

Abstract
Circadian clock components oscillate in cells of the cardiovascular system. Disruption of these oscillations has been observed in cardiovascular diseases. We hypothesized that obstructive sleep apnea, which is associated with cerebrovascular diseases, disrupts the cerebrovascular circadian clock and rhythms in vascular function. Apneas were produced in rats during sleep. Following two weeks of sham or obstructive sleep apnea, cerebral arteries were isolated over 24 h for mRNA and functional analysis. mRNA expression of clock genes exhibited 24-h rhythms in cerebral arteries of sham rats (p < 0.05). Interestingly, peak expression of clock genes was significantly lower following obstructive sleep apnea (p < 0.05). Obstructive sleep apnea did not alter clock genes in the heart, or rhythms in locomotor activity. Isolated posterior cerebral arteries from sham rats exhibited a diurnal rhythm in sensitivity to luminally applied ATP, being most responsive at the beginning of the active phase (p < 0.05). This rhythm was absent in arteries from obstructive sleep apnea rats (p < 0.05). Rhythms in ATP sensitivity in sham vessels were absent, and not different from obstructive sleep apnea, following treatment with L-NAME and indomethacin. We conclude that cerebral arteries possess a functional circadian clock and exhibit a diurnal rhythm in vasoreactivity to ATP. Obstructive sleep apnea attenuates these rhythms in cerebral arteries, potentially contributing to obstructive sleep apnea-associated cerebrovascular disease.
Keywords: Cerebrovascular circulation, obstructive sleep apnea, circadian clock, diurnal rhythm, vasodilation
Introduction
The presence of cell autonomous circadian clocks offers a cell/tissue/organ the selective advantage of anticipation, and thereby the ability to respond to potential stimuli and stresses in an appropriate timing and intensity.1 Components of the circadian clock have been identified in both prokaryotes and eukaryotes.1–3 Nearly all mammalian cell types investigated possess functional circadian clocks. In mammals, circadian clocks can be subdivided into two main groups, namely central and peripheral clocks.4 The central clock is located in the suprachiasmatic nucleus (SCN), a subset of approximately 20,000 neurons found in the hypothalamus.4 The key time keeper or zeitgeber (ZT) for the central clock is light.5,6 Entrainment of peripheral clocks, present in all other cell types, is thought to be achieved either indirectly by the SCN via neural/humoral stimulation, and/or by behavioral events (e.g. feeding/fasting or physical activity).7–10
The mammalian clock mechanism is comprised of a set of proteins that generate self-sustained transcriptional positive and negative feedback loops with a free-running period of approximately 24 h.1 At the heart of the clock mechanism are the transcription factors BMAL1 (brain and muscle ARNT like protein 1) and CLOCK (circadian locomotor output cycle kaput).2,11,12 Upon heterodimerization BMAL1/CLOCK binds to E-boxes of target genes and activates their transcription.11–13 Many targets of BMAL1/CLOCK are themselves clock components, feeding back on BMAL1/CLOCK in a positive or negative manner, resulting in an ∼24 h rhythmic expression of target genes.11,14 It is estimated that ∼10–15% of genes exhibit diurnal rhythms in any given tissue.15
Time-of-day-dependent oscillations of clock genes have been identified in vascular smooth muscle and endothelial cells from aorta.16–19 Through genetic disruption of clock genes, the circadian clock has been shown to influence the time-of-day-dependent oscillations in aortic response to various vasoactive compounds.20 Similarly, cerebral arteries have been shown to possess oscillating circadian clock components.16,21 However, it is unknown if, or how, the cerebrovascular circadian clock may influence cerebrovascular function or reactivity over the course of the day. We hypothesized that cerebral vessels possess a functional circadian clock mechanism that regulates vasoreactivity in a time-of-day-dependent manner. Additionally, we examined the effects of obstructive sleep apnea (OSA) on the cerebrovascular circadian clock mechanism and vasoreactivity over the course of the day.
Obstructive sleep apnea is a highly prevalent disorder involving collapse of the upper airway during sleep to significantly reduce (hypopnea) or completely block (apnea) the movement of air into and out of the lungs.22–24 Episodes of OSA generally occur repetitively throughout sleep with each episode producing transient hypoxia and hypercapnia.22 Recent estimates suggest that 5–25% of the adult Western population suffers from clinically significant OSA.22,23 Risk factors for OSA include obesity and aging, suggesting that the prevalence of OSA is likely to increase in the future given the current demographic trends.22
OSA alters cerebrovascular responsiveness and is strongly associated with cardiovascular and metabolic diseases that significantly impact the brain.22–24 Patients suffering from OSA reportedly show decreases in cerebral blood flow, impaired autoregulation, and attenuated dilatory responses to hypoxia and hypercapnia.23 As a result, it is not surprising that OSA is strongly associated with numerous disease states involving the cerebrovascular circulation. Cross-sectional and longitudinal studies have identified OSA as an independent risk factor for stroke, with moderate and severe OSA increasing the risk of stroke by three-fold.23 In addition, OSA augments the damage following a stroke and increases the likelihood of subsequent strokes.23
By isolating cerebral arteries at different times-of-day, we demonstrate the presence of a functioning circadian clock mechanism present in the cerebral vasculature. Using isolated pressurized perfused posterior cerebral arteries (PCAs), we demonstrate that the sensitivity to the vasodilator ATP exhibits profound time-of-day-dependent oscillations, with the greatest sensitivity observed at the beginning of the active phase. Finally, we demonstrate that OSA attenuates the oscillation of circadian clock genes in cerebral arteries, and this is associated with a loss of diurnal variation in sensitivity to ATP-induced vasodilation.
Materials and methods
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals, 8th edition, published by the National Institutes of Health (NIH) and were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine, Houston, TX. Experiments have been reported in compliance with the ARRIVE (Animal Research: Reporting in Vivo Experiments) guidelines. Animals were housed in a satellite facility with a 12 h light (6 a.m.–6 p.m.): dark (6 p.m.–6 a.m.) cycle, and access to normal chow diet and water ad libitum.
Endotracheal obstruction device implantation
Long Evans rats (Charles River Laboratories, Houston, TX), eight to nine weeks old, were anesthetized with CCM DEAIII-Rodent Cocktail (Ketamine 37.5 mg/mL, Xylazine 1.9 mg/mL, Acepromazine 0.7 mg/mL; 1 µL/g body weight). An obstruction device, fabricated from silicone tubing, was inserted into the lumen of the trachea caudal to the thyroid.25,26 The silicone obstruction device was attached to PE-50 tubing that was tunneled under the skin and exited the back of the neck. The obstruction device did not significantly impede the movement of air when deflated but completely occluded the airway in the trachea when inflated by pressurizing the system.25 Beginning the day of surgery, the animals were treated for three days with an analgesic (5 mg/kg Ketoprofen, Fort Dodge Animal Health, Fort Dodge, IA) and an antibiotic (5 mg/kg Baytril, Bayer HealthCare LLC, Shawnee Mission, KS).
Computer controlled inflation of obstruction device
Following a one week recovery from surgery, rats were subjected to 60 episodes of apnea (10 s in duration) per hour for 8 h a day (8 a.m.–4 p.m.) during the sleep cycle for two weeks. Durations between apneas was varied to prevent the rats from predicting when an apnea would occur. Apneas were produced by inflating the balloon using compressed air.26 The timing and duration of the inflations were controlled by an AD Instruments Power Lab stimulator output using LabChart 7 (ADInstruments; Colorado Springs, CO) and a custom-built solenoid valve/gas regulator. The balloon pressure was monitored at all times using a pressure transducer.
Sham rats underwent identical surgical procedures and device implantation, but endotracheal obstruction devices were never inflated. In the present study, the experimental rats undergoing periods of apnea are referred to as “OSA rats” and the sham-controls are referred to as “sham rats”. Only rats which had a burst obstruction device before the completion of two weeks of OSA were excluded.
Isolated posterior cerebral arteries
Following two weeks of apneas (60 apneas/h) for the OSA group or the equivalent time for the sham group, rats were anesthetized with isoflurane and decapitated at one of four time points, 6 a.m. (lights on, Zeitgeber Time 0 (ZT0)), 12 p.m. (ZT6), 6 p.m. (lights off, ZT12), or 12 a.m. (ZT18). For nocturnal organisms, such as rats, the sleep phase is from ZT0–12 and the active phase from ZT12–24. Each brain was rapidly removed and placed in cold Krebs Buffer. Posterior cerebral arteries were carefully harvested and placed in a vessel chamber that was filled with Krebs buffer consisting of: (mM) 119 NaCl, 4.7 KCl, 1 MgSO4, 1.2 KH2PO4, 0.026 EDTA, 1.6 CaCl2, 5.5 glucose, and 25 NaHCO3. The buffer was gassed with 20% O2, 5% CO2, balanced with N2 to obtain a pH of 7.4 and recirculated at a rate of 9 mL/min. The ends of each PCA were mounted on glass micropipettes and secured with 12-0 nylon sutures. Care was taken to ensure that the mounted vessel segments did not have side branches or leaks. The PCAs were warmed to 37℃, and pressurized to 85 mmHg by raising buffer-filled reservoirs connected to the glass micropipettes. After 40 min of pressurization, luminal flow (150 µL/min) was initiated by adjusting the heights of the inflow and outflow reservoirs. PCAs were allowed an additional 20 min to develop spontaneous tone before the experimental protocol began.
PCAs were magnified 450× and digitally recorded by a CCD camera connected to a DVR. Data were analyzed offline using edge detection software to follow changes in vessel outer wall diameter. Pressurized and perfused PCAs were used to investigate endothelium mediated dilations by exposing the lumen of the artery to increasing concentrations of ATP. Percent tone, percent dilation, and percent constriction were calculated by:
where DMax = maximal PCA diameter before tone, DTone = PCA diameter with steady tone (after 20 min with flow), and DResp = PCA diameter after 15 mi exposure to agonist.
In some studies, ATP dose response curves were performed after incubating the pressurized perfused vessel (luminally and abluminally) with the nitric oxide synthase inhibitor nitro-L-arginine methyl ester (L-NAME; 100 µM) and the cyclooxygenase inhibitor indomethacin (10 µM) for 30 min.
Quantitative RT-PCR
Initial characterization of circadian clock genes in the cerebral vasculature (Figure 1) was performed using pial arteries from naïve rats. Rats were anesthetized with isoflurane, transcardially perfused with cold Krebs Buffer to remove any blood from the cerebral arteries, and decapitated at ZT0, 6, 12, or 18. The Circle of Willis and pial arteries were quickly removed, stripped of any adventitia, and snap frozen.
Figure 1.

Rhythmic expression of the circadian clock genes bmal1 (a), per1 (b), per2 (c), and the clock output gene dbp (d), in rat cerebral arteries isolated over the course of 24 h. R2 values resulting from cosinor analysis, assuming 24-h periodicity, are presented for each gene. Data are shown as the mean ± SD; n = 8 for ZT0/12/ 24, n = 9 for ZT6, and n = 7 for ZT18.
Circadian clock gene expression was examined in the heart and cerebral vasculature following two weeks of sham or OSA (Figures 3 and 4). In each tissue, gene expression is the result of multiple cell types (i.e. vascular smooth muscle and endothelial for cerebral vessels and cardiomyocytes, fibroblasts, and vascular cell types for heart). Rats were anesthetized with isoflurane and decapitated at ZT0, 6, 12, or 18. The PCA from these rats was removed and used in pressurized perfused vessel experiments (see above). The Circle of Willis and remaining pial arteries and heart were quickly harvested and snap frozen. Frozen tissues were ground with a pestle and total RNA was isolated using the RNeasy Micro Kit (Qiagen; Valencia, CA). Isolated RNA was treated with DNAse I to eliminate potentially contaminating genomic DNA. RNA purity and concentration were assessed using a NanoDrop UV-Vis spectrophotometer (Thermo Scientific; Wilmington, DE). All samples were diluted to 30 ng/µL.
Figure 3.

OSA significantly attenuated the rhythmic expression of clock and clock output genes in cerebral arteries. OSA reduced bmal1 expression at ZT6 (a). Peak expression of per1 (b), per2 (c), and dbp (d) were significantly attenuated at ZT12 in OSA cerebral arteries. Data are shown as the mean ± SD, n = 3 for sham ZT6/18 and OSA ZT0/12/18/24, n = 4 for sham ZT0/12/24 and OSA ZT6. *p = 0.039, #p = 0.046, ψp = 0.017 for sham vs. OSA at respective ZT.
OSA: obstructive sleep apnea; ZT: zeitgeber.
Figure 4.

Level of spontaneous tone developed by rat PCAs exhibits a diurnal variation, peaking during the active phase. OSA phase-shifts the rhythm in spontaneous tone developed, peaking during the middle of the sleep phase. Data are shown as the mean ± SD, n = 3 for sham ZT6/12/18 and OSA ZT0/12/18/24, n = 4 for sham ZT0/24 and OSA ZT6. *p < 0.001 and #p = 0.004 for sham vs. OSA at respective ZT.
PCAs: posterior cerebral arteries; OSA: obstructive sleep apnea; ZT: zeitgeber.
Original TaqMan assays were designed from rat sequences available in GenBank using Primer Express 2.0 (Applied Biosystems; Grand Island, NY). Primer and probe sequences were: forward 5′-TCCGATGACGAACTGAAACAC-3′, reverse 5′-CTCGGTCACATCCTACGACAA-3′, and probe 5′-FAM-CAAAAATCCATCTGCTGCCCTGAGAAT-TAMRA-3′ for brain and muscle ARNT like protein1 (bmal1); forward 5′-GGTTCAGGATCCCACGAAG-3′, reverse 5′-AAGAGTCGATGCTGCCAAAG-3′, and probe 5′-FAM-AGCACCTCAGCCAGCATCACCC-TAMRA-3′ for period 1 (per1), forward 5′-GCAGCCTTTCGATTATTCTCC-3′, reverse 5′-GCTCCACGGGTTGATGAAG-3′, and probe 5′-FAM-ATTCGATTCCGCACACGCAACG-TAMRA-3′ for period 2 (per2), forward 5′-AGGCAAGGAAAGTCCAGGTG-3′, reverse 5′-TCTTGCGTCTCTCGACCTCTT-3′, and probe 5′-FAM-CCGAGGAACAGAAGGATGAGAAGTACTGGAG-TAMRA-3′ for D-site of albumin binding promoter (dbp). The reverse primer for each gene was used in reverse transcriptase reactions containing 45 ng of mRNA, resulting in gene-specific first-strand synthesis. Quantitative RT-PCR reactions were performed with an Eppendorf Realplex (Hauppauge, NY) using the following conditions: 95℃ (1 min) followed by 40 cycles of 95℃ (12 s) and 60℃ (1 min). The number of PCR cycles required for the fluorescent signal to reach a detection threshold (Ct) was chosen in the linear phase of the logarithmic amplification curve. Since all samples contained the same amount of starting mRNA, we assumed 100% efficiency of the TaqMan assays in calculating mRNA levels from Ct values. Therefore, values are arbitrary and can only be compared within a given data set (i.e. OSA versus sham), and not between genes of interest (i.e. per1 versus per2).
Circadian activity measurements
From 6 a.m. on day 13 to 6 a.m. on day 14 of the experiment, sham and OSA rats were recorded constantly with an infra-red video monitoring system under normal 12 h light:12 h dark conditions. The live video recordings were fed into the motion detection software iSPYConnect ver. 2.0.7.7 (Margaret River, Western Australia). The trigger sensitivity of the program was set at 90%, which was shown to detect movement around the cage, grooming, rearing while eating, but did not detect head turns or tail movements. Timestamped motion events were exported to Microsoft Excel and total events were summed each hour.
Drugs and reagents
All reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Statistics
All data is expressed as mean ± SD. Required sample sizes for clock gene expression studies were calculated using the following parameters: minimum detectable difference in means = 2500 target mRNA/ng total mRNA, expected standard deviation = 500 target mRNA/ng total mRNA, number of groups = 8, power = 0.8, α = 0.05.27 Required sample sizes for PCA dilations to ATP were calculated using the following parameters: minimum detectable difference in means = 20 µm, expected standard deviation = 5 µm, number of groups = 8, power = 0.8, α = 0.05.25 Required sample sizes for vessel dilations dependent on the time-of-day were calculated using the following parameters: minimum detectable difference in means = 35%, expected standard deviation = 8%, number of groups = 8, power = 0.8, α = 0.05.20 Required sample sizes for activity studies dependent on the time-of-day were calculated using the following parameters: minimum detectable difference in means = 1600 events/h, expected standard deviation = 400 events/h, number of groups = 2, power = 0.8, α = 0.05.28 Rats were randomly allocated to experimental groups. For gene expression and activity studies, the experimenter was blinded to treatment groups. For functional vessel studies, the experiment was blinded to treatment group (sham versus OSA), but not to time-of-day.
Cosinor analysis was performed in R (Vienna, Austria) for initial characterization of oscillations in clock and clock output genes (Figure 1). The resulting R2 statistics that measures the percent variance accounted for by an oscillation with a period of 24 h, are presented in each graph. For analyzing dose–response dilations (Figures 5 and 6) and 24-h activity (Figure 7), two-way repeated measures ANOVA was used followed by a Holm–Sidak test for individual comparisons when appropriate. For sham versus OSA gene expression and tone-development (Figures 2 to 4), two-way ANOVA was used followed by a Holm–Sidak test for individual comparisons when appropriate. In addition, main effect comparisons found to be statistically significant with parametric ANOVA analysis were also statistically significant when compared with non-parametric ANOVA on Ranks, with the exception of Figure 6(a). All statistical analyses for Figures 2 to 7 were performed using SigmaPlot 13 (SyStat Software; San Jose, CA). All differences were considered statistically significant if p ≤ 0.05.
Figure 5.

Rat PCAs exhibit a diurnal rhythm in sensitivity to ATP induced vasodilation, peaking at the beginning of the active phase. OSA significantly decreases the sensitivity to ATP induced vasodilation (a–d), and abolishes the observed rhythm in ATP sensitivity (e). Data are shown as the mean ± SD, n = 3 for OSA ZT0/12/18/24, n = 4 for sham ZT0/6/12/18/24 and OSA ZT6. *p = 0.001, #p = 0.006, ψp = 0.041, @p = 0.003, for sham vs. OSA at respective ZT or ATP concentration, $p = 0.007 for sham ZT0 vs. ZT12.
OSA: obstructive sleep apnea; ZT: zeitgeber.
Figure 6.

Pretreatment of vessels with L-NAME and indomethacin abolished the diurnal rhythm in ATP sensitivity of sham PCAs. ATP sensitivity of OSA PCAs was not different than sham PCAs at any time point (a–e). Data are shown as the mean ± SD, n = 2 for OSA ZT0/24, n = 3 for sham ZT12 and OSA ZT6/12/18, n = 4 for sham ZT0/6/18/24.
PCAs: posterior cerebral arteries; OSA: obstructive sleep apnea.
Figure 7.
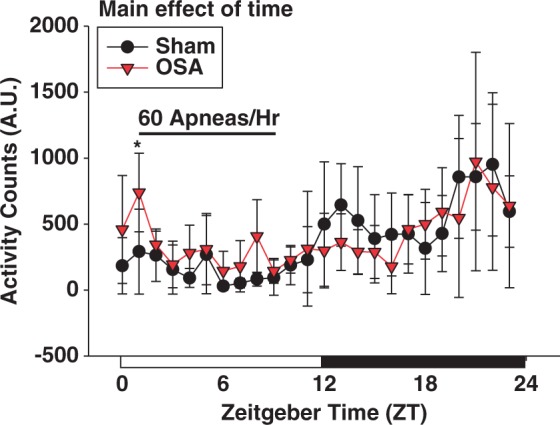
Two weeks of OSA, during the sleep cycle, did not significantly alter the diurnal variation in activity. Data are shown as the mean ± SD, n = 5 for sham, n = 6 for OSA, *p = 0.020 for sham vs. OSA at respective ZT.
OSA: obstructive sleep apnea; ZT: zeitgeber.
Figure 2.

Rhythmic expression of the circadian clock genes bmal1 (a), per1 (b), per2 (c), and the clock output gene dbp (d), were not altered in the heart following two weeks of OSA. Data are shown as the mean ± SD; n = 3 for sham ZT0/6/24 and OSA ZT0/6/12/18/24, and n = 4 for sham ZT12/18.
OSA: obstructive sleep apnea.
Results
Circadian clock and clock output genes oscillate over the course of 24 h in rat cerebral arteries
Seeing as the circadian clock is a transcriptionally based mechanism, we measured clock component mRNA levels in rat cerebral arteries isolated at 6-h intervals (beginning at zeitgeber time (ZT) 0; lights on) over the course of 24 h. Figure 1 illustrates profound diurnal variations in the core clock components bmal1, per1, and per2. Indicative of a functional circadian clock, the positive circadian clock component bmal1 peaks at a time when the negative clock components per1 and per2 are at a trough. Conversely, per1 and per2 peak when bmal1 is at a trough. The observed temporal patterns in expression, with positive clock components peaking at the beginning of the sleep phase and negative components peaking at the beginning of the active phase, are similar to what has been described in other tissues.10,29–33 In addition to the core clock components, we measured mRNA from a clock output gene, dbp. Figure 1(d) demonstrates that the clock output gene dbp also shows a diurnal variation in expression, peaking near the beginning of the active phase.
OSA attenuates oscillations of the circadian clock in cerebral arteries, but not the heart
To assess the effects of OSA on peripheral circadian clocks, the heart and cerebral arteries were isolated from sham and OSA rats every 6 h over the course of 24 h. Two-way ANOVA identified a significant time-of-day effect on the expression of the clock and clock output genes bmal1, per1, per2, and dbp in hearts isolated from rats (Figure 2; p < 0.001 for each gene). These rhythms were not significantly different in hearts of OSA rats (NS; two-way ANOVA and Holm–Sidak post hoc). Cerebral arteries isolated from sham rats also exhibited time-of-day dependent rhythms in clock related genes (Figure 3(a) to (d); p < 0.001, two-way ANOVA and Holm–Sidak post hoc). The rhythms in cerebral artery clock gene expression were similar in amplitude and temporal expression to rhythms observed in the heart. However, peak expression of per1, per2, and dbp at ZT12, were significantly lower in the OSA group compared to the sham group with OSA values being 32% (p = 0.046), 60% (p = 0.017), and 39% (p = 0.017) lower than the respective level in the sham group (Figure 3; two-way ANOVA and Holm–Sidak post hoc).
Posterior cerebral arteries exhibits a diurnal rhythm in sensitivity to ATP-induced dilation that is abolished by OSA
Maximum diameters for rat PCAs were 247 ± 23 µm and 244 ± 18 µm for sham and OSA rats, respectively, and not significantly different between groups at any individual time point (Supplemental Figure 1(a); two-way ANOVA). Two-way ANOVA indicates a significant effect of ZT*OSA for the percent of spontaneous tone developed by PCAs (Figure 4; p = 0.001). Sham PCAs isolated during the middle of the active phase (ZT18) developed 36 ± 4% tone, which was significantly greater than the 23 ± 1% tone developed by sham PCAs isolated during the middle of the sleep phase (ZT6; p = 0.016, two-way ANOVA and Holm–Sidak post hoc). Interestingly, PCAs isolated from rats following two weeks of OSA exhibited a time-of-day-dependent oscillation in the percent tone developed that was anti-phase to that observed in PCAs from sham rats. PCAs from OSA rats developed 37 ± 2% tone during the middle of the sleep phase (ZT6), which was significantly greater than the 24 ± 1% tone developed by OSA PCAs during the middle of the active phase (ZT18; p = 0.016, two-way ANOVA and Holm–Sidak post hoc).
Figure 5(a) to (d) shows concentration dependent dilations, to luminal ATP, of sham and OSA PCAs over the course of 24 h. Two-way repeated measures ANOVA revealed a significant effect of group (sham/OSA) at ZT0 (p = 0.008), ZT6 (p = 0.019), ZT12 (p < 0.001), and ZT18 (p = 0.023), as well a significant effect of ATP concentration at each timepoint (p < 0.001). Calculation of EC50 values revealed a significant time-of-day difference in the sensitivity of sham PCAs to ATP, with a 6.6-fold greater sensitivity at ZT12 (EC50 = 4.72e−7 M) as compared to ZT0 (EC50 = 3.13e−6 M; p = 0.025, t-test). Figure 5(e) demonstrates that the response to 10−6 M luminal ATP varied depending on the time-of-day, with significantly greater dilations at the beginning of the active phase (ZT12), as compared to the beginning of the sleep phase (ZT0; p = 0.007, two-way ANOVA and Holm–Sidak post hoc).
Independent of the time-of-day, there was a significant effect of OSA on dilations to ATP. At each timepoint PCAs isolated following two weeks of OSA showed no dilation to 10−8 to 10−6 M luminal ATP (Figure 5(a) to (d)). Compared to sham vessels, this resulted is significantly lower dilation of OSA vessels to 10−6 ATP at ZT0 (p = 0.006), ZT6 (p = 0.001), and ZT12 (p < 0.001; two-way ANOVA and Holm–Sidak post hoc). However, at each timepoint PCAs isolated from OSA rats maximally dilated, to a level not different than sham PCAs, in response to 10−5 M luminal ATP. When compared to sham PCAs, OSA completely abolished the time-of-day-dependent rhythm observed in response to 10−6 M luminal ATP (Figure 5(e)).
ATP has been shown to dilate cerebral arteries through a NO synthase cyclooxygenase independent mechanism known as endothelium dependent hyperpolarizations (EDH).34–36 To examine the EDH component of dilation contributing to the time-of-day-dependent rhythm observed in sham and OSA rats (Figure 5(e)), dilations to ATP in the presence of L-NAME (inhibits NO synthase; 100 µM) and indomethacin (inhibits cyclooxygenase; 10 µM) were examined over the 24-h period in PCAs. Supplemental Figure 1(b) illustrates the constriction of sham and OSA PCAs to L-NAME and indomethacin pretreatment over the course of 24 h. Although two-way ANOVA did not indicate any significant differences, sham PCAs exhibited greater constrictions to L-NAME and indomethacin at ZT0 versus ZT12. Sham PCA dilations to ATP following NOS and COX inhibition were similar to OSA dilations without NOS and COX inhibition (compare Figures 5 and 6(a) to (d)), with no significant response to 10−8 to 10−6 M luminal ATP and maximal dilations in response to 10−5 M ATP. Following inhibition of NOS and COX, dilations to luminal ATP were not different between groups (sham/OSA) at any timepoint (NS, two-way repeated measures ANOVA). Inhibition of NOS and COX in sham PCAs abolished the time-of-day dependent rhythm in dilation to 10−6 M ATP observed previously in sham PCAs (Figure 5(e)), and was not different than the PCA dilations after OSA (Figure 6(e); NS, two-way ANOVA).
Two weeks of OSA does not alter the central circadian clock
To examine the effect of OSA on the central clock, we measured activity in rats after two weeks of sham or OSA. As expected, Figure 7 demonstrates a diurnal rhythm in activity of rats, with increased activity during the active (dark) phase of the cycle (p < 0.001 main effect of ZT; two-way repeated measures ANOVA). Compared to sham rats, OSA rats exhibited increased activity during the first h of apneas (p = 0.020 at ZT1; two-way repeated measures ANOVA and Holm–Sidak post hoc) but had normal activity during the remainder of the day (Figure 7). Increased activity at ZT1 is likely due to the adjustments to balloon pressures in OSA rats during this time. These studies suggest that OSA does not significantly alter the central circadian clock.
Discussion
The goal of this study was to examine the role of the cerebrovascular circadian clock in regulating cerebral artery function over the course of the day. In addition we sought to determine if OSA, known to be associated with adverse cerebrovascular outcomes, alters the cerebrovascular circadian clock and clock controlled processes. We report three major findings from this study. (1) Circadian clock and clock output gene expression exhibit robust diurnal variations in cerebral arteries. (2) PCA sensitivity to ATP-induced vasodilation exhibits a diurnal rhythm. (3) OSA, attenuates oscillations of the cerebrovascular circadian clock, and abolishes the rhythm in PCA sensitivity to ATP.
Various parameters of cardiovascular physiology and pathophysiology exhibit diurnal variations. For example, in mammals heart rate and blood pressure peak at the beginning of the wake phase and trough during the sleep phase.30 Similarly there are diurnal variations in the onset of most pathological cardiovascular events. This includes increased incidence of stroke and myocardial infarction onset in the morning for humans.37 For the most part these rhythms have been attributed as secondary to rhythms in neuro-humoral stimulation. However, identification of peripheral circadian clocks in cardiovascular relevant cell types raises the question of what role these molecular clocks may play in cardiovascular physiology and pathophysiology.
Peripheral clocks have been identified in all cardiovascular relevant cell types; this includes cardiomyocytes, vascular smooth muscle cells, endothelial cells, and fibroblasts.17,18,38,39 Previous identification and characterization of the circadian clock mechanism in vascular smooth muscle and endothelial cells has largely been performed in immortalized cell lines and aorta. Davidson et al. first identified oscillating clock activity in the cerebral vasculature by culturing the basilar artery of the per1 luciferase rat (measurement of luciferase activity driven by the clock gene per1 promoter).16 Oscillations of core clock components and clock output genes have also been identified in the middle cerebral artery.21 Similarly, we observed robust 24-h oscillations in circadian clock and clock regulated gene expression in cerebral arteries (Figures 1 and 3). Importantly, the temporal distribution of these rhythms is similar to what has been observed in other tissues; with positive clock components (i.e. bmal1) peaking near the beginning of the sleep phase, and negative components (i.e. per1 and 2) peaking near the beginning of the active phase, when positive components are at a trough.
With the identification of functional circadian clocks in the vasculature, comes the question of “What is the biological relevance of the clock in vascular physiology?”. It has long been recognized that multiple aspects of blood vessel function exhibit diurnal variations.40 In animal models, diurnal variations have been observed in cardiac, skin, and muscle perfusion.40–42 Elegant studies by Wauschkuhn et al. demonstrate significant diurnal rhythms (∼20% change) in cerebral blood flow, in awake freely-ranging rats, that appear to be independent of rhythms in arterial blood pressure and activity.43 Similarly, diurnal variations have been described in human cerebral vessel blood flow velocity.44 Potential clock regulated mechanisms involved in vascular function have been explored in the aorta, where diurnal variations in the sensitivity to numerous vasoactive agents have been described.45–47 Relaxation of aortic rings in response to acetylcholine exhibits a time-of-day-dependence with increased relaxation during the active phase. Genetic disruption of the circadian clock abolished many of the rhythms observed in aortic responses, providing further evidence of circadian clock regulation.20,48,49 By isolating and perfusing PCAs ex vivo, thereby removing any extrinsic factors potentially influencing vessel responsiveness, we demonstrate that multiple aspects of vascular function exhibit diurnal rhythms. Both the developed tone and sensitivity to ATP-induced dilations exhibit diurnal rhythms, peaking during the active phase (Figures 4 and 5). This corresponds to the time-of-day when the brain is more metabolically active and cerebral blood flow is elevated.43 The active phase also represents a time-of-day when the cerebral circulation is exposed to greater fluctuations in blood pressure. Increased tone and sensitivity to vasodilators could allow cerebral arteries to respond to changes in pressure more precisely during the active phase. The time-of-day-dependent oscillation in ATP-induced dilation sensitivity was abolished by inhibiting NOS and COX (Figure 6). As we have never observed a role for COX in cerebral artery vasodilation, these findings suggest that the observed rhythm in ATP-induced vasodilation is NO-mediated. Circadian clock regulation of multiple genes (eNOS, caveolin1 and 3) and proteins (tetrahydrobiopterin, phospho-Akt, phospho-eNOS) involved in NO-induced vasodilation have been described in various tissues, but need to be explored in cerebral arteries.20,32,50,51
The onset of various pathological events, such as myocardial infarction, stroke, and sudden cardiac death have all been shown to be time-of-day-dependent in humans, with a primary peak in the early morning, a secondary peak in the evening, and the lowest vulnerability during the night.37 Evidence suggests that circadian clock regulated processes contribute not only to the timing of pathologic events, but also the susceptibility and damage. For example, outcome severity of myocardial ischemia and ischemic stroke both exhibit time-of-day-dependence in rats, with greatest infarct volumes observed following ischemia at the beginning of the active phase, and greatest protection when the ischemia occurred at the beginning of the sleep phase.52,53 In addition, certain stresses can disrupt peripheral circadian clocks, leading to exacerbation of the adverse outcome. For example, rhythms in circadian clock genes are attenuated in peripheral tissues following pressure overload induced cardiac hypertrophy, aging, and myocardial ischemia/reperfusion.53–56 Oscillations of the core clock gene per1 were abolished in white blood cells of OSA patients.57 We observed that, as compared to sham PCAs, two weeks of OSA lowered the amplitude of rhythmic clock genes as well as the sensitivity to ATP-induced vasodilation (Figures 3 and 5). As mentioned previously, OSA is strongly associated with increased risk of stroke and poorer outcomes following stroke.23 It is possible that loss of the normal rhythm in vasodilation sensitivity, caused by OSA, contributes to these outcomes. Interestingly, the normal morning prevalence of stroke is shifted in OSA patients to the sleep phase, further evidence of OSA disruption of diurnal rhythms.58,59 In addition, the clock mechanism has been shown to regulate numerous cellular processes in a time-of-day-dependent manner, including metabolism, apoptosis, immune response, and membrane potential.30,60–63 OSA induced dysregulation of these processes in the cerebral vasculature could exacerbate or potentially play a causal role in the adverse cerebrovascular outcomes associated with OSA.
In our model, OSA did not significantly alter the normal 24-h rhythm in activity, suggesting the central circadian clock was unaffected by OSA (Figure 7). Oscillation of core clock and clock output genes in the heart were also unaffected by OSA (Figure 2). As mentioned previously, the presence of circadian clocks in peripheral tissues allow for the cell/tissue/organ to anticipate and respond to stimuli in an appropriate manner.1 One hypothesis is that disruption of the circadian clock mechanism in one peripheral tissue and not others, removes temporal synchronization between tissues and organs, and may contribute to disease.64 One can see that it would be advantageous to have the clock in the heart, shown to influence rhythms in heart rate and blood pressure, in synchrony with the clock of the cerebral vasculature to properly respond to changes in flow and pressure. Additionally, peripheral circadian clocks have been identified in brain regions outside the SCN, as well as in distinct cell types of the brain such as astrocytes.21 Insults that impair the cerebrovascular clock (e.g. OSA), but not surrounding cell types, such as those in the neurovascular unit, may also setup a scenario leading to or exacerbating cerebrovascular diseases.
In conclusion, we demonstrate the presence of a functional circadian clock mechanism in the rat cerebral vasculature. We observe that isolated PCAs exhibit time-of-day-dependent oscillations in sensitivity to ATP-induced vasodilation. Additionally, two weeks of OSA attenuates the oscillation of circadian clock components and ATP sensitivity of cerebral arteries. The effects of OSA on the cerebrovascular clock, and clock regulated processes, may contribute to the adverse cerebrovascular outcomes associated with OSA.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke (R01NS080531, R21NS094806); and National Heart Lung and Blood Institute (T32 HL07812).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
David J. Durgan: Made a substantial contribution to the concept and design, acquisition of data, analysis and interpretation of data, drafting/revising/approving the article.
Randy F. Crossland: Made a substantial contribution to the acquisition of data and manuscript revisions.
Robert M. Bryan: Made a substantial contribution in data interpretation and manuscript revisions.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Edery I. Circadian rhythms in a nutshell. Physiol Genom 2000; 3: 59–74. [DOI] [PubMed] [Google Scholar]
- 2.Wilsbacher LD, Takahashi JS. Circadian rhythms: Molecular basis of the clock. Curr Opin Genet Dev 1998; 8: 595–602. [DOI] [PubMed] [Google Scholar]
- 3.Dunlap JC. Molecular bases for circadian clocks. Cell 1999; 96: 271–290. [DOI] [PubMed] [Google Scholar]
- 4.Young ME. The circadian clock within the heart: Potential influence on myocardial gene expression, metabolism, and function. Am J Physiol Heart Circ Physiol 2006; 290: H1–H16. [DOI] [PubMed] [Google Scholar]
- 5.Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science 2002; 295: 1070–1073. [DOI] [PubMed] [Google Scholar]
- 6.Berson DM. Strange vision: Ganglion cells as circadian photoreceptors. Trends Neurosci 2003; 26: 314–320. [DOI] [PubMed] [Google Scholar]
- 7.Ripperger JA, Schibler U. Circadian regulation of gene expression in animals. Curr Opin Cell Biol 2001; 13: 357–362. [DOI] [PubMed] [Google Scholar]
- 8.Schibler U, Ripperger JA, Brown SA. Circadian rhythms. Chronobiology—Reducing time. Science 2001; 293: 437–438. [DOI] [PubMed] [Google Scholar]
- 9.Le Minh N, Damiola F, Tronche F, et al. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. EMBO J 2001; 20: 7128–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Damiola F, Le Minh N, Preitner N, et al. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev 2000; 14: 2950–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi JS. Circadian-clock regulation of gene expression. Curr Opin Genet Dev 1993; 3: 301–309. [DOI] [PubMed] [Google Scholar]
- 12.Gekakis N, Staknis D, Nguyen HB, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998; 280: 1564–1569. [DOI] [PubMed] [Google Scholar]
- 13.Hogenesch JB, Gu YZ, Jain S, et al. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA 1998; 95: 5474–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shearman LP, Sriram S, Weaver DR, et al. Interacting molecular loops in the mammalian circadian clock. Science 2000; 288: 1013–1019. [DOI] [PubMed] [Google Scholar]
- 15.Ptitsyn AA, Gimble JM. True or false: All genes are rhythmic. Ann Med 2011; 43: 1–12. [DOI] [PubMed] [Google Scholar]
- 16.Davidson AJ, London B, Block GD, et al. Cardiovascular tissues contain independent circadian clocks. Clin Exp Hypertens 2005; 27: 307–311. [PubMed] [Google Scholar]
- 17.McNamara P, Seo SB, Rudic RD, et al. Regulation of CLOCK and MOP4 by nuclear hormone receptors in the vasculature: A humoral mechanism to reset a peripheral clock. Cell 2001; 105: 877–889. [DOI] [PubMed] [Google Scholar]
- 18.Nonaka H, Emoto N, Ikeda K, et al. Angiotensin II induces circadian gene expression of clock genes in cultured vascular smooth muscle cells. Circulation 2001; 104: 1746–1748. [DOI] [PubMed] [Google Scholar]
- 19.Takeda N, Maemura K, Horie S, et al. Thrombomodulin is a clock-controlled gene in vascular endothelial cells. J Biol Chem 2007; 282: 32561–32567. [DOI] [PubMed] [Google Scholar]
- 20.Anea CB, Zhang M, Stepp DW, et al. Vascular disease in mice with a dysfunctional circadian clock. Circulation 2009; 119: 1510–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carver KA, Lourim D, Tryba AK, et al. Rhythmic expression of cytochrome P450 epoxygenases CYP4x1 and CYP2c11 in the rat brain and vasculature. Am J Physiol Cell Physiol 2014; 307: C989–C998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Somers VK, White DP, Amin R, et al. Sleep apnea and cardiovascular disease: An American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing in collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation 2008; 118: 1080–1111. [DOI] [PubMed] [Google Scholar]
- 23.Durgan DJ, Bryan RM., Jr Cerebrovascular consequences of obstructive sleep apnea. J Am Heart Assoc 2012; 1: e000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dempsey JA, Veasey SC, Morgan BJ, et al. Pathophysiology of sleep apnea. Physiol Rev 2010; 90: 47–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crossland RF, Durgan DJ, Lloyd EE, et al. A new rodent model for obstructive sleep apnea: Effects on ATP-mediated dilations in cerebral arteries. Am J Physiol Regul Integr Comp Physiol 2013; 305: R334–R342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durgan DJ, Crossland RF, Lloyd EE, et al. Increased cerebrovascular sensitivity to endothelin-1 in a rat model of obstructive sleep apnea: A role for endothelin receptor B. J Cereb Blood Flow Metab 2015; 35: 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durgan DJ, Pat BM, Laczy B, et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem 2011; 286: 44606–44619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durgan DJ, Tsai JY, Grenett MH, et al. Evidence suggesting that the cardiomyocyte circadian clock modulates responsiveness of the heart to hypertrophic stimuli in mice. Chronobiol Int 2011; 28: 187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durgan DJ, Hotze MA, Tomlin TM, et al. The intrinsic circadian clock within the cardiomyocyte. Am J Physiol Heart Circ Physiol 2005; 289: H1530–H1541. [DOI] [PubMed] [Google Scholar]
- 30.Durgan DJ, Young ME. The cardiomyocyte circadian clock: Emerging roles in health and disease. Circ Res 2010; 106: 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolff G, Esser KA. Scheduled exercise phase shifts the circadian clock in skeletal muscle. Med Sci Sports Exerc 2012; 44: 1663–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudic RD, McNamara P, Reilly D, et al. Bioinformatic analysis of circadian gene oscillation in mouse aorta. Circulation 2005; 112: 2716–2724. [DOI] [PubMed] [Google Scholar]
- 33.Reilly DF, Curtis AM, Cheng Y, et al. Peripheral circadian clock rhythmicity is retained in the absence of adrenergic signaling. Arterioscler Thromb Vasc Biol 2008; 28: 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryan RM, Jr, You J, Golding EM, et al. Endothelium-derived hyperpolarizing factor: A cousin to nitric oxide and prostacyclin. Anesthesiology 2005; 102: 1261–1277. [DOI] [PubMed] [Google Scholar]
- 35.You J, Johnson TD, Childres WF, et al. Endothelial-mediated dilations of rat middle cerebral arteries by ATP and ADP. Am J Physiol 1997; 273: H1472–H1477. [DOI] [PubMed] [Google Scholar]
- 36.Bryan RM, Jr, Marrelli SP. Endothelium-dependent hyperpolarization: Out of the dish and into the brain. J Cereb Blood Flow Metab 2011; 31: 1173–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation 1989; 79: 733–743. [DOI] [PubMed] [Google Scholar]
- 38.Durgan DJ, Trexler NA, Egbejimi O, et al. The circadian clock within the cardiomyocyte is essential for responsiveness of the heart to fatty acids. J Biol Chem 2006; 281: 24254–24269. [DOI] [PubMed] [Google Scholar]
- 39.Takeda N, Maemura K. Circadian clock and vascular disease. Hypertens Res 2010; 33: 645–651. [DOI] [PubMed] [Google Scholar]
- 40.Kaneko M, Zechman FW, Smith RE. Circadian variation in human peripheral blood flow levels and exercise responses. J Appl Physiol 1968; 25: 109–114. [DOI] [PubMed] [Google Scholar]
- 41.Delp MD, Manning RO, Bruckner JV, et al. Distribution of cardiac output during diurnal changes of activity in rats. Am J Physiol 1991; 261: H1487–H1493. [DOI] [PubMed] [Google Scholar]
- 42.Fujita M, Franklin D. Diurnal changes in coronary blood flow in conscious dogs. Circulation 1987; 76: 488–491. [DOI] [PubMed] [Google Scholar]
- 43.Wauschkuhn CA, Witte K, Gorbey S, et al. Circadian periodicity of cerebral blood flow revealed by laser-Doppler flowmetry in awake rats: Relation to blood pressure and activity. Am J Physiol Heart Circ Physiol 2005; 289: H1662–H1668. [DOI] [PubMed] [Google Scholar]
- 44.Lewis NC, Atkinson G, Lucas SJ, et al. Diurnal variation in time to presyncope and associated circulatory changes during a controlled orthostatic challenge. Am J Physiol Regul Integr Comp Physiol 2010; 299: R55–R61. [DOI] [PubMed] [Google Scholar]
- 45.Keskil Z, Gorgun CZ, Hodoglugil U, et al. Twenty-four-hour variations in the sensitivity of rat aorta to vasoactive agents. Chronobiol Int 1996; 13: 465–475. [DOI] [PubMed] [Google Scholar]
- 46.Saito T, Hirano M, Ide T, et al. Pivotal role of rho-associated kinase 2 in generating the intrinsic circadian rhythm of vascular contractility. Circulation 2013; 127: 104–114. [DOI] [PubMed] [Google Scholar]
- 47.Hodoglugil U, Uluoglu C, Guney HZ, et al. Twenty-four-hour variations in the effect of nitrodilators in rat aorta: Lack of influence of the endothelium. J Pharm Pharmacol 1997; 49: 1102–1108. [DOI] [PubMed] [Google Scholar]
- 48.Viswambharan H, Carvas JM, Antic V, et al. Mutation of the circadian clock gene Per2 alters vascular endothelial function. Circulation 2007; 115: 2188–2195. [DOI] [PubMed] [Google Scholar]
- 49.Carvas JM, Vukolic A, Yepuri G, et al. Period2 gene mutant mice show compromised insulin-mediated endothelial nitric oxide release and altered glucose homeostasis. Front Physiol 2012; 3: 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Panda S, Antoch MP, Miller BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002; 109: 307–320. [DOI] [PubMed] [Google Scholar]
- 51.Kunieda T, Minamino T, Miura K, et al. Reduced nitric oxide causes age-associated impairment of circadian rhythmicity. Circ Res 2008; 102: 607–614. [DOI] [PubMed] [Google Scholar]
- 52.Tischkau SA, Cohen JA, Stark JT, et al. Time-of-day affects expression of hippocampal markers for ischemic damage induced by global ischemia. Exp Neurol 2007; 208: 314–322. [DOI] [PubMed] [Google Scholar]
- 53.Durgan DJ, Pulinilkunnil T, Villegas-Montoya C, et al. Short communication: Ischemia/reperfusion tolerance is time-of-day-dependent: Mediation by the cardiomyocyte circadian clock. Circ Res 2010; 106: 546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kung TA, Egbejimi O, Cui J, et al. Rapid attenuation of circadian clock gene oscillations in the rat heart following ischemia-reperfusion. J Mol Cell Cardiol 2007; 43: 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young ME, Bray MS. Potential role for peripheral circadian clock dyssynchrony in the pathogenesis of cardiovascular dysfunction. Sleep Med 2007; 8: 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Young ME, Razeghi P, Taegtmeyer H. Clock genes in the heart: Characterization and attenuation with hypertrophy. Circ Res 2001; 88: 1142–1150. [DOI] [PubMed] [Google Scholar]
- 57.Burioka N, Koyanagi S, Endo M, et al. Clock gene dysfunction in patients with obstructive sleep apnoea syndrome. Eur Respir J 2008; 32: 105–112. [DOI] [PubMed] [Google Scholar]
- 58.Shepard JW., Jr Hypertension, cardiac arrhythmias, myocardial infarction, and stroke in relation to obstructive sleep apnea. Clin Chest Med 1992; 13: 437–458. [PubMed] [Google Scholar]
- 59.Martinez Garcia MA, Salt CL, Cataluna JJ, et al. Prevalence of sleep-disordered breathing in patients with acute ischemic stroke: Influence of onset time of stroke. Arch Bronconeumol 2004; 40: 196–202. [DOI] [PubMed] [Google Scholar]
- 60.Durgan DJ, Young ME. Linking the cardiomyocyte circadian clock to myocardial metabolism. Cardiovasc Drugs Ther 2008; 22: 115–124. [DOI] [PubMed] [Google Scholar]
- 61.Young ME. Anticipating anticipation: Pursuing identification of cardiomyocyte circadian clock function. J Appl Physiol 2009; 107: 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ko GY, Shi L, Ko ML. Circadian regulation of ion channels and their functions. J Neurochem 2009; 110: 1150–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Virag JA, Anderson EJ, Kent SD, et al. Cardioprotection via preserved mitochondrial structure and function in the mPer2-mutant mouse myocardium. Am J Physiol Heart Circ Physiol 2013; 305: H477–H483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bray MS, Young ME. The role of cell-specific circadian clocks in metabolism and disease. Obes Rev 2009; 10: 6–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.