

Abstract
HIV Gag (Pr55Gag), a multidomain polyprotein that orchestrates the assembly and release of the human immunodeficiency virus (HIV), is an active target of antiretroviral inhibitor development. However, highly pure, stable, recombinant Pr55Gag has been difficult to produce in quantities sufficient for biophysical studies due to its susceptibility to proteolysis by cellular proteases during purification. Stability has been improved by using a construct that omits the p6 domain (Δp6). In vivo, p6 is crucial to the budding process and interacts with protein complexes in the ESCRT (Endosomal Sorting Complexes Required for Transport) pathway, it has been difficult to study its role in the context of Gag using in vitro approaches. Here we report the generation of a full length Gag construct containing a tobacco etch virus (TEV)-cleavable C-terminal hexahistidine tag, allowing a detailed comparison of its nucleic acid binding properties with other constructs, including untagged, Δp6, and C-terminally tagged (TEV-cleavable and non-cleavable) Gags, respectively. We have developed a standard expression and purification protocol that minimizes nucleic acid contamination and produces milligram quantities of full length Gag for in vitro studies and compound screening purposes. We found that the presence of a carboxyl-terminal hexahistidine tag changes the nucleic binding properties compared to the proteins that did not contain the tag (full length protein that was either untagged or reulted from removal of the tag during purification). The HIV Gag expression and purification protocol described herein provides a facile method of obtaining large quantities of high quality protein for investigators who wish to study the full length protein or the effect of the p6 domain on the biophysical properties of Gag.
Keywords: HIV, Gag, Pr55Gag, Nucleic acid binding, p6, Proteolysis, His-tag
1. Introduction
The HIV-1 Gag protein directs the assembly of virus particles from infected cells. It is synthesized as a polyprotein precursor (Pr55Gag) and selectively binds to the viral genomic RNA to form a viral ribonucleoprotein (RNP) complex. The viral RNP complex traffics to the plasma membrane where Gag multimerizes to form a spherical shell that buds from the plasma membrane. During or just after budding, Gag is proteolytically processed into the MA, CA, SP1, NC, SP2, and p6 proteins, a step that is required for the production of mature, infectious virions [1].
One of the challenges of studying the HIV-1 virus particle assembly and maturation processes in vitro has been the difficulty in producing high quality protein in sufficient amounts for biochemical analysis. Previously, individual domains or deletion mutants of the Gag polyprotein have been expressed for biophysical studies, rather than the full-length protein, which is susceptible to C-terminal proteolysis [2]. Removal of the p6 domain prevents C-terminal proteolysis therefore, constructs without p6 have been the most widely used for studying Gag. More recently, Carlson and Hurley reported the development of a myristoylated full length Gag fused to a tobacco etch virus (TEV)-cleavable C-terminal maltose binding protein (MBP) tag that was used to identify the minimal set of components needed for Endosomal sorting complexes required for transport (ESCRT) assembly at HIV budding sites [3]. In addition, McKinstry et al. reported the development of a bacterial heterologous protein expression system to produce soluble recombinant wild type full length HIV-1 Pr55Gag using a C-terminal hexahistidine tag (Pr55Gag-His) to facilitate purification [4]. Although this approach was an important step forward and improved the purity of the protein, the possible effect of a non-cleavable C-terminal His tag on the function of the p6 domain was not examined.
Using the Pr55Gag construct as a reference point, we sought to undertake a systematic comparison of HIV-1 Pr55Gag expression constructs containing cleavable or non-cleavable carboxyl terminal hexahistidine tags grown in various expression media to examine the quality and behavior of the protein under an assortment of purification schemes. We designed constructs and a purification strategy to minimize the differences in sequence and the levels of trace metals or protein contaminants, respectively. Comparison of the properties of wild type Pr55Gag, with a non-cleavable C-terminal hexahistidine tag (Pr55Gag-His) and with a tobacco etch virus (TEV) cleavable hexahistidine tag (Pr55Gag-TEV-His) revealed that the presence of the tag altered nucleic acid binding properties. By contrast, the Pr55Gag-TEV-His construct, which contains six additional amino acids derived from the TEV recognition sequence (ENLYFQ), produced a protein that, after TEV cleavage, exhibited nucleic acid binding properties indistinguishable from untagged Pr55Gag. This TEV-cleavable construct will be useful for biochemical studies in which the presence of the hexahistidine tag could adversely impact functional studies.
2. Materials and methods
2.1. Bacterial strains and media
All transformations were performed using the E. coli strains DH5α and BL21(DE3) pRIL and standard heat shock (42 °C for 40 s) of RbCl2 competent cells. Plasmid DNA was produced in DH5α cells cultured in LB media supplemented with the antibiotic kanamycin (50 μg/ml). BL21(DE3) pRIL cells containing plasmid DNA were cultured in MDG media [5] supplemented with the antibiotics kanamycin (50 μg/ml) and chloramphenicol (35 μg/ml).
2.2. Cloning of Gag constructs into pET-based expression vectors
The plasmid containing the full length wild type Pr55Gag gene in a pET28a vector engineered with a non-cleavable hexahistidine sequence at the C-terminus of the gene was a kind gift from Johnson Mak [4] and was used as the template for all other full length constructs. For simplicity, it is referred to herein as Pr55Gag-His. The TEV cleavable Pr55Gag-TEV-His construct was generated using the Q5 site directed mutagenesis kit (New England Biolabs), and the first oligonucleotides suggested by the NEBasechanger software therein. To generate constructs without specific domains, such as MA, p6 or SP2p6, Q5 mutagenesis was again used with templates Pr55Gag, Pr55Gag-His or Pr55Gag-TEV-His, as appropriate. In all cases, the Gag sequence in the expression plasmids was verified by DNA sequencing. For clarity, when multiple constructs are being discussed as a group, they will be referred to as Gag whereas individual constructs, shown schematically in Fig. 1, will be written explicitly.
Fig. 1. Schematic showing constructs used and nomenclature within the text.

The rectangular blocks represent the domains named within. The black rectangle labeled “2” represents SP2. The numbers above each block of constructs correspond to the amino acid number of the amino and carboxyl terminus of the protein fragment.
2.3. Test inductions
For test inductions, BL21(DE3) pRIL cells containing the appropriate HIV Gag T7-based expression plasmid were inoculated from a 8% glycerol MDG frozen stock by scratching cells from the top layer with a sterile pipet and then dipping the pipet into 2 mL MDG media [5] containing 100 μM kanamycin. The cultures were grown at 37 °C with shaking (300 rpm, New Brunswick) until they reached saturation, typically 12–18 h. Generally, MDG starter cultures are viable for at least 2 weeks when stored at 4 °C [6]. For test inductions, the saturated MDG cultures were diluted 1:1000 into 1 mL each of LB, ZYP5052 and ZYM5052 media supplemented with 100 μg/mL kanamycin. The LB culture was induced at an OD600nm of 0.8 by addition of IPTG to a final concentration of 0.4 mM, allowed to grow for 3 more hours at 37 °C and then harvested by centrifugation. The two autoinduction cultures (ZYP502 and ZYM5052) were allowed to grow for ~18 h at 37 °C before harvesting. The OD of each culture was measured (in cases where OD was expected to be > 2, for example in the autoinduction cultures, a 20-fold dilution was read). In each case, a 1 ml sample containing 1 OD of cells was centrifuged at 16 k×g in a microcentrifuge. The supernatant was removed and the pellet resuspended and lysed in 60 μL of Bugbuster (Novagen) containing 500 mM NaCl, 0.6 U/mL benzonase and rLysozyme (Novagen), or equivalent enzymes. After incubation on ice for 15 min, a 20 μL aliquot from the lysate was removed and mixed 1:1 with 2x SDS-PAGE sample buffer to make the “Total Cell Extract” gel sample. The remainder of the sample was centrifuged at 16 k×g for 5 min. 20 μL of the supernatant was removed and mixed 1:1 with 2x SDS-PAGE sample to make the “Soluble Fraction” gel sample and the remainder discarded. The pellet was resuspended in 40 μL of the Bugbuster reagent and 40 μL of 2x SDS-PAGE buffer to make the “Insoluble Fraction” gel sample. All samples were heated to 75 °C for 3 min and analyzed on a 12.5% SDS-PAGE gel.
2.4. Large-scale protein expression
For protein purification, a saturated MDG starter culture was diluted 1:250 to 1:1000 fold into 250 ml of LB, ZYP5052 or ZYM5052 media supplemented with 100 μg/mL kanamycin. Induction of IPTG and autoinduction cultures were carried out as test inductions and the cell pellets frozen and stored at −20 °C until used. For IPTG induction, a typical yield of cell mass was ~4 gm/L wet weight. For autoinduction, the cultures were grown to stationary phase, as judged by lack of change in OD600nm in two successive readings taken 30 min apart, to ensure maximal protein production. The typical yield of cell mass for autoinduction cultures was 3–4 gm/250 mL culture. Significantly higher or lower cell yields were associated with poor or no expression of the target protein.
2.5. Optimization of DNA removal by PolyminP
Frozen E. coli BL21(DE3) pRIL cells containing the overexpressed wild type HIV Pr55Gag-TEV-His protein were thawed by resuspension (~0.4 gm) in 5 mL of ice cold Lysis buffer (25 mM HEPES pH 7.5; 500 mM NaCl; 20 mM imidazole; 0.1 mM EDTA; 0.1 mM TCEP) with 7000 units of rLysoszyme (Novagen) or Redilyse (Epicenter Technology). Lysis was aided by brief sonication (5 × 5 s pulses) with an ice cold microprobe tip using a Missonic Ultrasonic Liquid Processor set at an amplitude of 75%. The cells were transferred into a small beaker in an ice water bath and stirred for 10 min. Subsequently, 20 μL aliquots of 10% PolyminP, pH 7.8 (Sigma Aldrich) were added. After each addition, 20 μL of cell slurry was removed and 2 μL used for absorbance measurements at 280 and 260 nm with a nanodrop spectrophotometer (ThermoFisher). The remainder was diluted to 50 μL with lysis buffer followed by addition of 50 μL of 2x PAGE protein sample buffer. PolyminP additions were continued until the absorbance at 260 nm reached a stable value. Lysate samples from each addition were heated to 75 °C for 3 min and then the proteins resolved on a 12% SDS-PAGE gel and visualized by Coomassie staining.
2.6. Cell lysis and removal of DNA in large scale cultures
Cells (3–4 gm wet weight) containing overexpressed Gag constructs were resuspended in 50 mL of lysis buffer (25 mM HEPES pH 7.5; 500 mM NaCl; 20 mM imidazole; 0.1 mM EDTA; 0.1 mM TCEP; and 0.1% NP-40 or Triton X-100) with 72,000 units of rLysoszyme (Novagen) or Redilyse (Epicenter Technology). The container of cell suspension was placed in an ice water bath for lysis by sonication using a Missonic Ultrasonic Liquid Processor fitted with a standard size tip that had been cooled on ice. The sonicator was set to an amplitude of 100 and sonication performed with 4 × 30 s pulses over a total processing time of 7 min, corresponding to a total power input of ~10,000 W. Following sonication, the cell lysate was allowed to stir for 5 min in an ice water bath after which 0.7 mL of polyethylenimine (PEI) was added slowly from a 10% PEI, pH 7.8, stock (final concentration ~0.14% PEI). The suspension was allowed to stir for an additional 2 min and the debris removed by centrifugation at 21 k×g for 15 min. The supernatant was removed, its volume determined and then transferred to a beaker in an ice water bath. The Gag protein was precipitated by slow addition to a final concentration of 30% (vol:vol) of saturated ammonium sulphate (pH 7.0) that had been sat at room temperature. This solution was allowed to stir 15–30 min on ice and the precipitate collected by centrifugation at 21 k×g for 30 min. The supernatant was then carefully decanted and the pellets dried by inversion for 5 min at room temperature. Any residual supernatant was removed from the rim of the centrifuge tubes with a Kimwipe (Kimberly-Clark).
2.7. Strong cation exchange chromatography
Ammonium sulfate pellets from 3 to 4 g of cells were dissolved in final volume of 10 mL resuspension buffer (10 mM HEPES pH 7.5, 500 mM NaCl, 0.1 mM TCEP and 0.1 mM EDTA). Once the pellets were fully resuspended, the solution was diluted 1:5 with buffer containing 10 mM HEPES pH 7.5, 0.1 mM TCEP and 0.1 mM EDTA. The final solution was centrifuged at 21,000g for 15 min and then loaded onto a strong cation exchange column (either Poros HS50, Fractogel EMD SO3, Eshmuno CPX or GE SP FF resins) equilibrated with HS A buffer (25 mM HEPES pH 7.5; 100 mM NaCl; 0.1 mM EDTA; 0.1 mM TCEP). The column was washed with ~10 column volumes (CV) of HS-A (or until the A280 nm returned to baseline) and the bound protein was eluted in a 15 CV linear gradient from HS-A to HS-B buffer (25 mM HEPES pH 7.5; 1 M NaCl; 0.1 mM EDTA; 0.1 mM TCEP). The peak Gag-containing fractions were identified by the absorbance profile of the chromatogram and Coomassie-stained SDS-PAGE. All of the Gag constructs eluted between 0.5 and 0.7 M NaCl.
2.8. Ni-NTA affinity chromatography
The ammonium sulfate pellets from 3 to 4 gm of cells were dissolved in 50 mL of Ni-Affinity A (10 mM HEPES, pH 7.5; 500 mM NaCl; 20 mM imidazole; 0.1 mM TCEP and 0.1 mM EDTA) and then loaded onto a 4 mL Nickel Sepharose HP column (GE Healthcare). Unbound protein was removed in a 20 CV wash with 8% Ni-Affinity B (10 mM HEPES, pH 7.5; 500 mM NaCl; 400 mM imidazole; 0.1 mM TCEP and 0.1 mM EDTA) and the bound protein eluted in a 10 CV step gradient to 100% Ni-Affinity B.
2.9. Removal of the carboxyl-terminal hexahistidine-tag
Where appropriate, TEV cleavable hexahistidine-tags were removed by incubation with TEV protease [7] using 1 mg TEV for 25 mg of target protein. Briefly, the approximate Pr55Gag-TEV-His protein concentration was estimated from the absorbance of the pooled sample at 280 nm (at this stage the extinction coefficient was assumed to be 1 OD280/mg·mL−1). Cleavage was performed overnight by dialysis at 4 °C against Ni-Affinity buffer A. The TEV protease was removed by re-chromatography of the dialyzed solution on a 1 mL Ni-Sepharose FF column (GE) and the column eluted as described above. The cleaved HIV Gag protein eluted in the flow through from this column, while any uncleaved material and the TEV protease were found in the 100% Ni-Affinity buffer B bump fractions.
2.10. Preparative size exclusion chromatography (SEC)
Peak fractions from the cation exchange or nickel affinity column (before or after TEV cleavage) were concentrated to 5–10 mg/mL and then chromatographed in 2 mL aliquots on a 120 mL preparative Superdex 200 column equilibrated in Sizing Column buffer (10 mM HEPES pH 7.5; 400 mM NaCl; 0.1 mM EDTA, 0.1 mM TCEP and 10 μM ZnCl2). The peak fractions were identified by the absorbance profile at 280 nm for the chromatogram and by SDS-PAGE analysis of peak fractions. The pooled fractions were concentrated to ~5–10 mg/mL (depending upon the construct) using an Amicon 30 k centrifugal concentrator, dispensed in 100 μL aliquots at ~0.5 mg/mL, snap-frozen in liquid nitrogen and stored at −80 °C.
2.11. Analytical size exclusion chromatography and molecular mass determination
The molecular mass of each construct was estimated by size exclusion chromatography either by comparison to molecular weight standards or by multiangle light scattering (SEC-MALS) with a semi-analytical S200 column and in-line Optilab ReX refractive index and Helios multiangle light scattering detectors (Wyatt). Aliquots (100 μL at ~0.5 mg/mL) of each protein were injected onto the column and the sample eluted at 0.4 mL/min in Sizing column, or Low Salt Arginine (10 mM HEPES pH 7.5; 150 mM NaCl; 100 mM Arginine; 0.1 mM EDTA and 0.1 mM TCEP) buffers. For SEC-MALS the molecular mass was determined using the Wyatt software.
2.12. Fluorescence anisotropy-based DNA-binding assay
Binding of HIV Gag constructs to a fluorescein labeled oligonucleotide DNA 24mer (FL-(TG)12) was followed by the change in the anisotropy of fluorescence polarization as a function of added protein [8,9]. These measurements were conducted by serial dilution from a Gag construct stock into low salt ITC (20 mM HEPES pH 7.5; 140 mM KCl and 10 mM NaCl) or high salt (20 mM HEPES pH 7.5; 340 mM KCl and 10 mM NaCl) buffer. Typically, 40 μL from 1 mL Gag solution (initial concentration between 200 and 1000 nM) in the desired buffer, was added to the first well of a black half-area 96-well microtiter plate (Corning 3686). 40 μl containing 2 nM of the labeled DNA was added to each well to give a final concentration in the binding reaction of 1 nM oligonucleotide. The 40 μL removed from the Gag stock was replaced with buffer, the solution mixed gently and then 40 μL of this mixture was added to the next well. This pattern was repeated for the next 39 wells (Samples 1–40). Wells 41–44 contained 80 μL of 1 nM FL-(TG)12 and 45–48 buffer alone. The plates were read in a Paradigm (Molecular Dynamics) plate reader with a fluorescein FP cartridge. Wells 45–48 were averaged and used as a plate blank. The anisotropy and total fluorescence intensity was computed using the standard formulas from the intensity of the perpendicular and parallel light using a g-factor determined from the manufacturers protocol.
The observed anisotropies as a function of protein concentration were fit to a variety of models using an in-house non-linear least squares fitting MATLAB (Mathworks) script that minimized the difference in chi-square (χ2) between the observed (Aobs) and calculated (Acalc) anisotropy values [10]:
The constrained optimizer lsqnonlin was used and the free concentrations of Gag and nucleic acid were estimated using the fmincon routines in MATLAB.
2.13. Determination of zinc content
Protein samples (20 μM) were dialyzed exhaustively against 3 changes of 10 mM HEPES pH 7.5, 400 mM NaCl, 0.1 mM EDTA and 0.1 mM TCEP buffer. Following dialysis, the protein concentrations were determined from their absorbance at 280 nm and using extinction coefficients estimated from their sequence using Prot-Param (http://web.expasy.org/protparam/). The zinc content was determined either by atomic absorption spectrometry or colorimetrically using 4-(2-pyridylazo)-resorcinol (PAR) and protein denatured in 5M Guanidinium-HCl or digesting with proteinase K [11]. The reported values and standard deviations are the average of at least three independent samples.
3. Results & discussion
To study the biophysical properties of HIV Pr55Gag nucleic acid binding, it is crucial to have reproducibly highly pure protein samples with negligible nucleic acid contamination. Previous studies have shown that HIV Pr55Gag can be obtained in an untagged form using cation exchange chromatography followed by antibody-based affinity chromatography with a non-commercially-available antibody directed against the last 21 amino acids of the p6 domain at the authentic C-terminus [2]. Although this protocol selected only full-length protein, production of antibodies was time-consuming and expensive. Without the antibody affinity column, preparations tended to suffer from truncation of amino acids at the C-terminus [2]. Subsequently, researchers have moved away from using this construct to one in which p6 has been removed because it does not produce significant truncated protein species and was considered to be an acceptable substitute in most binding and assembly studies [2]. Concerned by the limitation of previous studies using Gag proteins lacking p6, the Mak group developed a construct that produces in E. coli recombinant full length Gag fused to a non-cleavable C-terminal hexahistidine affinity tag [4]. However, in our unpublished studies with the Gag protein from the alpharetrovirus Rous sarcoma virus, we have observed differences in the conformation and nucleic acid binding properties of the N-terminally hexahistidine tagged protein (personal communication) and therefore, we examined whether this might also be the case for HIV Pr55Gag-His.
To compare the binding properties of untagged HIV Pr55Gag and Pr55Gag-His, we overexpressed and purified both proteins using the published methods, with the exception that the Pr55Gag protein was not further purified by an antibody affinity column directed against the authentic Gag C-terminus [2]. Consistent with published observations, this method resulted in variable amounts of prematurely terminated untagged protein (Fig. 2A; [2]). To investigate whether there were any differences in the nucleic acid binding properties between the two HIV Gag constructs, we performed a fluorescence anisotropy-based oligonucleotide-binding assay. Fig. 2B shows the nucleic acid binding properties of the Pr55Gag and Pr55Gag-His constructs. Both proteins bound with high affinity to the FL-(TG)12 oligonucleotide, as seen by the changes in anisotropy with increasing protein concentrations. However, a binding curve for the Pr55Gag-His construct was shifted at lower protein concentration, suggesting that it had a higher affinity (Pr55Gag-His KD,app ~1 nM vs ~3 nM for the Pr55Gag) and a sharper transition in our standard buffer with physiological salt and pH, suggesting that its binding was more cooperative than Pr55Gag. Initial quantitative analysis of Pr55Gag binding data indicated the existence of at least two binding events, whereas for Pr55Gag-His, only models involving cooperative binding of 4 Pr55Gag-His molecules fit the experimental data. Both protein-nucleic acid complexes reached the same limiting anisotropy, suggesting that their limiting complexes were similar in size. One possible explanation for the difference in binding of these constructs is that in preparations of Pr55Gag, ~10–20% of the protein molecules were truncated to slightly shorter species lacking 2–4 kDa of the C-terminal p6 sequence and, in some cases, likely extending through the SP2 domain, whereas these truncated products were not seen in the final purified Pr55Gag-His protein (Fig. 2A). Alternatively, the differences in anisotropy may reflect non-physiological contributions of the hexahistidine sequence to nucleic acid binding, as has been observed for other proteins [12–14]. To investigate this possibility further, we examined the construct containing a C-terminal TEV protease cleavage site placed upstream of the hexahistidine tag (Pr55Gag-TEV-His), allowing the consequence of the added histidine residues to be probed.
Fig. 2. Nucleic acid binding properties of Pr55Gag and Pr55Gag-His proteins.

(A) Coomassie-stained 12.5% SDS-PAGE of Gag proteins. Coomassie stained SDS PAGE of final purified proteins, as labeled above each lane. (B) The Pr55Gag-His (black square) construct binds cooperatively to DNA, whereas the Pr55Gag (gray circle) does not.
3.1. Expression and purification of untagged and carboxyl-terminal hexahistidine-tagged full-length HIV Gag constructs
To optimize the expression of all Gag constructs (Pr55Gag, Pr55Gag-His, Pr55Gag-TEV-His), we characterized their growth, induction and solubility properties in three different media: LB media with IPTG induction and two types of autoinduction medium, ZYP5052 and ZYM5052 [5]. On a per-cell basis, the expression, and solubility of all three constructs were approximately equal. However, in autoinducing medium, the total cell mass was approximately four fold greater than the IPTG inductions with LB (Supplemental Fig. 1). All three growth conditions resulted in a family of smaller forms of Gag that appeared to be the result of premature termination, as they were each detected by anti-p24 antibodies in Western blotting experiments. Initial experiments suggested that some growth conditions produced lesser amounts of these species; however, none of the parameters tested (different IPTG concentrations in LB inductions, a range of growth temperatures using each medium, or altering the length of growth in autoinduction medium) reproducibly affected their intensities. Based upon the higher per liter yields and the equivalence between the solubility and purification properties of protein produced in each of the media, the autoinducing medium ZYP5052 was used in all subsequent experiments.
3.2. Optimization of DNA removal and initial purification
HIV Pr55Gag binds strongly and nonspecifically to cellular nucleic acids. To ensure that the final preparations of each Gag construct lacked nucleic acids, we used polyethyleneimine (PEI) to precipite DNA, RNA, ribosomes and other cellular debris that otherwise could be carried through the purification. Cells grown in 50 ml ZYP5052 culture were lysed by sonication in 10 ml ice-cold lysis buffer. In all cases, addition of increasing concentrations of PEI, up to 0.14%, resulted in decreased absorption at 260 nm of the cleared lysate (Supplemental Fig. 2). At higher concentrations of PEI, no further reduction in the absorbance at 260 nm was observed. Analysis of the total protein by SDS-PAGE indicated that the majority (> 70%) of the Pr55Gag remained in solution following centrifugation (Fig. 3). Excess cationic PEI in the lysate can adversely affect some downstream chromatographic steps, especially in cation-exchange chromatography. To minimize PEI carryover and to partially purify the Gag proteins, the PEI supernatant was precipitated with 33% ammonium sulphate solution in an ice water bath. This step significantly reduced contaminating proteins (Fig. 3). Together, these two steps provided a consistent starting material for the purification of all the constructs tested to date, with the exception of the isolated MA and NC domains, which were not precipitated by 33% ammonium sulphate.
Fig. 3. Addition of a 33% ammonium sulphate solution after nucleic acid removal by PEI visibly reduced contaminating proteins.
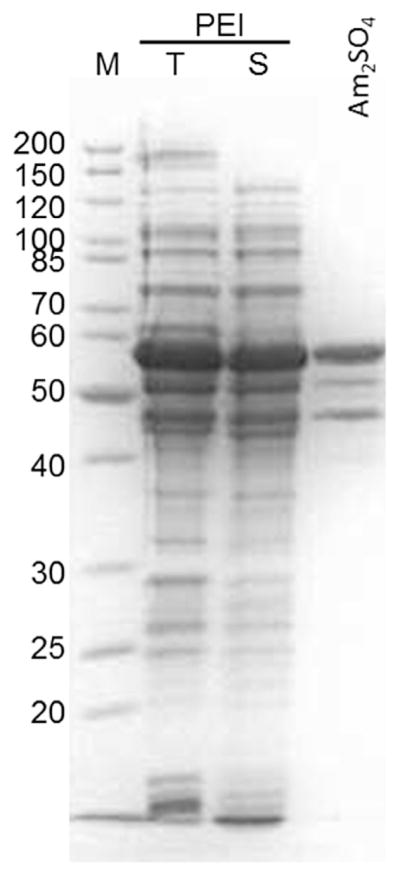
SDS-PAGE of Pr55Gag: Lane 1, markers; lanes 2 and 3, total and supernatant fraction after PEI precipitation, respectively; lane 4, supernatant after addition of ammonium sulphate.
3.3. Purification of Gag constructs for nucleic acid binding studies
Initially, we sought to develop a purification protocol to minimize differences between nickel affinity-based purification, which potentially exposes the histidine-tagged constructs to nickel or other metal ions, and the ion-exchange-based purification of untagged Pr55Gag. Fig. 4A shows a schematic of the protocol developed here; a key feature is that the final two steps expose all constructs to sequential strong cation-exchange and size exclusion chromatography. Starting with resuspended ammonium sulphate pellets, this purification protocol resulted in proteins with >90% purity, irrespective of the construct used.
Fig. 4. Overview of purification scheme.

(A) Purification scheme. (B–D) SDS-PAGE of the three Gag construct proteins. (B) Top, Pr55Gag; middle, Pr55Gag-His; bottom, Pr55Gag-TEV-His. In each case, lane 1, markers; lanes 2 and 3, respectively, total and supernatant fraction after PEI precipitation; lane 4, supernatant after addition of ammonium sulphate; lanes 5–7, wash fractions; lanes 8–13, relevant fractions after CPX column; lane 14, total pooled fractions. (C) Size exclusion chromatography (SEC) showed that all of the constructs had similar behavior on the resin regardless of the presence or absence of a His tag. The traces were offset for clarity and colored as follows: black - Pr55Gag-His6; red - Pr55Gag; blue -Pr55Gag-TEV-His; cyan - Pr55Gag-ENLYFQ and (D) Coomassie stained SDS PAGE of final purified proteins, as labeled above each lane. Pr55Gag had poorer purity than the other constructs due to the presence of smaller molecular weight products.
For nickel affinity purification, Gag constructs Pr55Gag-His and Pr55Gag-TEV-His were eluted in a linear gradient from 40 to 400 mM imidazole, rather than a step gradient, to minimize over-concentration. This step is consistent with a wide range of nickel resins including GE-HP, FF, FF-crude, Qiagen and SIGMA. In all cases, the prematurely terminated forms of the Pr55Gag-His or Pr55Gag-TEV-His protein eluted within the initial 40 mM wash (Fig. 4B upper and middle gels). By contrast, these species were not effectively removed using Roche Complete His-tag resin and the manufacturer's suggested conditions. Although this resin has a much lower rate of nickel leaching, it provided little selectivity between the C-terminally hexahistidine-tagged species and truncated, untagged species, which presumably bind the resin through their zinc finger domains in NC. Therefore, removal of the prematurely terminated contaminants was inefficient using this column. All of the cation exchange resins tested (Poros HS50, Eshmuno CPX, EMD SO3 and GE SP) resulted in proteins of similar purity, although none was able to separate full-length from truncated Pr55Gag.
Removal of the hexahistidine tag from the Pr55Gag-TEV-His protein with TEV protease (20:1 mg:mg) to generate Pr55Gag-ENLYFQ was very efficient (Fig. 4B bottom panel). Although a significant amount of the protein precipitated during overnight dialysis with TEV protease at 4 °C, on warming the protein to room temperature, the majority of precipitate resolubilized. By contrast, cleavage of this construct with TEV protease at room temperature resulted in significant proteolysis at non-TEV sites, reducing the quality of the final preparation. For SEC, all of the constructs were concentrated to 4–10 mg/ml using an ultrafiltration device with a 30 kDa cut off at 15 °C. Attempts to concentrate the protein at lower temperatures resulted in measurable losses of full-length protein with apparent enrichment of the residual truncated species. In the case of Pr55Gag, enrichment of smaller species was enhanced.
We noted that the solubility of the full-length protein at 4 °C varied with the nature of the construct; Pr55Gag-His was more soluble than Pr55Gag-TEV-His and Pr55Gag-ENLYFQ; Pr55Gag was the least soluble. Following cation exchange purification, all proteins were >95% pure by SDS-PAGE and eluted in a similar manner after SEC (Fig. 4C). This final chromatography step increased the purity to >98% and did not contain any measurable nucleic acid contamination as judged by the ratios of absorbance at 260 and 280 nm. All protein constructs purified in this manner were stable from degradation during incubation at room temperature for 24 h (Fig. 4D).
3.4. Characterization of Gag constructs
To characterize the purified Gag proteins, we measured the zinc content from various preparations (biological replicates). Initially, we used zinc atomic absorption spectroscopy from a single preparation of Pr55Gag and found that it contained 1.8 ± 0.3 mol zinc/protein, consistent with it containing two zinc finger motifs. For more routine analysis of all protein constructs, we used a chromogenic assay that gave a value of 1.8 ± 0.4 (8 biological replicates each containing 3 technical replicates; Table 1). Similar zinc content was found for the Pr55Gag-His and Pr55Gag-TEV-His constructs. For the TEV-cleaved Pr55Gag-ENLYFQ construct, the measured zinc content ranged from 1.5 to 2.0 atoms per protein with the majority of outliers (<1.7 or >1.9 Zn/protein), corresponding to our earliest preparations of this construct. The variation most likely arose from the solubility behavior of the protein impacting the protein concentration estimate based on the observation that the five most recent preparations, in which the protein was held at room temperature during protein and zinc concentration measurement, had a zinc content of 1.9 ± 0.3 atoms/protein. This value is consistent with measurements for the other three protein constructs.
Table 1.
Summary of purification data.
| Gag Construct | Independent preparations | Yield mg/250 mLa | Zn/protein | Mw SEC | c1/2 (nM)b 140 mM KCl | c1/2 (nM)b 340 mM KCl |
|---|---|---|---|---|---|---|
| Pr55Gag | 9 | 16 ± 2 | 1.8 ± 0.3 | 110 kDa | 2.8 ± 0.2 | 22 ± 5 |
| Pr55 Gag–His | 6 | 12 ± 4 | 1.8 ± 0.4 | 112 kDa | 1.1 ± 0.3 | 19 ± 3 |
| Pr55 Gag–TEV-His | 4 | 11 ± 4 | 1.7 ± 0.3 | 110 kDa | 1.0 ± 0.1 | 21 ± 4 |
| P55 Gag–ENLYFQ | 8 | 6–10 | 1.5–0.2 | 107 kDa | 2.9 ± 0.4 | 24 ± 3 |
| Pr55Gag Δp6 | 3 | 22 ± 4 | ND | 95 kDa | 3.5 ± 0.5 | 21 ± 3 |
| Pr55Gag ΔSP2Δp6 | 3 | 21 ± 3 | ND | 95 kDa | ND | ND |
ND Not determined.
Final yield of purified protein per 250 mL of ZYP5052.
Protein concentration at ½ maximal anisotropy. Average of titrations from at least three independent preparations of three technical replicates each.
All four Gag constructs had identical hydrodynamic radii as judged by size exclusion chromatography (Fig. 4A). Based upon their elution volumes, their molecular weights were ~110 kDa, consistent with that of a dimer, as previously reported for Pr55Gag-His [15]. Also consistent with this value, mass determination by SEC-MALS for Pr55Gag was 105 ± 10 kDa for the peak, with decreasing molecular weight for species eluting in the tail. This behavior is consistent with the formation of a relatively weak dimer [15], but not of the trimer described by Mak [4], which might have resulted from the higher salt concentration used in their assay (1M).
3.5. Effect of carboxyl-terminal hexahistidine tag on nucleic acid binding
Titration of FL-(TG)12 with the four purified proteins, Pr55Gag, Pr55Gag-His, Pr55Gag-TEV-His and Pr55Gag-ENLYFQ, in buffer containing physiological salt concentrations showed two types of binding behavior (Fig. 5A). Both constructs containing an intact hexahistidine tag bound more tightly to this oligonucleotide (concentration at half maximal anisotropy, C½, ~1 nM; Table 1) and the binding curve was visibly steeper compared to untagged protein (C½ ~ 3 nM; Table 1). Removal of the tag to create Pr55Gag-ENLYFQ resulted in binding behavior indistinguishable from Pr55Gag (Fig. 5A). Fitting of these titration curves to simple 1- or 2-state models or cooperative dimer or tetramer models (accounting for ligand depletion), suggest that the binding model for Pr55Gag was different from Pr55Gag-His. The Pr55Gag and Pr55Gag-ENLYFQ curves could only be modeled as two independent sequential binding events requiring either Gag monomers or one Gag dimer, whereas Pr55Gag-His and Pr55Gag-TEV-His required a cooperative binding model with a stoichiometry of 4. A more detailed analysis of binding stoichiometry is beyond the scope of this work and will be presented elsewhere. At higher salt concentrations, the affinity of each of the proteins for nucleic acids was reduced, as expected for an interaction involving highly charged oligonucleotides. At 340 mM KCl, all protein titrations were essentially identical (Table 1), demonstrating that the hexahistidine tag altered the affinity of Pr55Gag for oligonucleotides at physiological salt concentrations.
Fig. 5. Nucleic acid binding properties of full length and deletion constructs.

A. Pr55Gag-TEV-His (blue triangle) and Pr55Gag-ENLYFQ (cyan inverted triangle) overlaid on data from Fig. 2B Pr55Gag-His (black square), Pr55Gag (red circle). B. Pr55GagΔp6 (green) and Pr55GagΔp6-His (purple).
3.6. The presence of a C-terminal His tag perturbed nucleic acid binding in Pr55GagΔp6 constructs
To test whether the perturbed nucleic acid binding in the presence of a His-tag was specific to full length Pr55Gag, we compared the binding of untagged Pr55GagΔp6 (residues 1–432) and Pr55GagΔp6-His to the fluorescently labeled 24-mer. Fig. 5B shows the resultant binding curves. Similar to the full-length constructs, the presence of the C-terminal His-tag resulted in an apparent increase in cooperativity compared with Pr55GagΔp6 (residues 1–450). However, in this case, the limiting anisotropy of Pr55GagΔp6-His was significantly larger than Pr55GagΔp6. This finding could be due to a difference in the size of the limiting complex or could result from perturbation of the mobility of the fluorescein label by the His-tag in the shorter Gag constructs. A similar situation was observed with the Pr55GagΔSP2Δp6 construct. These observations further highlight the potential functional differences between Gag constructs with and without a His-tag in certain assays.
4. Conclusions
Studies have shown that Pr55Gag containing a variety of C-terminal tags can be assembled into virus-like particles (VLPs) in tissue culture cells [16,17]. Although Pr55Gag-His forms VLPs in vitro [4], demonstrating it is assembly competent, the results herein indicate that non-native histidine residues fused to the C-terminus can alter the biophysical properties of nucleic acid binding, one of the key early events in assembly. Therefore, it is important that care be taken when interpreting nucleic acid binding or nucleic acid promoted Pr55Gag associations using the Pr55Gag-His protein. For these studies, the use of the Pr55Gag-ENLYFQ protein or another cleavable His-tag construct may be preferable to ensure that the majority of the sample contains the entire p6 region. For constructs in which the p6 regions had been removed (residues 1–450; Pr55Gag-Δp6) or SP2p6 (residues 1–432; Pr55Gag-ΔSP2Δp6), we observed little or no shorter products even with untagged constructs (Fig. 2A), and the desired proteins were obtained in high yields and purity using only cation exchange and SEC chromatography. Thus, for these preparations there seems to be little, if any, benefit in the addition of a C-terminal hexahistidine tag. Of note, others have also found benefit to inserting a cleavable tag at the Gag C-terminus for in vitro studies. For example, Carlson and Hurley examined the in vitro interaction of Gag with purified ESCRT proteins, which required the p6 domain for binding [3]. To facilitate purification of the full length construct, the authors produced a myristoylated Gag protein fused to a TEV-cleavable C-terminal MBP tag [3]. However, in that study, only the cleaved version of the protein was used in experiments, and there was no comparison of the nucleic acid binding properties of the C-terminally tagged Gag versus the TEV-cleaved constructs. Based on our findings that a carboxyl-terminal tag can alter the biophysical properties of the Gag protein in certain assays, therefore, if a carboxyl-terminal hexahistidine tag were to be used to purify Gag, it would seem prudent to remove the tag before performing functional assays.
In summary, we have developed a set of TEV protease cleavable carboxyl-terminal hexahistidine tagged Gag expression constructs that lower the potential for artifacts when performing in vitro studies. Moreover, our purification protocol provides a rapid and facile means of purifying a range of His-tagged or untagged Pr55Gag constructs in which particular domains of Gag have been deleted (CANC, ΔSP2, Δp6). For studies in which the p6 domain is not essential, untagged Gag constructs can be obtained in very high yield and purity, and are therefore preferable for biochemical studies. However, in other cases, where the p6 domain is required, the full-length Pr55Gag construct should either be isolated using an antibody column against the authentic C-terminus or, when this approach is not possible, a cleavable hexahistidine tag should be considered.
Supplementary Material
Acknowledgments
We appreciate the technical contributions of Malgorzata Sudol. This work was supported in part by NIH grant P50GM103297 (LJP), a Collaborative Development Pilot Grant from the Center for HIV RNA Studies (JMF; P50GM103297, A. Telesnitsky, PI), and under a grant with the Pennsylvania Department of Health using Tobacco Settlement Funds (JMF). SNH was supported by NIH T32 CA060395.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.pep.2016.10.001.
Footnotes
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
References
- 1.Lingappa JR, Reed JC, Tanaka M, Chutiraka K, Robinson BA. How HIV-1 Gag assembles in cells: putting together pieces of the puzzle. Virus Res. 2014;193:89–107. doi: 10.1016/j.virusres.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell S, Rein A. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J Virol. 1999;73:2270–2279. doi: 10.1128/jvi.73.3.2270-2279.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlson LA, Hurley JH. In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proc Natl Acad Sci U S A. 2012;109:16928–16933. doi: 10.1073/pnas.1211759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKinstry WJ, Hijnen M, Tanwar HS, Sparrow LG, Nagarajan S, Pham ST, Mak J. Expression and purification of soluble recombinant full length HIV-1 Pr55(Gag) protein in Escherichia coli. Protein Expr Purif. 2014;100:10–18. doi: 10.1016/j.pep.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Studier FW. Stable expression clones and auto-induction for protein production in E. coli. Methods Mol Biol. 2014;1091:17–32. doi: 10.1007/978-1-62703-691-7_2. [DOI] [PubMed] [Google Scholar]
- 7.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 8.Heyduk T, Lee JC. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc Natl Acad Sci U S A. 1990;87:1744–1748. doi: 10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LeTilly V, Royer CA. Fluorescence anisotropy assays implicate protein-protein interactions in regulating trp repressor DNA binding. Biochemistry. 1993;32:7753–7758. doi: 10.1021/bi00081a021. [DOI] [PubMed] [Google Scholar]
- 10.Woll MP, De Cotiis DA, Bewley MC, Tacelosky DM, Levenson R, Flanagan JM. Interaction between the D2 dopamine receptor and neuronal calcium sensor-1 analyzed by fluorescence anisotropy. Biochemistry. 2011;50:8780–8791. doi: 10.1021/bi200637e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfister T, Jones KW, Wimmer E. A cysteine-rich motif in poliovirus protein 2C(ATPase) is involved in RNA replication and binds zinc in vitro. J Virol. 2000;74:334–343. doi: 10.1128/jvi.74.1.334-343.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milojevic T, Sonnleitner E, Romeo A, Djinovic-Carugo K, Blasi U. False positive RNA binding activities after Ni-affinity purification from Escherichia coli. RNA Biol. 2013;10:1066–1069. doi: 10.4161/rna.25195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chant A, Kraemer-Pecore CM, Watkin R, Kneale GG. Attachment of a histidine tag to the minimal zinc finger protein of the Aspergillus nidulans gene regulatory protein AreA causes a conformational change at the DNA-binding site. Protein Expr Purif. 2005;39:152–159. doi: 10.1016/j.pep.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 14.Misselwitz R, de la Hoz AB, Ayora S, Welfle K, Behlke J, Murayama K, Saenger W, Alonso JC, Welfle H. Stability and DNA-binding properties of the omega regulator protein from the broad-host range Streptococcus pyogenes plasmid pSM19035. FEBS Lett. 2001;505:436–440. doi: 10.1016/s0014-5793(01)02865-4. [DOI] [PubMed] [Google Scholar]
- 15.Datta SA, Zuo X, Clark PK, Campbell SJ, Wang YX, Rein A. Solution properties of murine leukemia virus gag protein: differences from HIV-1 gag. J Virol. 2011;85:12733–12741. doi: 10.1128/JVI.05889-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutierrez-Granados S, Cervera L, Godia F, Carrillo J, Segura MM. Development and validation of a quantitation assay for fluorescently tagged HIV-1 virus-like particles. J Virol Methods. 2013;193:85–95. doi: 10.1016/j.jviromet.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Ono A, Orenstein JM, Freed EO. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol. 2000;74:2855–2866. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.