

Abstract
Mascarenhas et al. report that TRPV4 expression is upregulated in mast cells in response to the proteolytic cathelicidin fragment LL37 in a murine rosacea model and that TRPV4 loss of function attenuates mast cell degranulation. These findings render TRPV4 a translational-medical target in rosacea. However, signaling mechanisms causing increased expression of TRPV4 await elucidation. Moreover, we ask whether TRPV4-mediated Ca++-influx evokes mast cell degranulation.
Rosacea translational target TRPV4
Mascarenhas et al. from Anna DiNardo's group communicate in a Letter to the Editor an exciting new finding on mast cells' inflammatory response mechanisms in the chronic facial inflammatory dermatitis, rosacea (Mascarenhas et al., 2017) (Figure 1). The antibacterial peptide, cathelicidin LL-37, is highly expressed in the skin in rosacea, where it becomes cleaved from the 18 KD cathelicidin peptide (Yamasaki et al., 2007). Mascarenhas et al. now show that LL37 evokes upregulation of the Ca++-permeable TRPV4 ion channel in a murine rosacea model in which skin-associated mast cells colocalize with TRPV4 in response to intradermally injected LL37. Isolated mast cells from the mouse and humans share upregulation of TRPV4 evoked by LL37, whereas upregulation of TRPV2 affects only murine mast cells. Blocking TRPV4 with a selective inhibitor, HC-067, for 24 hours before stimulating cultured mast cells with compound 48/80 attenuates degranulation. CRISPR-mediated knockdown of TRPV4 in cells of human mast cell lineage reduces mast cell degranulation by both compound 48/80 and LL37. Mascarenhas et al. go on to show that inhibiting the small G-protein, Gi/o, using pertussis toxin, eliminates upregulation of mast cell TRPV4 by LL37 completely. CRISPR-mediated knockdown of the 48/80 receptor (a G-protein coupled receptor), MRGX2, has a similar effect.
Figure 1. Mast cell degranulation and TRPV4 Ca++-permeable ion channels in rosacea.
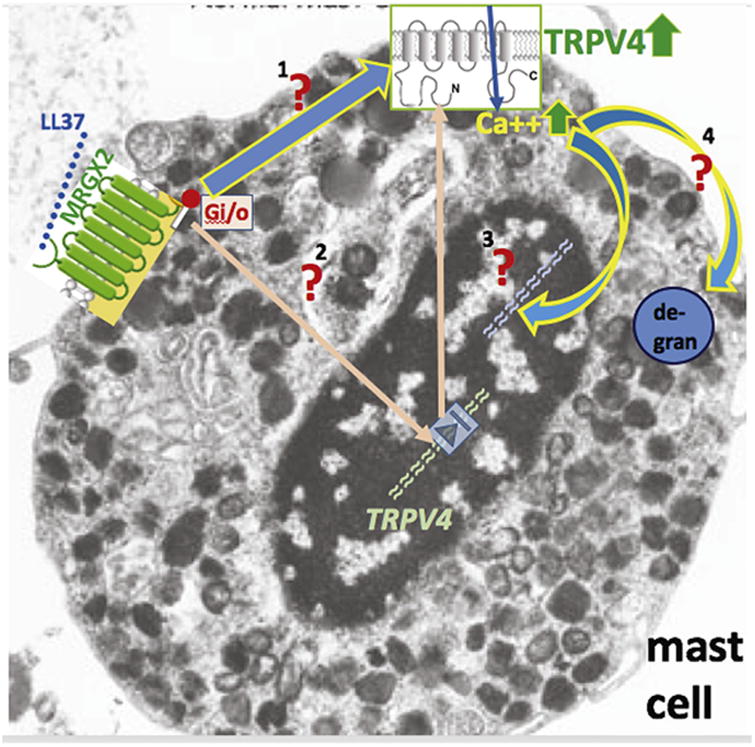
Schematic summarizing findings from Mascarenhas et al. and highlighting pathways of interest, awaiting experimental testing, in mast cells. Cathelicidin cleavage product LL37 binds to and activates its cognate receptor, MRGX2. This leads to increased expression of Ca++-permeable TRPV4 ion channels, via Gi/o signaling. Red “?” means that this pathway will have to be assessed experimentally. 1—direct membrane-bound signaling MRGX2 to TRPV4 (presumably via Gi/o); 2—intracellular signal transduction, via activated MRGX2-Gi/o, leading to increased expression of TRPV4 in the nucleus (the pink arrow pointing out of the nucleus refers to translation and protein trafficking of the TRPV4 channel protein); 3—Ca++-influx via TRPV4 leading to altered gene expression in the nucleus, for example, for the genes of the degranulation machinery; 4—Ca++-influx via TRPV4 constituting the intracellular Ca++ that is needed for degranulation.
This is an exciting brief report that puts TRPV4 fair and square into therapeutic cross-hairs as a translational-medical target for rational and improved treatment of rosacea-mediated skin inflammation. With relevance for skin biology and disease, the TRPV4 channel has been found to be expressed in keratinocytes in the epidermis, with a significant role in barrier function, UVB-overexposure-evoked pain and injury responses, itch, thermoregulation, in skin innervating sensory neurons with roles in pain and itch, as well as in the roles of immune cells such as macrophages and neutrophils (Akiyama et al., 2016; Alessandri-Haber et al., 2005; Chen et al., 2016; Chung et al., 2003; Denda et al., 2007; Kim et al., 2016; Moore et al., 2013). Although the LL37-MRGX2-TRPV4 pathway has a rational foundation, and thus considerable appeal, we await additional critical detail and mechanistic insight.
Four key questions regarding signaling mechanisms
LL37 binds to and activates MRGX2, which signals via Gi/o to increase the abundance of TRPV4 mRNA in mast cells. This finding needs to be deconstructed, from the cell membrane to the TRPV4 gene locus in the nucleus. Could MRGX2→Gi/o signaling possibly be coupled to Ca++ signaling, which would lead to Ca++-dependent reprogramming of transcriptional programs in mast cells to sustain the degranulation response? This is a timely and medically relevant question that is now tractable. As for Ca++ signaling, Gi/o signaling in human bronchial epithelia has been found to be coupled to TRPV4 in response to airborne irritants from diesel combustion engines (Li et al., 2011), but, of course, other Ca++-influx pathways in mast cells have to be examined, as well.
Directly related to the first question, the study raises the key issue of whether increased mRNA abundance of TRPV4 correlates with increased and/or more rapid degranulation of mast cells. In terms of the stimuli that evoke mast cell degranulation, we can benefit from a broader spectrum of degranulating agents, beyond the use of compound 48/80 and LL37. Rosacea is a chronic dermatitis of facial skin, so thermal cues as well as sunlight/UVB radiation exposure have to be considered as provoking factors. The senior authors' group demonstrated that epidermal TRPV4 channels were activated in response to UVB overexposure, leading to sunburn pain and tissue damage (Moore et al., 2013). Epidermal keratinocytes show stronger expression of proalgesic, propruritogenic, and mast cell-degranulating endothelin-1 (ET-1) via an epidermal autocrine/paracrine feedforward loop that involves increased ET-1 expression/secretion and activation of TRPV4 channels expressed in keratinocytes, downstream of ET(R)-A and -B. In other words, TRPV4 in keratinocytes is involved in responding to UVB, leading to Ca++-influx, which evokes increased expression/secretion of ET-1. According to Mascarenhas et al., LL37 “spring-loads” mast cells by increasing the expression of TRPV4, and it is known that ET-1 can degranulate mast cells via ET(R)-A. Thus, the question is whether TRPV4 is a critical part of the ET-1/ET(R)-A mast cell degranulation pathway, which is facilitated by excess ET-1 made by skin keratinocytes (in a TRPV4-dependent manner) in response to UVB exposure (Moore et al., 2013). Moreover, given TRPV4′s thermal response profile, can thermal cues enhance skin inflammation in rosacea via TRPV4, by affecting both TRPV4 signaling in keratinocytes and/or mast cells? Interestingly, LL37 has been reported to increase UVB-evoked inflammasome activation in cultured human keratinocytes and organotypic skin culture (Salzer et al., 2014).
This specific question presented above can be framed more generally, namely by asking whether TRPV4-mediated Ca++ influx functions as a signaling mechanism to reprogram mast cells' transcription, involving regulated expression of the degranulation apparatus so that degranulation becomes more potent/rapid. The alternative, not mutually exclusive, is that TRPV4-mediated Ca++-influx is part of mast cells' Ca++-response that is critical for degranulation itself, a more rapid signaling mechanism that does not involve the genomic response (Figure 1). In both cases, targeting TRPV4 for therapeutic benefit would be rational.
Additional important tasks are to identify the participants in TRPV4-independent mechanisms that are suggested by the attenuation, not elimination of degranulation in response to TRPV4 loss-of-function experiments, as reported by Mascarenhas et al.
Importance of the facial localization of rosacea—external cues and innervation
The Mascarenhas et al. (2017) paper is exciting because it casts a critical element of rosacea pathogenesis in a new light. The increased presence of antibacterial cathelicidin is a hallmark of rosacea facial dermatitis. The suggested “revving-up” of a mast cell degranulation mechanism by a cleavage product of cathelicidin sheds new light on an evolutionary well-honed antibacterial defense mechanism that in rosacea obviously overshoots to induce chronic cutaneous inflammation. As argued above, we have to remain mindful of its link to UVB exposure on the human face, but facial skin as the most common location of rosacea also implies another critical biological variable that is, perhaps, underappreciated. All facial skin is innervated by the trigeminal nerve in terms of sensory innervation. The trigeminal nerve, with its three branches, is at the center of a vital neural system that has evolved as a powerful sentinel for detecting danger/damage/noxious cues that might affect a vertebrate's head-face. Trigeminal sensory malfunction in humans can result in craniofacial pain, including migraine, temporomandibular joint pain, and trigeminal nerve pain. As a functional and structural system that underpins these medically relevant conditions, the trigeminal system can generate neurogenic inflammation powerfully. Bearing in mind this important feature, we should consider whether the chronic facial dermatitis of rosacea can be facilitated by neurogenic inflammation via trigeminal sensory afferents and also whether chronically inflamed skin in rosacea can sensitize trigeminal sensory afferents. From the senior author's own clinical practice (with now >1,000 cases encountered suffering from trigeminally mediated pain and sensory disorders), the clinical impression is affirmative, as he has observed worsening of facial pain in patients with poorly controlled rosacea. Clearly, this clinical impression awaits controlled assessment through clinical studies.
Last but not least, the link between chronic facial skin inflammation in rosacea, constitutive facial innervation by the trigeminal system, and possible involvement of trigeminal neuroinflammation and neural sensitization mechanisms in rosacea should prompt one to think more proactively about why the presence and severity of rosacea could act as a risk factor for Alzheimer's disease, as recently reported (Egeberg et al., 2016).
Acknowledgments
Work in the authors' laboratories has been supported by NIH grants DE018549 (WL), UL1TR001117 (pilot award to WL), P30AR066527 (Duke PI RPH, pilot award to WL), F33DE024668 (YC), K12DE022793 (YC), 5K08AR063729-04 (ASM), DoD W81XWH-13-1-0299 (WL), and a Harrington Discovery Institute (Cleveland, OH) Scholar-Innovator Award (WL).
Small molecules were generated in the senior author's laboratory at Duke University, with subsequent patent filings by the Duke University Office of Licensing and Ventures, one of them granted in 2016 (US Patent 9290489).
Footnotes
Conflict of Interest: Aside from this, there is no conflict of interest on behalf of any of the authors.
References
- Akiyama T, Ivanov M, Nagamine M, Davoodi A, Carstens MI, Ikoma A, et al. Involvement of TRPV4 in serotonin-evoked scratching. J Invest Dermatol. 2016;136:154–60. doi: 10.1038/JID.2015.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri-Haber N, Joseph E, Dina OA, Liedtke W, Levine JD. TRPV4 mediates pain-related behavior induced by mild hypertonic stimuli in the presence of inflammatory mediator. Pain. 2005;118:70–9. doi: 10.1016/j.pain.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Chen Y, Fang Q, Wang Z, Zhang JY, MacLeod AS, Hall RP, et al. Transient receptor potential vanilloid 4 ion channel functions as a pruriceptor in epidermal keratinocytes to evoke histaminergic itch. J Biol Chem. 2016;291:10252–62. doi: 10.1074/jbc.M116.716464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MK, Lee H, Caterina MJ. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem. 2003;278:32037–46. doi: 10.1074/jbc.M303251200. [DOI] [PubMed] [Google Scholar]
- Denda M, Sokabe T, Fukumi-Tominaga T, Tominaga M. Effects of skin surface temperature on epidermal permeability barrier homeostasis. J Invest Dermatol. 2007;127:654–9. doi: 10.1038/sj.jid.5700590. [DOI] [PubMed] [Google Scholar]
- Egeberg A, Hansen PR, Gislason GH, Thyssen JP. Patients with rosacea have increased risk of dementia. Ann Neurol. 2016;79:921–8. doi: 10.1002/ana.24645. [DOI] [PubMed] [Google Scholar]
- Kim S, Barry DM, Liu XY, Yin S, Munanairi A, Meng QT, et al. Facilitation of TRPV4 by TRPV1 is required for itch transmission in some sensory neuron populations. Sci Signal. 2016;9:ra71. doi: 10.1126/scisignal.aaf1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Kanju P, Patterson M, Chew WL, Cho SH, Gilmour I, et al. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ Health Perspect. 2011;119:784–93. doi: 10.1289/ehp.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas NL, Wang Z, Chang YL, Di Nardo A. TRPV4 mediates mast cell activation in cathelicidin-induced rosacea inflammation. J Invest Dermatol. 2017;137:972–5. doi: 10.1016/j.jid.2016.10.046. [DOI] [PubMed] [Google Scholar]
- Moore C, Cevikbas F, Pasolli HA, Chen Y, Kong W, Kempkes C, et al. UVB radiation generates sunburn pain and affects skin by activating epidermal TRPV4 ion channels and triggering endothelin-1 signaling. Proc Natl Acad Sci USA. 2013;110:E3225–34. doi: 10.1073/pnas.1312933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer S, Kresse S, Hirai Y, Koglin S, Reinholz M, Ruzicka T, et al. Cathelicidin peptide LL-37 increases UVB-triggered inflammasome activation: possible implications for rosacea. J Dermatol Sci. 2014;76:173–9. doi: 10.1016/j.jdermsci.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975–80. doi: 10.1038/nm1616. [DOI] [PubMed] [Google Scholar]