Figure 2A, p<0.005). In addition, COPD smokers had elevated levels of total EMPs compared to nonsmokers (p<0.007), but lower than those of healthy smokers (p<0.02). Twenty-two (36%) healthy smokers and 8 (16%) COPD smokers had high levels of total EMPs (p<0.03). There was no correlation between the level of total EMPs and any pulmonary function or demographic parameters (r2<0.08, all correlations, Supplemental Figure 2). For the COPD group, total circulating EMP levels were independent of drugs used for treatment, including inhaled and systemic corticosteroids and bronchodilators.
Figure 2.
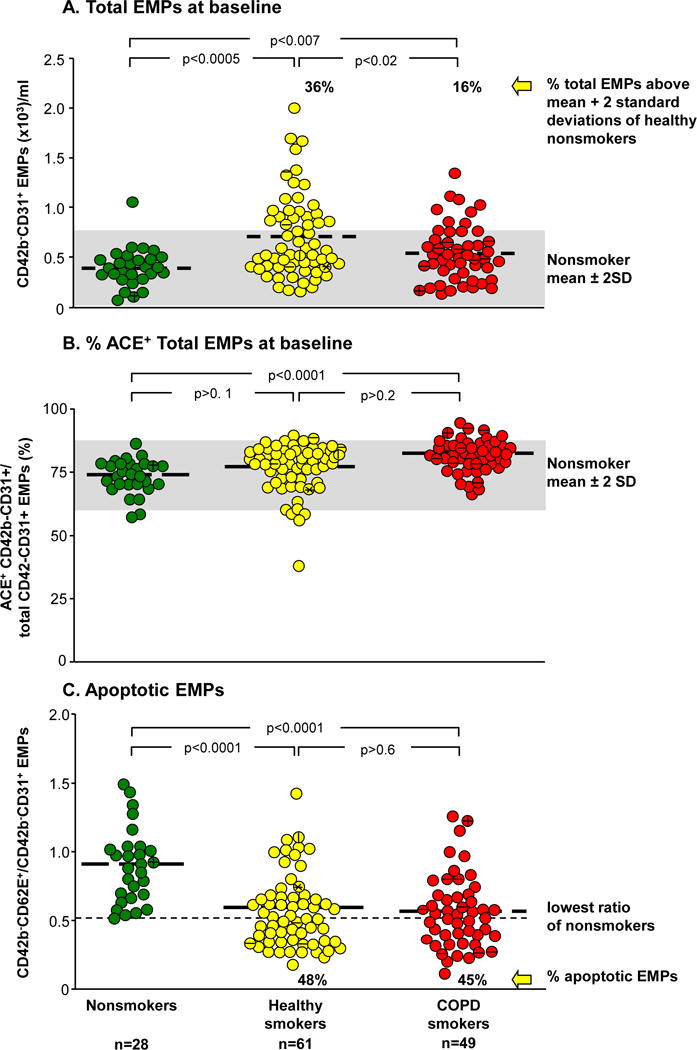
Levels of total circulating endothelium microparticles (EMPs), ACE+ EMPs, and apoptotic EMPs in plasma at baseline. Shown is data for nonsmokers (n=28, green circles), healthy smokers (n=61, yellow circles) and smokers with COPD (n=49, red circles). A. Total CD42b−CD31+ EMPs. The gray shaded area indicates the nonsmoker mean ± 2 standard deviations. The % value above the smoker populations represents the proportion of smokers with EMP levels above that mean. B. CD42b−CD31+ACE+ EMPs. Proportion of total CD42b−CD31+ EMPs in plasma that express angiotensin converting enzyme (ACE+). The gray shaded area represents the nonsmoker mean ± 2 standard deviations. C. Ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs. The dashed line indicates the lowest ratio of CD42b−CD62E+/CD42b−CD31+ EMPs in nonsmokers. The % value below the smoker populations represents the proportion of smokers with a ratio below that level. A–C; Bold dashed lines represent the mean for each group. Symbols inside the dots: A horizontal line indicates subjects with systemic hypertension; a vertical line indicates subjects with type 2 diabetes mellitus; a star indicates subjects with type 1 diabetes.
Origin of the Circulating EMPs
In our prior study of circulating EMPs12, we demonstrated that most of the circulating CD42b−CD31+ EMPs were positive for angiotensin converting enzyme (ACE), a surface protein more highly expressed on pulmonary capillary endothelium than in other endothelial beds13. In the present study, an average of 75% of all subjects had CD42b−CD31+ACE+ EMPs. There were similar levels of ACE+ EMPs in the healthy smoker group compared to the nonsmokers or COPD smokers (Figure 2B, p>0.1, both comparisons), and higher levels of ACE+ EMPs in the COPD smoker group compared to the nonsmokers (p<0.0001).
EMPs Derived from Apoptotic Endothelium
To quantify the proportion of EMPs originating from apoptotic endothelium, we assessed the ratio of CD42b−CD62E+/CD42b−CD31+ EMPs in all groups. The CD42b−CD62E+/CD42b−CD31+ EMP ratio in nonsmokers was distributed around a mean of 0.9, significantly higher than in healthy smokers (mean 0.6, a lower ratio indicates greater number of apoptotic EMPs) and COPD smokers (mean 0.55, p<0.0001, both groups compared to nonsmokers, p>0.6, COPD smokers compared to healthy smokers). CD42b−CD62E+/CD42b−CD31+ EMPs below the lowest level in nonsmokers were defined as apoptotic EMPs with 48% of healthy smokers and 45% of COPD smokers having increased levels of apoptotic EMPs (p>0.7), i.e., even though there are less subjects with total circulating EMPs in COPD smokers compared to healthy smokers (Figure 2A), the relative proportion of subjects with apoptotic EMPs was similar (Figure 2C), implying that there is active pulmonary capillary apoptosis ongoing in both the healthy smokers and COPD smokers. There was no correlation of CD42b−CD62E+/CD42b−CD31+ EMP ratio with any lung function or demographic parameter (r2=0.09, all correlations, Supplemental Figure 3). Within the COPD smoker group, there was no correlation of total CD42b−CD31+ EMP levels or CD42b−CD62E+/CD42b−CD31+ EMP ratio to the DLCO (% predicted) or % emphysema on HRCT (r2=0.04, all comparisons; not shown), suggesting these parameters are likely measuring different aspects of the destruction process.
Effect of Smoking Cessation on Total EMP Levels
The levels of total EMPs were followed for a period of one year at baseline, 3, 6, and 12 months in nonsmokers, healthy smokers and COPD smokers (Figure 3A, B). In nonsmokers, total EMP levels were stably low throughout the duration of the study (p>0.6, each time point compared to baseline). In healthy smokers who continued smoking, the levels were stably high at each time point (p>0.1, each time point compared to baseline and p<0.05, each time point compared to the nonsmokers at the same time point). In contrast, in the healthy smokers who quit smoking, total EMP levels significantly decreased following smoking cessation to the levels of nonsmokers (p<0.002, each time point compared to baseline and p>0.4, compared to nonsmokers at the same time point at 3, 6 and 12 months, Figure 3A). At 12 months, the total EMP levels in healthy smokers who continued smoking were significantly higher compared to healthy smokers who quit (p<10−3). Total EMP levels in COPD smokers who continued smoking were stably elevated compared to nonsmokers at each time point (p<0.05, all comparisons). In contrast to the healthy smokers, in COPD smokers who quit smoking, total EMP levels initially decreased following smoking cessation at month 3 but became elevated again at 6 and 12 months (Figure 3B). The levels were not significantly different compared to nonsmokers at 3 and 6 months (p>0.1, both comparisons), but were significantly elevated at 12 months (p<0.05) and similar to those of COPD smokers who continued smoking (p>0.08).
Figure 3.
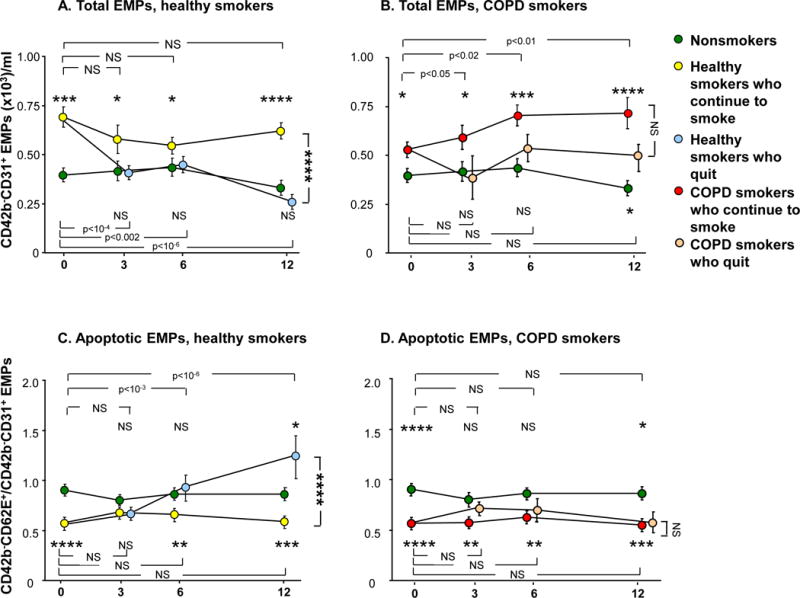
Total circulating CD42b−CD31+ endothelial microparticles (EMPs) and ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs over time in nonsmokers (n=28, green circles), healthy smokers who continue to smoke (n=44, yellow circles), healthy smokers who quit smoking following baseline (n=17, light blue circles), smokers with COPD (COPD smokers) who continue smoking (n=31, red circles) and COPD smokers who quit smoking following baseline (n=18, tan circles). A, B. Total CD42b−CD31+ EMPs. C, D. Ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs A, C. Healthy smokers who continue to smoke and healthy smokers who quit smoking vs nonsmokers. B, D. COPD smokers who continue to smoke and COPD smokers who quit smoking vs nonsmokers. A–D. Data represent mean ± standard error. P values comparing each time point to baseline within the same group are shown at the top of the panel (for the group that is above the nonsmokers at month 12) and at the bottom of the panel (for the group that is below the nonsmokers at month 12). P values comparing each time point in a smoker group to the same time point in the nonsmoker group are shown above the group if the group is above the nonsmokers at month 12 and below the group if the group is below the nonsmokers at month 12. P values comparing the subjects who continue to smoke with those who quit smoking at month 12 are to the right of the panel. NS = not significant; *, **, ***, **** indicate p<0.05, p<0.01, p<0.001 and p<0.0001, respectively.
Effect of Smoking Cessation on Apoptotic EMP Levels
The ratio of CD42b−CD62E+/CD42b−CD31+ EMPs was stably high in nonsmokers and stably low (i.e., EMPs were apoptotic-derived) in healthy smokers who continued smoking (p>0.1, each time point compared to baseline within the nonsmoker group and within the healthy smoker group and p<0.01, healthy smokers who continued smoking compared to nonsmokers, at baseline, months 6 and 12; Figure 3C). Interestingly, in the healthy smokers who quit smoking, the ratio increased following smoking cessation (p<10−3, within the healthy smokers who quit group, months 3 and 6 compared to baseline, a p value significant even with correction for multiple tests) to the level of nonsmokers at months 3 and 6 (p>0.1, both comparisons compared to nonsmokers at the same time point) and superseded that of nonsmokers at month 12 (p<0.05). The ratio was significantly higher (i.e., less apoptotic-derived EMPs) in healthy smokers who quit smoking compared to smokers who continued smoking at month 12 (p<10−3, a p value significant even with correction for multiple tests). There were no significant changes in the CD42b−CD62E+/CD42b−CD31+ ratio in COPD smokers who continued smoking (p>0.1, within the COPD smoker group, each time point compared to baseline) and it remained significantly low compared to nonsmokers at each time point (p<0.01, all comparisons, Figure 3D). In contrast to the healthy smoker group, in COPD smokers who quit smoking there was no change in the ratio (p>0.1, within the COPD who quit group, each time point compared to baseline) and the ratio remained significantly low compared to nonsmokers at baseline and month 12 (p<0.05, both comparisons). The ratio at month 12 was similarly low (i.e., more apoptotic-derived EMPs) in COPD smokers who continued smoking and in COPD smokers who quit (p>0.4).
Discussion
COPD is a chronic, debilitating disease that is caused primarily by cigarette smoking1,3–5. Cigarette smoke is very complex, with 1014 oxidants and >4000 compounds stressing the lung with each puff22. The apoptotic loss of pulmonary capillaries in association with smoking is well recognized8–10, although it is not known whether this represents the primary mechanism of lung destruction associated with smoking, a subtype of lung destruction, or is secondary to other mechanisms, such as inflammatory cell-mediated processes5,6,9,10,23. Using circulating, total and apoptotic-endothelial cell microparticles as biomarkers for pulmonary capillary apoptosis, the data in the present study documents that pulmonary capillary endothelial apoptosis is a persistent process in smokers with and without COPD. In healthy smokers who quit smoking, the levels of total and apoptotic EMPs return to the levels of nonsmokers over time. In contrast, in COPD smokers who quit smoking, the levels of total and apoptotic EMPs remain abnormal over 1 year and were still significantly different compared to nonsmoker levels at 12 months. The majority of the COPD subjects assessed for EMPs in this study were GOLD I and GOLD II, providing evidence for ongoing pulmonary endothelial apoptosis even in the earliest stages of COPD. Importantly, the FEV1/FVC ratio of the COPD smokers was 0.63 ± 0.06, on average, well below the 0.7 ratio threshold definition of COPD GOLD I.
These observations were not altered by the removal of subjects with diseases known to be associated with elevated EMPs, suggesting that smoking has a much stronger effect on EMP levels than hypertension or diabetes.
Endothelial Microparticles
Different cell types respond to cell activation, injury, and/or apoptosis by shedding submicron membrane vesicles, called microparticles, from their plasma membranes16,24. Microparticles detected in plasma are of various cellular origins, predominantly derived from platelets, leukocytes and endothelial cells24. Endothelial microparticles, defined as CD42b−CD31+ or CD42b−CD62E+ microparticles, can be generally distinguished from microparticles of other cell types by their size (0.1 to 1.5 μm), constitutive expression of the platelet-endothelial cell adhesion marker CD31 (PECAM), and the absence of the platelet-specific glycoprotein Ib marker CD42b24,25. Apoptosis-induced EMPs express CD31 whereas activation-induced EMPs express CD62E. In this regard, a low ratio of CD42b−CD62E+ to CD42b−CD31+ EMPs can be used as an index of apoptosis24,25. EMPs variably co-express phosphatidylserine (annexin V)25,26.
EMPs can be found in the plasma of healthy subjects24, however, increased levels are associated with vascular disease and endothelial dysfunction in atherosclerosis and acute coronary syndrome24,27, acute ischemic stroke24,28, end stage renal failure29, preeclampsia and gestational hypertension30, hypertension21,24,29, pulmonary hypertension31, metabolic syndrome32, venous thromboembolism33, and obstructive sleep apnea34. Consistent with our prior study12 and the data in the present study, Heiss and colleagues35 have shown healthy nonsmokers exposed for 30 min to low levels of cigarette smoke had increased circulating EMP levels.
Clinical Measures of Alveolar Capillary Destruction
The observation of endothelial apoptosis in the lungs of humans with emphysema is well documented. There is increased DNA fragmentation in the pulmonary capillaries and arteriolar endothelium of subjects with COPD, and increased alveolar endothelial and epithelial cell death in human emphysematous lungs compared with lungs of nonsmokers or smokers without emphysema9,10. Lung levels of alveolar epithelial-derived vascular endothelial growth factor are decreased in emphysema, contributing to the complex mechanisms of pulmonary capillary endothelial destruction10.
The data in the present study adds total and apoptotic EMP levels to a growing list of biomarkers that may be useful in helping to assess active destruction and to define subclinical molecular phenotypes of lung disease17,36,37. As described in the editorial by Chandra and colleagues14, accompanying our study of apoptotic EMPs in healthy smokers and smokers with normal spirometry but low DLCO, the observation of elevated levels of apoptotic EMPs may not only represent early lung destruction, but could represent a sub-phenotype of subjects with lung destruction. The observation in the present study that, on the average, a fraction of smokers with COPD also have elevated levels of total and apoptotic EMPs is consistent with the Chandra et al14 discussion of the concept of vascular sub-phenotypes of COPD. In this context, the data on circulating and apoptotic EMPs supports the idea that, while the global concept of COPD as an FEV1-defined disorder is useful for epidemiologic studies and as a paradigm for routine clinical care, it is likely masking the concept that there are several sub-phenotypes of COPD38–42. Consistent with this concept, correlation of the total EMP levels and apoptotic EMP levels with the conventional measures of lung destruction (DLCO and HRCT) was, at best, very weak. This may imply that, while overlapping, each parameter is measuring a somewhat different aspect of the same process and/or that each parameter is assessing a different subpopulation of what is globally referred to as “lung destruction”.
Our longitudinal study demonstrates that total and apoptotic EMP levels remain stable over a period of one year in subjects with no change to their smoking habits. Intervention with smoking cessation can normalize the levels of total and apoptotic EMPs in healthy smokers, but in contrast, cessation did not lead to significant changes in total and apoptotic EMP levels in COPD smokers in our study. This observation provides a biological correlate for epidemiologic data showing that smoking cessation only has a moderate effect to slow the decline of lung function in COPD smokers, i.e., we believe that these data show that EMP levels might serve as a useful biomarker to follow smoking-associated endothelial apoptosis. The longitudinal aspect of this study demonstrates persistent endothelial stress in subjects with COPD despite smoking cessation, and may help to explain the irreversible lung destruction associated with most cases of COPD, as evidenced by lung function of COPD smokers following smoking cessation that does not return to normal15 and may serve as a basis for additional studies of the mechanisms of the continuous pulmonary damage leading to this persistent EMP release.
Supplementary Material
What is the key question?
Is pulmonary capillary apoptosis, as measured by plasma levels of microparticles released from apoptotic endothelial cells (EMPs), reversible in healthy smokers and COPD smokers following smoking cessation?
What is the bottom line?
Pulmonary capillary apoptosis is reversible in healthy smokers who quit, but not in COPD smokers despite 12 months of smoking cessation, suggesting that the apoptosis continues to play a role in COPD pathogenesis in smokers who progressed to airflow obstruction despite smoking cessation.
Why read on?
Our longitudinal study demonstrates persistent endothelial stress in subjects with COPD despite smoking cessation and provides a biologic correlate to the epidemiologic data showing that smoking cessation only has a moderate effect on the continues decline of lung function in COPD smokers, suggesting EMP levels might serve as a useful biomarker to follow smoking-associated endothelial apoptosis.
Acknowledgments
We thank M. Elnashar, A. Rogalski, E. Blass, S. Mootoo, and T. Wilson for help in processing blood samples; the Clinical Operations and Regulatory Affairs, Department of Genetic Medicine for help with these studies; and N Mohamed for help in preparing this manuscript. These studies were supported, in part, by P50HL084936, R01HL107882, U01HL121828, UL1 TR000457, UL1 RR02414, and Hoffmann-La Roche, Inc.
Footnotes
Financial disclosures: Hans Bitter, Sriram Sridhar, Sreekumar G. Pillai, Holly Hilton, Gerhard Wolff, Christopher S. Stevenson, Sudha Visvanathan and Jay S. Fine were all former employees of Hoffmann-La Roche. No conflicts to declare for all the other authors.
Author contribution: Conception and design: RGC
Acquisition and data interpretation: YSB, MRS, AK, CG, AET, BG-H, RJK, CH, JGM, HB, SS, SP, HH, GW, CSS, SV, JSF RGC
Drafting of the manuscript: YSB, MRS, AK, AET, RGC
References
- 1.Global Initiative for Chronic Obstructive Lung Disease. 2011 http://www.goldcopd.com/[last accessed 07/30/2015]
- 2.Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1:1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, Pare PD, Sin DD, Pierce RA, Woods JC, McWilliams AM, Mayo JR, Lam SC, Cooper JD, Hogg JC. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365:1567–1575. doi: 10.1056/NEJMoa1106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hogg JC, Senior RM. Chronic obstructive pulmonary disease - part 2: pathology and biochemistry of emphysema. Thorax. 2002;57:830–834. doi: 10.1136/thorax.57.9.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56:515–548. doi: 10.1124/pr.56.4.2. [DOI] [PubMed] [Google Scholar]
- 6.Spurzem JR, Rennard SI. Pathogenesis of COPD. Semin Respir Crit Care Med. 2005;26:142–153. doi: 10.1055/s-2005-869535. [DOI] [PubMed] [Google Scholar]
- 7.Cosio Piqueras MG, Cosio MG. Disease of the airways in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2001;34:41s–49s. doi: 10.1183/09031936.01.00234601. [DOI] [PubMed] [Google Scholar]
- 8.Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364:709–721. doi: 10.1016/S0140-6736(04)16900-6. [DOI] [PubMed] [Google Scholar]
- 9.Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol. 2003;28:555–562. doi: 10.1165/rcmb.2002-0090OC. [DOI] [PubMed] [Google Scholar]
- 10.Plataki M, Tzortzaki E, Rytila P, Demosthenes M, Koutsopoulos A, Siafakas NM. Apoptotic mechanisms in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:161–171. doi: 10.2147/copd.2006.1.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rennard SI. Pathogenesis of chronic obstructive pulmonary disease. Pneumonol Alergol Pol. 2011;79:132–138. [PubMed] [Google Scholar]
- 12.Gordon C, Gudi K, Krause A, Sackrowitz R, Harvey BG, Strulovici-Barel Y, Mezey JG, Crystal RG. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am J Respir Crit Care Med. 2011;184:224–232. doi: 10.1164/rccm.201012-2061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danilov SM, Gavrilyuk VD, Franke FE, Pauls K, Harshaw DW, McDonald TD, Miletich DJ, Muzykantov VR. Lung uptake of antibodies to endothelial antigens: key determinants of vascular immunotargeting. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1335–L1347. doi: 10.1152/ajplung.2001.280.6.L1335. [DOI] [PubMed] [Google Scholar]
- 14.Chandra D, Sciurba FC, Gladwin MT. Endothelial chronic destructive pulmonary disease (E-CDPD): is endothelial apoptosis a subphenotype or prequel to COPD? Am J Respir Crit Care Med. 2011;184:153–155. doi: 10.1164/rccm.201104-0758ED. [DOI] [PubMed] [Google Scholar]
- 15.Scanlon PD, Connett JE, Waller LA, Altose MD, Bailey WC, Buist AS, Tashkin DP. Smoking cessation and lung function in mild-to-moderate chronic obstructive pulmonary disease. The Lung Health Study. Am J Respir Crit Care Med. 2000;161:381–390. doi: 10.1164/ajrccm.161.2.9901044. [DOI] [PubMed] [Google Scholar]
- 16.Chironi GN, Boulanger CM, Simon A, Gnat-George F, Freyssinet JM, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–151. doi: 10.1007/s00441-008-0710-9. [DOI] [PubMed] [Google Scholar]
- 17.Paige M, Burdick MD, Kim S, Xu J, Lee JK, Michael SY. Pilot analysis of the plasma metabolite profiles associated with emphysematous Chronic Obstructive Pulmonary Disease phenotype. Biochem Biophys Res Commun. 2011;413:588–593. doi: 10.1016/j.bbrc.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strulovici-Barel Y, Omberg L, O’Mahony M, Gordon C, Hollmann C, Tilley AE, Salit J, Mezey J, Harvey BG, Crystal RG. Threshold of biologic responses of the small airway epithelium to low levels of tobacco smoke. Am J Respir Crit Care Med. 2010;182:1524–1532. doi: 10.1164/rccm.201002-0294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koga H, Sugiyama S, Kugiyama K, Watanabe K, Fukushima H, Tanaka T, Sakamoto T, Yoshimura M, Jinnouchi H, Ogawa H. Elevated levels of VE-cadherin-positive endothelial microparticles in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol. 2005;45:1622–1630. doi: 10.1016/j.jacc.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 20.Tushuizen ME, Nieuwland R, Rustemeijer C, Hensgens BE, Sturk A, Heine RJ, Diamant M. Elevated endothelial microparticles following consecutive meals are associated with vascular endothelial dysfunction in type 2 diabetes. Diabetes Care. 2007;30:728–730. doi: 10.2337/dc06-1473. [DOI] [PubMed] [Google Scholar]
- 21.Preston RA, Jy W, Jimenez JJ, Mauro LM, Horstman LL, Valle M, Aime G, Ahn YS. Effects of severe hypertension on endothelial and platelet microparticles. Hypertension. 2003;41:211–217. doi: 10.1161/01.hyp.0000049760.15764.2d. [DOI] [PubMed] [Google Scholar]
- 22.MacNee W. Oxidative stress and lung inflammation in airways disease. Eur J Pharmacol. 2001;429:195–207. doi: 10.1016/s0014-2999(01)01320-6. [DOI] [PubMed] [Google Scholar]
- 23.Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis. 2008;12:361–367. [PubMed] [Google Scholar]
- 24.Horstman LL, Jy W, Jimenez JJ, Ahn YS. Endothelial microparticles as markers of endothelial dysfunction. Front Biosci. 2004;9:1118–1135. doi: 10.2741/1270. [DOI] [PubMed] [Google Scholar]
- 25.Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res. 2003;109:175–180. doi: 10.1016/s0049-3848(03)00064-1. [DOI] [PubMed] [Google Scholar]
- 26.Werner N, Wassmann S, Ahlers P, Kosiol S, Nickenig G. Circulating CD31+/annexin V+ apoptotic microparticles correlate with coronary endothelial function in patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2006;26:112–116. doi: 10.1161/01.ATV.0000191634.13057.15. [DOI] [PubMed] [Google Scholar]
- 27.Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet JM, Tedgui A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation. 2000;101:841–843. doi: 10.1161/01.cir.101.8.841. [DOI] [PubMed] [Google Scholar]
- 28.Simak J, Gelderman MP, Yu H, Wright V, Baird AE. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J Thromb Haemost. 2006;4:1296–1302. doi: 10.1111/j.1538-7836.2006.01911.x. [DOI] [PubMed] [Google Scholar]
- 29.Amabile N, Guerin AP, Leroyer A, Mallat Z, Nguyen C, Boddaert J, London GM, Tedgui A, Boulanger CM. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol. 2005;16:3381–3388. doi: 10.1681/ASN.2005050535. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez-Quintero VH, Smarkusky LP, Jimenez JJ, Mauro LM, Jy W, Hortsman LL, O’Sullivan MJ, Ahn YS. Elevated plasma endothelial microparticles: preeclampsia versus gestational hypertension. Am J Obstet Gynecol. 2004;191:1418–1424. doi: 10.1016/j.ajog.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 31.Amabile N, Heiss C, Real WM, Minasi P, McGlothlin D, Rame EJ, Grossman W, De MT, Yeghiazarians Y. Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension. Am J Respir Crit Care Med. 2008;177:1268–1275. doi: 10.1164/rccm.200710-1458OC. [DOI] [PubMed] [Google Scholar]
- 32.Arteaga RB, Chirinos JA, Soriano AO, Jy W, Horstman L, Jimenez JJ, Mendez A, Ferreira A, de ME, Ahn YS. Endothelial microparticles and platelet and leukocyte activation in patients with the metabolic syndrome. Am J Cardiol. 2006;98:70–74. doi: 10.1016/j.amjcard.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 33.Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, Horstman LL, Soriano AO, Zambrano JP, Ahn YS. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol. 2005;45:1467–1471. doi: 10.1016/j.jacc.2004.12.075. [DOI] [PubMed] [Google Scholar]
- 34.Ayers L, Ferry B, Craig S, Nicoll D, Stradling JR, Kohler M. Circulating cell-derived microparticles in patients with minimally symptomatic obstructive sleep apnoea. Eur Respir J. 2009;33:574–580. doi: 10.1183/09031936.00107408. [DOI] [PubMed] [Google Scholar]
- 35.Heiss C, Amabile N, Lee AC, Real WM, Schick SF, Lao D, Wong ML, Jahn S, Angeli FS, Minasi P, Springer ML, Hammond SK, Glantz SA, Grossman W, Balmes JR, Yeghiazarians Y. Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: sustained vascular injury and blunted nitric oxide production. J Am Coll Cardiol. 2008;51:1760–1771. doi: 10.1016/j.jacc.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 36.Luisetti M, Ma S, Iadarola P, Stone PJ, Viglio S, Casado B, Lin YY, Snider GL, Turino GM. Desmosine as a biomarker of elastin degradation in COPD: current status and future directions. Eur Respir J. 2008;32:1146–1157. doi: 10.1183/09031936.00174807. [DOI] [PubMed] [Google Scholar]
- 37.Comandini A, Rogliani P, Nunziata A, Cazzola M, Curradi G, Saltini C. Biomarkers of lung damage associated with tobacco smoke in induced sputum. Respir Med. 2009;103:1592–1613. doi: 10.1016/j.rmed.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Criner GJ, Cordova F, Sternberg AL, Martinez FJ. The National Emphysema Treatment Trial (NETT): Part I: Lessons learned about emphysema. Am J Respir Crit Care Med. 2011;184:763–770. doi: 10.1164/rccm.201103-0454CI. [DOI] [PubMed] [Google Scholar]
- 39.Kim V, Han MK, Vance GB, Make BJ, Newell JD, Hokanson JE, Hersh CP, Stinson D, Silverman EK, Criner GJ. The chronic bronchitic phenotype of COPD: an analysis of the COPDGene Study. Chest. 2011;140:626–633. doi: 10.1378/chest.10-2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishimura M, Makita H, Nagai K, Konno S, Nasuhara Y, Hasegawa M, Shimizu K, Betsuyaku T, Ito YM, Fuke S, Igarashi T, Akiyama Y, Ogura S. Annual Change in Pulmonary Function and Clinical Phenotype in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2012;185:44–52. doi: 10.1164/rccm.201106-0992OC. [DOI] [PubMed] [Google Scholar]
- 41.Salzman SH. Which Pulmonary Function Tests Best Differentiate Between COPD Phenotypes? Respir Care. 2012;57:50–60. doi: 10.4187/respcare.01585. [DOI] [PubMed] [Google Scholar]
- 42.Agusti A, Celli B. Avoiding confusion in COPD: from risk factors to phenotypes to measures of disease characterisation. Eur Respir J. 2011;38:749–751. doi: 10.1183/09031936.00062211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.