

Abstract
The effects of global mitochondrial calcium (Ca2+) uniporter (MCU) deficiency on hypoxic-ischemic (HI) brain injury, neuronal Ca2+ handling, bioenergetics and hypoxic preconditioning (HPC) were examined. Forebrain mitochondria isolated from global MCU nulls displayed markedly reduced Ca2+ uptake and Ca2+-induced opening of the membrane permeability transition pore. Despite evidence that these effects should be neuroprotective, global MCU nulls and wild-type (WT) mice suffered comparable HI brain damage. Energetic stress enhanced glycolysis and depressed Complex I activity in global MCU null, relative to WT, cortical neurons. HI reduced forebrain NADH levels more in global MCU nulls than WT mice suggesting that increased glycolytic consumption of NADH suppressed Complex I activity. Compared to WT neurons, pyruvate dehydrogenase (PDH) was hyper-phosphorylated in MCU nulls at several sites that lower the supply of substrates for the tricarboxylic acid cycle. Elevation of cytosolic Ca2+ with glutamate or ionomycin decreased PDH phosphorylation in MCU null neurons suggesting the use of alternative mitochondrial Ca2+ transport. Under basal conditions, global MCU nulls showed similar increases of Ca2+ handling genes in the hippocampus as WT mice subjected to HPC. We propose that long-term adaptations, common to HPC, in global MCU nulls compromise resistance to HI brain injury and disrupt HPC.
Keywords: Glycolysis, hypoxic preconditioning, mitochondrial calcium uniporter, neuronal bioenergetics, stroke
Introduction
Stroke is the second leading cause of death in the world population.1 Approximately 16 million first-ever strokes occur globally each year, causing a total of 5.7 million deaths.1 Nearly half of stroke survivors suffer disabilities that force them to seek the assistance of others for daily living which places a considerable burden on their family, friends and the health care system.2 The staggering human and economic costs inflicted by stroke have driven an intensive effort to understand how the brain can be rendered more resistant to damage by a stroke.3,4 Hypoxic preconditioning (HPC) activates powerful adaptations that dramatically protect the brain from subsequent damage by cerebral ischemia.5–7 Clinical studies suggest that these protective mechanisms render patients which have experienced a transient ischemic attack more resistant to brain damage by a subsequent stroke.8 Identification of the signaling events that mediate HPC are therefore of considerable therapeutic interest.3,4
Mitochondria are strategically positioned to sense and respond to elevations in cytosolic Ca2+ concentrations [Ca2+]c that range from the physiological to toxic.9,10 Each neuron contains about 1000–2000 of these dynamic organelles that are rapidly transported by kinesin motors to synaptic sites in urgent need of metabolic support for neurotransmission.11,12 Mitochondria also play a pivotal role in orchestrating the Ca2+ signaling events that activate complex genetic networks which protect neurons by enhancing mitochondrial Ca2+ handling, anti-oxidant defense, energy production and biogenesis.7,13 These key features enable mitochondria to exert exquisite positive control over the neuroprotective events implicated in HPC.5,14,15 However, mitochondria can also promote neuronal cell death. Excessive mitochondrial Ca2+ uptake triggers the formation of a mitochondrial membrane permeability transition pore (mPTP) that executes both apoptotic16,17 and necrotic18–20 cell death. Identification of the transport mechanisms for increased mitochondrial Ca2+ uptake that promote neuronal cell death and survival may thus open new therapeutic avenues for the prevention of ischemic brain injury.6,21,22
The mitochondrial Ca2+ uniporter (MCU) is responsible for rapid and high capacity mitochondrial Ca2+ uptake in the heart.23 Genetic identification of the MCU in 20112,4,25 has enabled the generation of various genetic mouse lines in which MCU activity is blocked by either global MCU deletion,23 cardiac-specific expression of a dominant-negative MCU (DN-MCU)26,27 or inducible cardiac MCU ablation at maturity.28,29 Experimentation with these genetic lines has clearly demonstrated that the MCU is required for both adaptive and destructive increases of mitochondrial Ca2+ uptake in the heart.23,26–29
In view of the considerable implications of these findings for ischemic brain damage, we have examined the effects of global MCU deficiency on Ca2+ uptake and Ca2+-induced mPTP opening by forebrain mitochondria, resistance of primary cortical neuron cultures to oxygen-glucose deprivation-induced viability loss, various aspects of neuronal energy production, and the ability of HPC to prevent central neuron loss and sensorimotor deficits in a model of hypoxic-ischemic (HI) brain injury.
Materials and methods
Ethical statement for animal experimentation
All animal procedures were approved by the University Committee on Laboratory Animals at Dalhousie University that follows the Canadian Council on Animal Care guidelines for the ethical use of laboratory animals. All experiments have been reported in compliance with the ARRIVE guidelines.30
Animal breeding
Heterozygous MCU nulls were kindly provided by Dr Toren Finkel and genotyped according to methods described by his laboratory.23 These animals were breed as trios (one male and two females) to generate a sufficient number of global MCU nulls and wild-type (WT) littermates for experimentation. Mice had ad libitum access to food and water and were maintained on a 12-h light–cycle.
Forebrain mitochondrial isolation
Mice were euthanized by intra-peritoneal (i.p.) injection of sodium pentobarbital (150 mg/kg) followed by decapitation. Brains were immediately removed and the forebrain (cortex, hippocampus, thalamus, striatum and hypothalamus) placed in chilled isolation buffer [300 mM sucrose, 5 mM Tris-HCl, 2 mM EDTA, 0.5 mg/ml bovine serum albumin (BSA)]. The forebrains were then cut into small pieces and immediately homogenized with a dounce homogenizer. The forebrain homogenate was spun at 1000 × g for 10 min. The supernatant was then transferred to a new tube of isolation buffer and spun at 6500 × g for 10 min. The supernatant was then discarded and the pellet was homogenized in 300 mM sucrose and 5 mM Tris-HCl, and spun at 6500 × g for 10 min. The supernatant was then discarded and the pellet containing mitochondria was homogenized in 300 mM sucrose and 5 mM Tris-HCl, creating a concentrated stock mitochondrial preparation which was used for subsequent experiments.
Mitochondrial Ca2+ uptake
Mitochondria were diluted in 2 ml of measuring buffer composed of mannitol (210 mM), sucrose (70 mM), MgCl2 (5 mM), KH2PO4 (0.1 mM), calcium green 5 -N (2 µM), rotenone (1 µM), succinate (10 µM), Tris-HCl (5 mM), pH 7.4, with or without ruthenium red (RR; 5 µM, data not shown). Uptake was monitored by calcium green 5 -N fluorescence with excitation at 506 nm and emission at 532 nm. Fluorescence was monitored over sequential additions of calcium to raise free Ca2+ concentrations by 5 µM increments. Each experiment was terminated with the addition of carbonyl cyanide p-triflouromethoxyphenylhydrazone (FCCP, 10 µM).
Ca2+-induced mPTP opening
Forebrain mitochondria were reconstituted in the measuring buffer that contained CGP-37157 (10 µM; Tocris Bioscience) to inhibit the mitochondrial Na+/Ca2+ exchanger. Tetramethylrhodamine methyl ester (TMRM, Sigma-Aldrich) was loaded at a concentration of 0.125 μM. Opening of the mPTP in WT mitochondria was induced by sequential additions of Ca2+ to raise the free Ca2+ concentrations by 10, 25, 100, and 100 µM. Since MCU null mitochondria did not show Ca2+-induced mPTP opening, FCCP (10 µM) was added to confirm depolarization of the mitochondrial membrane potential.
Hypoxic preconditioning
WT or MCU null mice were placed in a chamber maintained at 36.5℃ and vented with 8% oxygen balanced with nitrogen flowing at a rate of 4 l/min for 50 min to induce HPC. Mice that served as sham controls for HPC were placed in the same apparatus maintained at 36.5℃ except the chamber was vented at 4 l/min for with normal atmospheric air (20% oxygen) for 50 min.
Hypoxic-ischemic brain injury model
All procedures were ethically approved by the University Committee on Laboratory Animals at Dalhousie University. Adult (9–11 weeks old; 25–30 g) male and female mice were anesthetized using isoflurane (3% with medical oxygen at a flow rate of 3 l/min). HI brain injury was performed according to a modified version of the methods described by Levine, 1960.31 The ventral portion of the neck was shaved and cleaned with soluprep and betadine (SoluMed Inc; Laval, QC Canada and Purdue Frederick Inc; Pickering, ON, Canada, respectively). Anesthesia was maintained with 3% isoflurane with an oxygen flow rate of 1.0 l/min. To alleviate pain following recovery, mice were injected with ketoprofen (5 mg/kg, i.p.). An incision (1 cm) was made with a scalpel to expose the sternohyoid and sternomastoid muscles. The left carotid artery was isolated and removed from the vagus nerve by blunt dissection. The carotid artery was then permanently occluded using a high temperature electrocautery pen (Bovie Instruments; St. Petersburg, FL, USA). Incomplete occlusions and/or animals displaying blood loss were immediately euthanized and excluded from the study. Following a 2–3-h recovery period, animals were placed in a low oxygen chamber (8% oxygen balanced with nitrogen, flow rate 4 l/min) maintained at 36.5℃. Following 40 min in the low oxygen chamber, mice were removed and individually housed to recover for 24 h. The total number of animals required to reach a group size of 12 mice for each of the four experimental treatments are as follows: WT-HI (16); WT-HPC+HI (14); MCU-HI (17); MCU-HPC+HI (19). Additional animals were required because of mortality during the HI procedure.
Neuroscore scale: assessment of general condition and neurological deficits
A comprehensive behavioural assessment of sensorimotor deficits produced by HI brain injury was performed using a neuroscore scale developed by Dr Ulrich Dirnagl (Charité – Universitätsmedizin Berlin, Germany). Scores ranged from 0 (healthy) to 56 (the worst performance in all categories) and represented the sum of scores for six general deficit categories (hair, ears, eyes, posture, spontaneous activity and epileptic behaviour categories) and seven focal deficits categories (body symmetry, gait, climbing on angled surface, circling behaviour, front limb symmetry, compulsory circling, whisker response to light touch). Mice were scored by an observer unaware of the experimental conditions. A detailed description of the methods used to assess the general (Part 1) and focal (Part 2) behavioural deficits following HI brain injury is provided in the Supplementary information.
Detection and quantification of HI-induced neuronal damage
Mice were injected with an overdose of pentobarbital (150 mg/kg, i.p.) and perfused transcardially with saline (10 ml) followed by 0.1 M phosphate buffer containing 4% paraformaldehyde (pH = 6.5; 10 ml) to fix the brain. The brain was then removed and post-fixed in 0.1 M phosphate buffer containing 2% paraformaldehyde for 24 h followed by cryoprotection for 48 h in 0.1 M phosphate buffer containing 30% sucrose. The brain was then frozen and coronal sections 30 µm thick were cut at the level of dorsal hippocampus (−1.7 to −1.9 mm from bregma) and striatum (1.18 to 0.98 from bregma) with a cryostat. Sections were stained with Fluoro-Jade (FJ; AG325-30MG; EMD Millipore Canada) according to the manufacturer’s recommendations. Slides were then cover-slipped with Fluorescence-preserving VECTASHIELD Mounting medium (Vector, H-1,000) and observed under a Zeiss fluorescence microscope equipped with a computer-assisted image analysis system. FJ staining was quantified by determining the area occupied by FJ-positive neurons in CA1 region of the dorsal hippocampus (150 × 300 µm), motor cortex (300 × 300 µm) and dorsolateral striatum (300 × 300 µm). Four serial sections per region for 12 animals per group were quantified by an individual unaware of the treatment conditions.
Transmission electron microscopy
Whole brains were fixed in 2.5% glutaraldehyde by perfusion in PBS (pH 7.4) and processed in the EM Core Facility (Dalhousie University). Briefly, the samples were rinsed three times in 0.1 M sodium cacodylate buffer and fixed in 1% osmium tetroxide for 2 h and, after dehydration, embedded in epon araldite resin. Sections 100 nm thick were cut with an ultramicrotome and placed on 300 mesh copper grids which were stained with 2% aqueous uranyl acetate, rinsed and treated with lead citrate, then rinsed and air dried. Images were captured with a Jeol Jem 1230 transmission electron microscope at 80 kV attached to a Hamatsu ORCA-HR digital camera.
Quantification of mitochondrial damage
The effects of HI brain injury on mitochondrial damage were assessed in the ipsilateral CA1 region of the dorsal hippocampus of WT and MCU null mice (n = 4/group). Mitochondria were considered damaged if the integrity of the cristae was compromised. Ten photomicrographs from each animal were captured from the ipsilateral dorsal hippocampus and mitochondria (categorized as either healthy or damaged) were manually counted by two observers unaware of the treatment conditions. A total of 1024 mitochondria were categorized as either normal or damaged for both WT and MCU null mice.
Primary cortical neuron cultures
Embryonic brains were aseptically removed from WT or MCU mice at E16 and placed in Neurobasal medium containing 10% fetal bovine serum (FBS) on ice. The meninges were then peeled back and the cortices transferred to 35 mm petri dishes containing Hanks Balanced salt solution (HBSS; Invitrogen) at 4℃. Cortices were pooled and treated with 2 ml of Stem-Pro® Accutase® cell dissociation reagent. The cortices were then triturated with a fire polished pipette 3–4 times and allowed to incubate for 15 min at 37℃. This procedure was repeated a second time. Protease activity was inhibited by the addition of 1 ml FBS and the cortical tissue was transferred to a 15 ml falcon tube containing 3 ml of neurobasal mix (Neurobasal 500 ml + B27 + 500 µM glutamine +20 µg/ml gentamicin) at 4℃. The solution was then triturated 3–4 more times and left to settle. The supernatant was then collected and passed through a 40 micron filter. The neurons were then centrifuged at 300 × g for 7 min and re-suspended in neurobasal mix, counted and plated for experiments. Media changes were 60% of the total volume and occurred on the fourth day and every third day thereafter.
Oxygen glucose deprivation
Primary cortical neuron cultures were used after 8–10 days in vitro for all oxygen glucose deprivation (OGD) testing. Glucose-free balanced salt solution (GBSS) was prepared by bubbling nitrogen through the solution to remove oxygen. The cultures were then washed twice with oxygen-free GBSS and placed in air-tight containers that were purged with a gas mixture of 90% nitrogen, 5% carbon dioxide and 5% hydrogen for 5 min. Next, the air-tight containers holding the cultures were placed in a 37℃ incubator for 2 h. OGD was terminated by aspirating the HBSS and replacing it with the original media. Cell viability was determined 24 h later.
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide] cell viability assay
Cell viability was assessed using an MTT (Sigma-Aldrich) assay performed in 48-well plates seeded at a density of 150,000 neurons/well. Briefly, 60 μl of MTT (10 mg/ml; dissolved in no phenol red neurobasal) was added to each well. After incubation at 37℃ for 1.5 h, 5% CO2, the supernatant was removed and the formazan crystals were then dissolved in 200 μl/well of dimethyl sulfoxide (DMSO, Sigma). The absorbance of each well was then measured at 592 nm with a plate reader (ELx 800, BioTek). The absorbances of blank wells (200 μl DMSO) were subtracted from the absorbance of each of the other wells. The net absorbance of untreated no-OGD control wells were defined as 100% cell viability.
Multiplexed enzyme-linked immunosorbent assays
The phosphorylation of pyruvate dehydrogenase was examined using a multi-species pyruvate dehydrogenase complex magnetic bead panel (cat# PDHMAG-13 K EMD Millipore). This kit is a multiplex enzyme-linked immunosorbent assay (ELISA) for total pyruvate dehydrogenase as well as three serine (Ser) phosphorylated isoforms: PDH (Ser 232), PDH (Ser 293) and PDH (Ser 300). Analyses were performed using primary neurons derived from WT or MCU null embryos cultured for 8–10 days treated with one of the three conditions: control (n = 10), glutamate (n = 6) or ionomycin (n = 6). Controls were naïve to any treatment and harvested directly from the neurobasal media. Glutamate samples were treated with glutamate (25 μM) for 30 min and immediately harvested. Ionomycin samples were treated with ionomycin (2 μM) for 30 min and immediately harvested. Samples were split into two aliquots – one for protein concentration determination and one for PDH analyses. Protein concentration was determined using the Bio-Rad protein assay Cat# 500-0006. All procedures were done in accordance with the manufacture’s instructions and analysed on a Bio-Plex® 200 system. Protease and phosphatase inhibitor cocktails (EMD chemicals cat # 535140 and 524629) were also added to the lysis buffer following the manufacture’s suggestions.
Seahorse analysis of mitochondrial function
Analysis of mitochondrial function in respiring cortical neurons was performed using a Seahorse XF24 instrument according to our previously described methods.32 Primary cortical neurons derived from WT and MCU null embryos (E16) were plated on 24-well Seahorse XF24 plates at a density of 100,000 cells/well. Experiments were performed on days 8–10 in vitro. Calibration sensors were prepared according to the manufacturer’s instructions. Following OGD, plates were placed in normal neurobasal media for 1 h. Following this short reperfusion, the media were changed to an artificial cerebral spinal fluid containing: NaCl (120 mM), KCl (3.5 mM), CaCl2 (1.3 mM), KH2PO4 (0.4 mM), MgCl2 (1 mM) HEPES (20 mM), glucose (15 mM), sodium pyruvate (2 mM) and fatty acid free BSA (4 mg/ml). Mitochondrial function was probed by the sequential addition of oligomycin (2 μM), FCCP (2 μM), rotenone (300 nm) and antimycin A (5 μM). The FCCP (2 μM) concentration was titrated by previous concentration–response experiments. Three measurements were performed for each condition. All experiments were normalized to total live cells, as determined by trypan blue.
NAD+/NADH and pyruvate measurements
WT or MCU null mice were subjected to sham conditions or HI-induced brain injury as previously described. Thirty min later, all animals were humanely euthanized with an overdose of sodium pentobarbital (150 mg/kg, i.p.) and then perfused transcardially with saline (10 ml). The brains were then rapidly removed and the ipsilateral striatum and hippocampus dissected on ice and snap frozen in liquid nitrogen. Striatal NAD+ and NADH levels (abcam ab65348, Toronto, ON, Canada) and hippocampal pyruvate concentrations (ab65342) were determined according to the manufacturer’s instructions.
Trypan Blue assay for cell viability
Immediately following the completion of the Seahorse analysis, cells were washed three times with PBS followed by trypsinization with 2.5% trypsin (Sigma) for 3 min. Enzymatic activity was arrested by the addition of neurobasal culture media containing 10% FBS. Neurons were then treated with 0.4% trypan blue (Sigma) and incubated at 37℃ for 10 min. Neurons were then counted using a Bio-Rad TC20TM automated cell counter.
Quantitative PCR (qRT-PCR)
Total RNA was extracted from hippocampal tissue 6 h after HPC. Both hippocampi from each animal were rapidly microdissected on ice and placed in RNA later (ThermoFischer Scientific, cat# AM7020) and snap frozen in liquid nitrogen. Hippocampi were then mechanically homogenized and RNA was extracted with an Aurun total RNA minikit (Bio-Rad, cat# 732-6820). The spin protocol was performed according to the manufacturer’s instructions. After elution, the quality and integrity of the RNA was assessed using a Bio-Rad Experion bioanalyser with an RNA StdSns analysis kit. Only RNAs with values above 7.5 were used for further analysis. RNA was quantified using an Epoch microplate spectrophotometer (BioTek Instruments, Vermont, USA). Reverse transcription was carried out with the iScript cDNA synthesis kit (Bio-Rad) using 750 ng of template RNA for each sample. Negative controls (without the reverse transcriptase present) were run in every reaction. qRT-PCR was performed with a SsoFast EvaGreen Supermix kit (Bio-Rad) with β-actin, GAPDH, B2M and HPRT1 evaluated as reference genes. Individual genes were optimized for both annealing temperature and concentration. The qRT-PCR program was: 95℃ × 30 s s + (95℃ × 5 s + 58℃ × 5 s + plate reading) × 40 cycles + melting curve. The melting curve program was 2-s stop with plate reading for every 0.5℃ increase from 65℃ to 95℃. All qRT-PCR experiments were performed with a Bio-Rad CFX 96 real time system C1000 Touch thermal cycler. All qRT-PCR protocols were done in accordance with the MIQE guidelines. Analysis was performed using the Bio-Rad CFX Manager 3.1 software using the ΔΔCq method. Experiments were performed in duplicate with triplicate measurements from each experiment. Primer sequences used for qRT-PCR were as follows: BCL-XL, Forward (F) AGCAGGTAGTGAATGAACTCTTTCG, Reverse (R) – CCATCCAACTTGCAATCCGACTC; BAX, (F) – TGCTGATGGCAACTTCAACTG, (R) – GATCAGCTCGGGCACTTTAGTG; MT-ND2, (F) – GGGCATGAGGAGGACTTAACCAAAC, (R) – TGAGGTTGAGTAGAGTGAGGGATGG; NCX-1, (F) – AAAGAGTGCAGTTTCTCCCTTG, (R) TGAAGCCACCTTTCAATCCTC; NCX-2, (F) – ATGGCTCCCTTGGCTTTGATG, (R) – CAGCGGTAGGAACCTTGGC; NCX-3, (F) – GTGAACCGAAATGGATGGAACG, (R) – TCACCCAATACTGGCTTTCCC; PMCA2, (F) – AAAGAGAAGTCGGTGCTTCAG, (R) – GTGTGCATTCCGTCAGCCA; β-actin, (F) – GTGACGTTGACATCCGTAAAGA, (R) – GCCGGACTCATCGTACTCC; GAPDH, (F) – AGGTCGGTGTGAACGGATTTG, (R) – GGGGTCGTTGATGGCAACA; B2M, (F) – TTCTGGTGCTTGTCTCACTGA, (R) – CAGTATGTTCGGCTTCCCATTC; HRPT, (F) – TCAGTCAACGGGGGACATAAA, (R) – GGGGCTGTACTGCTTAACCAG.
Statistical analyses
A two-way ANOVA followed by group comparisons with the Bonferroni's post hoc test was used to assess potential differences between WT and global MCU nulls for neuroscores (Figure 2), central neuron loss (Figures 3; Supplementary Figures 1 and 2), cortical neuron viability loss after OGD (Supplementary Figure 4), cortical neuron phosphorylation levels of PDH (Figure 5), cortical neuron OCRs and ECARs (Figure 6), NAD+/NADH ratios and pyruvate concentrations (Figure 6) and mRNA levels in the hippocampus (Figure 7). An unpaired t-test was used to assess potential differences for mitochondrial damage and complex I (MT-ND2) mRNA levels between WT and MCU null mice (Supplementary Figures 3 and 5).
Figure 2.
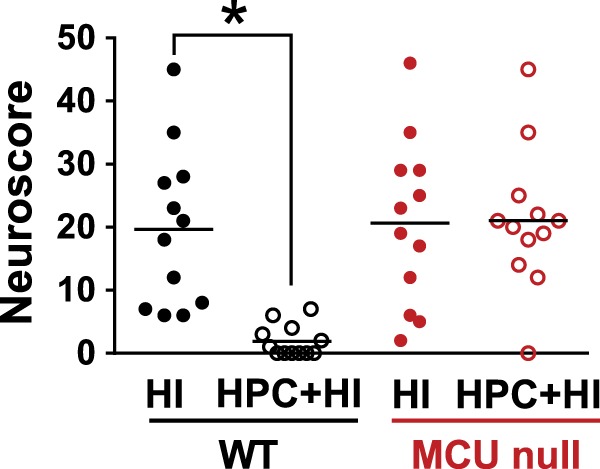
Neuroscores for wild-type (WT) and global MCU null mice after HI brain injury that were subjected to sham conditions or HPC. Mice were graded on a 56-point scale that rated increased levels of neurobehavioural impairment. *p < 0.05, Two-way ANOVA followed by Bonferroni's post hoc test.
Figure 3.

Fluorojade (FJ)-positive neurons damaged by HI brain injury in the dorsolateral striatum of WT (c) and MCU nulls (e) subjected to sham conditions (a) or HPC (d, f). Top right panel (b) – HI damage quantified by determining the area occupied by FJ-positive cells within the indicated sector of dorsolateral striatum (inset, top left panel). *p < 0.05, Two-way ANOVA followed by Bonferroni's post hoc test. Scale bar = 150 µm.
Figure 5.

PDH phosphorylation levels for pSer232 (a), pSer293 (B), pSer300 (c) and total pSer (d) in WT and MCU null cortical neurons under control conditions or 30 min after stimulation with glutamate (25 μM) or ionomycin (2 µM). *p < 0.001, Two-way ANOVA followed by Bonferroni's post hoc test (n = 7/group).
Figure 6.

OCRs (a, c) and ECARs (b, d) for WT and MCU null neurons under control conditions (a and b) or 3 h after OGD (c and d). *p < 0.05, Two-way ANOVA followed by Bonferroni's post hoc test (n = 3/group). Striatal NAD+/NADH ratios (e) and hippocampal pyruvate (f) concentrations isolated from the ipsilateral hemisphere of wild-type (WT) and MCU null mice 30 min following exposure to sham conditions or HI. *p < 0.05, Two-way ANOVA followed by Bonferroni's post-hoc test.
Figure 7.

Relative levels of BCL-XL (a), BAX (b), NCX-1 (c), NCX-2 (d), NCX-3 (e) and PMCA-2 (f) mRNAs detected in the dorsal hippocampi of WT or MCU null mice 4 h after exposure to sham conditions or HPC (50 min of global hypoxia). *p < 0.05, Two-way ANOVA followed by Bonferroni's post hoc test. Each bar represents the mean ± SEM (n = 7/group).
Power calculations for group sizes
Power calculations were performed to determine the group sizes required to detect statistical differences for the animal experimentation. A group size of 12 mice with a standard deviation of 75% was needed to detect an 80% difference between the means for measurements of neuroscores, neuronal cell counts, NAD+/NADH and pyruvate concentrations with 100% accuracy for an alpha (α) level of 0.05.
Results
MCU deficiency impairs Ca2+ uptake and inhibits Ca2+-induced mPTP opening by forebrain mitochondria
We first examined Ca2+ uptake and Ca2+-induced opening of the mPTP in forebrain mitochondria isolated from WT or MCU null mice. As expected, Ca2+ uptake into MCU deficient forebrain mitochondria was blocked (Figure 1(a)) in a similar manner to that observed for heart mitochondria isolated from these global MCU knockouts.23 Ca2+-induced mPTP opening in brain mitochondria that lacked the MCU was also inhibited (Figure 1(b)). MCU deficiency therefore suppressed both mitochondrial Ca2+ uptake and Ca2+-induced mPTP opening that are considered to be pivotal events in ischemic neuronal cell death.19,33
Figure 1.
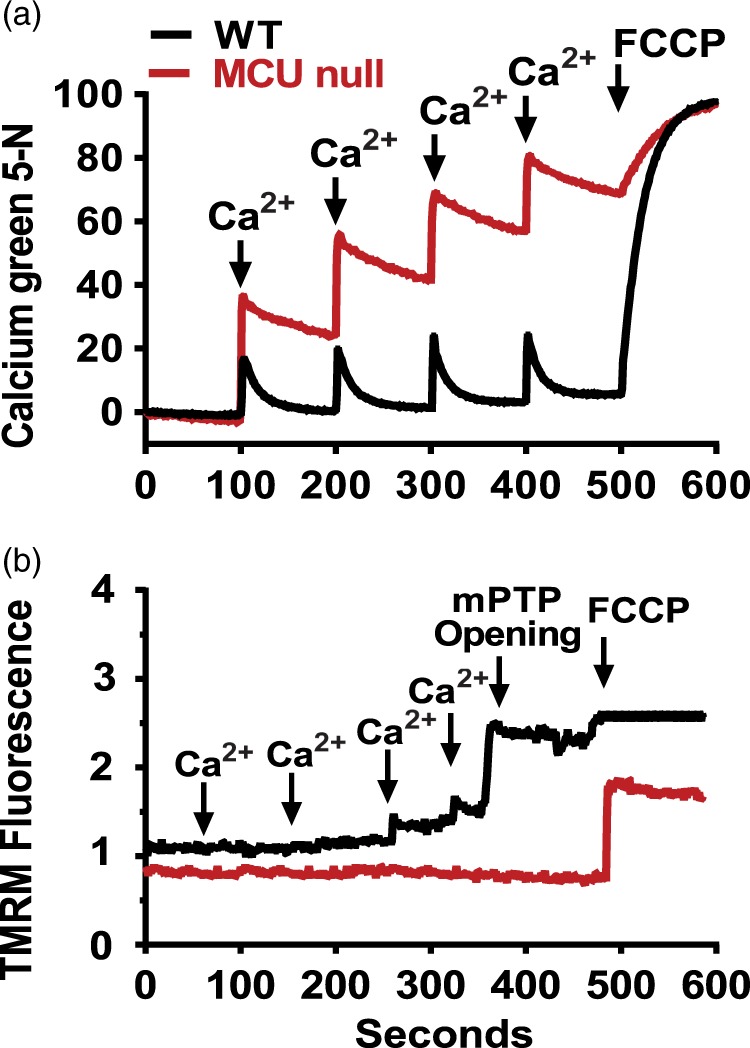
Mitochondrial calcium (Ca2+) uptake (a) and Ca2+-induced mitochondrial permeability transition pore (mPTP) opening (b) in brain mitochondria isolated from WT and MCU null mice. These data are representative of the results from three separate experiments.
MCU deletion blocks HPC but not HI brain injury
In a second series of experiments, we assessed the effects of global MCU deficiency on HPC using a model of HI brain injury that impairs sensory-motor function by damaging the motor cortex, dorsolateral striatum and dorsal hippocampus. HPC was induced 24 h before HI by exposure to a low oxygen atmosphere (8% O2) for 50 min. Control mice for HPC were simply placed in the experimental apparatus under normal atmospheric conditions (20% O2) for the same amount of time. HI brain injury was induced 24 h later. HI-induced behavioural deficits were measured 24 h later using a comprehensive scoring system of neurological function. Immediately afterwards, neuronal damage in the dorsolateral striatum, motor cortex and CA1 region of the dorsal hippocampus was measured by quantifying Fluoro-Jade-positive neurons in these brain regions using Image J. Despite the inhibition of mitochondrial Ca2+ uptake and Ca2+-induced mPTP opening in MCU deficient brain mitochondria, global MCU null mice displayed comparable HI-induced sensorimotor deficits (Figure 2) and central neuronal damage [dorsolateral striatum (Figure 3(a), (c) and (e)), motor cortex (Supplementary Figure 1) and CA1 region of the dorsal hippocampus (Supplementary Figure 2)] as WT mice. These findings demonstrate that MCU-mediated mitochondrial Ca2+ uptake is dispensable in the progression of HI brain injury. HPC profoundly attenuated HI-induced sensorimotor deficits (Figure 2) and central neuronal damage [dorsolateral striatum (Figure 3(d)), motor cortex and CA1 region of the dorsal hippocampus (Supplementary Figures 1 and 2)]. However, HPC failed to decrease HI-induced sensorimotor deficits and central neuronal damage for MCU nulls (Figure 3(f)) indicating that global MCU loss interfered with HPC.
Ultrastructural features of brain mitochondria are similar in WT mice and MCU nulls
Electron microscopic analyses showed that the morphology of mitochondria in the CA1 region of global MCU nulls appeared normal and comparable to that of WT mice (Figure 4). The disrupted morphology (loss of cristae integrity) and degree of mitochondrial damage in the dorsal hippocampus of WT and MCU null mice after HI brain injury were also the same (Supplementary Figure 3).
Figure 4.

Electron microscopic images of mitochondria in the CA1 region of the dorsal hippocampus of control or HI injured WT and MCU null mice.
MCU deletion does not alter the susceptibility of cortical neurons to ischemic cell death
To determine whether MCU deficiency alters the resistance of central neurons to ischemic injury, cell viability loss produced by oxygen glucose deprivation (OGD) was compared in primary cultures of cortical neurons derived from WT and global MCU null mice. Neurons exposed to OGD undergo rapid energetic decline, failure of ATP-dependent ion pumps, excessive reactive oxygen species generation, N-methyl-D-aspartate receptor hyper-stimulation and mitochondrial dysfunction resulting in both apoptotic and necrotic cell death.34 Consistent with our in vivo findings, OGD produced the same degree of cell viability loss for WT and MCU deficient cortical neurons (Supplementary Figure 4). Global MCU deletion therefore does not alter the susceptibility of central neurons to OGD-induced damage.
MCU null neurons exhibit hyper-phosphorylation of the pyruvate dehydrogenase complex
Consistent with findings using cardiac or skeletal myocytes, phosphorylation of the PDH complex at all three serine sites (pS233, pS292, pS300; Figure 5(a) to (c)) were dramatically increased in MCU null relative to WT cortical neurons.23,27–29 In total, PDH phosphorylation was elevated by three fold in MCU null relative to WT neurons (Figure 5(d)). Exposure to glutamate (25 µM; 30 min) and ionomycin (2 µM; 30 min) decreased the relative levels of PDH phosphorylation by comparable amounts in WT and MCU null cortical neurons (Figure 5(b) and (c)). However, by comparison to WT neurons, PDH in MCU null neurons was significantly hyper-phosphorylated across basal, glutamate and ionomycin stimulated conditions (Figure 5(a) to (d)). These findings indicate that mitochondria in MCU null neurons may utilize alternative Ca2+ transport and/or metabolic substrates to regulate energy metabolism.10,35
MCU null neurons have reduced spare respiratory capacity offset by increased glycolytic rates
Oxygen consumption rates (OCR) under control and oligomycin-inhibited conditions were comparable for both WT and MCU null neurons, indicating a similar reliance on mitochondrial oxidative phosphorylation for basal ATP production (Figure 6(a)). PDH hyper-phosphorylation is known to suppress tricarboxylic acid flux.36 Despite PDH hyper-phosphorylation in MCU null neurons, basal OCRs were comparable to WT neurons. Two possibilities may account for unaltered basal OCRs in MCU null neurons: (1) residual unphosphorylated PDH provides sufficient substrate supply36 and (2) alternative substrates and metabolic pathways generate the necessary reducing equivalents to maintain oxidative phosphorylation.10,35 When stimulated with FCCP, which induces maximal respiratory capacity by dissipating the mitochondrial membrane potential and uncoupling Complex I-IV activity from ATP synthase, MCU null cortical neurons showed a reduced spare respiratory capacity compared to WT neurons. This was accompanied by an increase in ECARs for MCU null relative to WT neurons indicating an enhanced reliance on glycolysis for ATP production during energetic stress (Figure 6(b)). Treatment with both rotenone and antimycin A resulted in matching OCR reductions for both WT and MCU nulls (Figure 6(a)). However, ECARs for MCU null neurons remained elevated above those for WT neurons after Complex I (rotenone) and Complex I/III (rotenone + antimycin A) inhibition (Figure 6(b)). These findings suggest that MCU null neurons employ glycolysis to meet increased metabolic demands.
Glycolytic induction by OGD is accompanied by Complex I suppression in MCU null neurons
OCRs were also measured 1 h post OGD (Figure 6(c) and (d)). This delay provided a measure of the early impact of in vitro ischemic/reperfusion injury on mitochondrial respiration and glycolysis. Relative to WT neurons after OGD, MCU null neurons displayed reduced OCRs under basal, oligomycin-inhibited and FCCP-stimulated conditions indicating a diminished capacity to produce energy by oxidative phosphorylation (Figure 6(c)). By contrast, MCU neurons showed a marked increase in ECARs compared to WT neurons after OGD (Figure 6(d)). This result further supports an increased dependence of MCU null neurons on glycolysis for energy production during metabolic stress. The formation of glycolytic compartments was recently discovered in neurons at sites of local synaptic activity, where we propose that these sites are enhanced in MCU nulls relative to WT.37 Following all measurement conditions after OGD, ECARs for MCU null neurons remained higher than those for WT neurons. Treatment with rotenone reduced OCRs in WT neurons to those seen in MCU neurons thus demonstrating that reduced Complex I activity was responsible for deficient mitochondrial respiration in MCU null neurons.
Forebrain NADH and pyruvate concentrations are reduced in MCU nulls subjected to HI
Forebrain NAD+/NADH ratios (striatum) and pyruvate (hippocampus) concentrations were measured in WT and MCU null 30 min after exposure to HI (Figure 6(e)). Relative to WT mice, striatal NAD+/NADH ratios were increased in MCU nulls indicating decreased NADH levels.
NADH is consumed by lactate dehydrogenase to convert pyruvate to lactate for ATP synthesis. The lower concentrations of pyruvate in the hippocampus of MCU nulls compared to WT mice after HI further supports the increased glycolytic capacity of MCU deficient mice (Figure 6(f)). The increased glycolytic consumption of NADH may thus suppress Complex I activity in MCU null neurons by depleting reducing equivalents needed to drive oxidative phosphorylation.38
Global MCU ablation and HPC increase the expression of apoptosis-related and Ca2+ handling genes in the hippocampus
In a final series of experiments, we compared mRNA levels for BCL-2 family members (BCL-XL and BAX), Na+/Ca2+ exhangers from the plasma membrane (NCX-1, NCX-2, NCX-3) and PMCA-2 in the hippocampus of WT and global MCU null mice subjected to sham conditions or HPC. Relative to WT mice, levels of mRNAs for BCL-XL, BAX, NCX-1, NCX-2, NCX-3 and PMCA-2 were elevated by 2–3 fold in global MCU nulls (Figure 7(a) to (f)). HPC increased levels for all of these mRNAs in the hippocampi of WT mice. Except for a modest elevation of PMCA-2 mRNA levels (50%; Figure 7(f)), HPC did not produce a further induction of these genes in global MCU nulls (Figure 7(a) to (e)).
Discussion
Unaltered HI brain injury in global MCU nulls despite massively reduced mitochondrial Ca2+ uptake and the inhibition of Ca2+-induced mPTP opening
We have observed that global MCU deletion suppressed the uptake of Ca2+ by forebrain mitochondria (Figure 1(a)). Ca2+-induced mPTP opening, that executes both apoptotic16 and necrotic19,39 neuronal cell death, was also blocked in forebrain mitochondria isolated from global MCU nulls (Figure 1(b)). Despite this inhibition of mPTP opening, global MCU nulls were not protected from HI-induced sensorimotor deficits (Figure 2) and brain injury (Figure 3; Supplementary Figures 1 and 2) nor were cortical neurons isolated from these animals rendered more resistant to cell viability loss after a lethal period of OGD (Supplementary Figure 4). Electron microscopic analyses showed that mitochondria in the CA1 region of global MCU nulls also displayed similar morphological features of damage as WT mice after HI brain injury (Figure 4; Supplementary Figure 3). These findings are consistent with evidence that complete MCU ablation during development activates alternative mitochondrial Ca2+ handling mechanisms that compensate for global MCU ablation.27–29
Metabolic regulation by mitochondrial Ca2+uptake is impaired in global MCU nulls
Since developmental inhibition of the MCU has been reported to increase the phosphorylation of PDH in cardiac myocytes,23,27 we measured the phosphorylation levels for S232, S293 and P300 of PDH in WT and MCU null cortical neurons (Figure 5(a) to (c)). Relative to WT neurons, all of these PDH sites were hyper-phosphorylated in MCU cortical neurons resulting in about a 300% increase of total PDH phosphorylation (Figure 5(d)). Phosphorylation of these three sites by at least four PDH kinases (PDKs) produces additive reductions in PDH activity.36 The hyper-phosphorylation of PDH in MCU null neurons is likely explained by decreased mitochondrial Ca2+ uptake required to stimulate dephosphorylation by PDH phosphatases.36 However, elevations of cytosolic Ca2+ by glutamate or ionomycin still triggered the dephosphorylation of PDH in MCU null cortical neurons (Figure 5(a) to (d)). Since dephosphorylation of each of these three sites is mediated by Ca2+-induced phosphatases, these results suggest that MCU null cortical neurons use alternative mitochondrial Ca2+ transport mechanisms to regulate metabolic activity. This line of reasoning is further supported by our measurements of forebrain NADH and pyruvate levels in WT and MCU nulls. Under control conditions, NADH and pyruvate were similar in WT and MCU nulls (Figure 6(e) and (f)). As a result, increased PDK activities driven by differences between the concentrations of these metabolic substrates cannot account for the hyper-phosphorylation of PDH.36
The induction of glycolysis by metabolic stress depletes NADH levels and suppresses Complex I activity in MCU null cortical neurons
In order to assess the impact of compensations for global MCU ablation on the metabolic function of cortical neurons, we compared the glycolytic rates and several aspects of mitochondrial performance in respiring cortical neurons derived from WT and global MCU nulls. These studies showed that basal respiration by MCU null neurons were unaltered despite a considerable reduction in mitochondrial Ca2+ uptake (Figures 1(a) and 6(a)). However, relative to WT neurons, maximal respiratory capacity stimulated with FCCP (2 µM) was reduced in MCU null neurons. This was accompanied by higher ECARs for MCU null than WT neurons (Figure 6(b)). Since increased lactate production is principally responsible for elevated neuronal ECARs,40 these results reflect an increased reliance of MCU null neurons on glycolysis to produce ATP during increased energetic demand. This finding is consistent with recent evidence that active synaptic sites are fueled by the dynamic formation of glycolytic compartments.37 We have also found that striatal NADH levels are reduced following HI in MCU nulls relative to WT mice (Figure 6(e)). This suggests that enhanced glycolysis depletes MCU null cortical neurons of NADH required to fuel oxidative phosphorylation causing a decrease in FCCP-induced maximal respiration (Figure 6(a)). This is supported by the pronounced inhibitory effects of OGD on mitochondrial respiration in MCU null neurons. Relative to WT neurons, MCU null neurons displayed greater reductions in basal respiration, ATP production and maximal respiration after OGD (Figure 6(c)) that were closely associated with increased ECARs under all of these conditions (Figure 6(d)). Interestingly, FCCP produced a further burst of ECARs in MCU null neurons that rapidly declined to levels below those seen at baseline. We also interpret these findings as being reflective of an increased dependence on glycolysis for metabolic support that rapidly consumed NADH resulting in energy depletion. Rotenone normalized OCRs in WT neurons to levels observed in MCU null neurons (Figure 6(c)) demonstrating that Complex I activity in MCU null neurons is suppressed after OGD. However, because our experiments were performed just 1 h post OGD, we cannot rule out the possibility that the metabolic shift towards glycolysis is transient in surviving MCU nulls neurons.
The OCR experiments were performed in the presence of 2 mM pyruvate. Standard neurobasal media contains only 0.22 mM pyruvate that corresponds closely with the concentration of pyruvate (0.175 mM) we have detected in brain (Figure 6(f)). Pyruvate concentrations should therefore not have been a limiting factor for the respiration and Complex I activity of MCU null neurons. We have also measured mRNA levels for the complex I member (MT-ND2) by qRT-PCR in MCU null and WT cortical neurons. There was no significant difference between WT and MCU deficient cortical neurons (Supplementary Figure 5). This suggests that reduced Complex I expression was not responsible for the lower OCRs of global MCU null relative to WT neurons following FCCP treatment or OGD.
Global MCU deficiency produces similar effects as HPC on apoptosis-related and Ca2+ handling gene expression in the hippocampus
Global MCU deficiency engaged mechanisms shared by HPC known to oppose impairments in mitochondrial function and cytosolic Ca2+ handling genes that promote ischemic brain damage.14,15,21 There was a remarkable concordance between hippocampal patterns of elevated gene expression for mitochondrial apoptosis-regulators (BCL-XL, BAX), Na+/Ca2+ exchangers from the plasma membrane (NCX-1, NCX-2, NCX-3) and the ATP-driven Ca2+ plasma membrane transporter (PMCA-2) in sham global MCU nulls and WT mice subjected to HPC (Figure 7(a) to (f)). These increases are consistent with adaptations in MCU nulls that compensate for chronic impairments in Ca2+ homeostasis.27
MCU nulls fail to benefit from HPC
Although HPC reduced HI-induced neurological deficits and brain injury in WT mice, MCU nulls failed to benefit from HPC. Apart from a modest 50% elevation of mRNA levels for PMCA-2 (Figure 7(f)), HPC failed to produce a further increase in the hippocampal expression of BAX, BCL-XL, NCX-1, NCX-2, NCX-3 in global MCU null mice (Figure 7(a) to (e)). These findings indicate that developmental compensations for constitutively impaired Ca2+ handling in global MCU nulls may have disrupted signaling mechanisms necessary for HPC. Similar to our findings, constitutive MCU inhibition in the heart increases the expression of mitochondrial apoptosis-regulators (BCL-2 and BAX), stimulates NCX activity and sensitizes extra-mitochondrial cell death pathways.27 This coupled with an increased reliance on glycolysis that supresses Complex I activity may have contributed to the inability of MCU nulls to benefit from HPC. Such adverse effects of these long-term compensations may also account for the failure of global MCU deletion to reduce mitochondrial damage, central neuron loss and neurological deficits following HI brain injury.
Conclusions and therapeutic implications
In summary, we have demonstrated that the MCU is primarily responsible for Ca2+ uptake by forebrain mitochondria. Consistent with this role, Ca2+-induced mPTP opening was blocked in MCU-deficient forebrain mitochondria. Although mPTP opening is considered to be a pivotal event in the execution of ischemic cell death,18–20 global MCU nulls were not resistant to HI brain injury. In what may be a resolution of these findings, we have shown that metabolic adaptations for global MCU deficiency render cortical neurons more susceptible to Complex I suppression following OGD that models ischemic/reperfusion damage. Relative to WT cortical neurons, global MCU null neurons depend more heavily on glycolysis to produce ATP following energetic stress. We propose that enhanced glycolysis after OGD deprives Complex I of reducing equivalents (NADH) required to drive oxidative phosphorylation. The resultant energetic collapse may thus promote ischemic/reperfusion injury despite reduced mitochondrial Ca2+ uptake. To withstand the detrimental effects of chronically impaired mitochondrial Ca2+ uptake, MCU nulls appear to employ genetic adaptations common to HPC. These adaptations included the induction of mitochondrial apoptosis regulators and Ca2+ transporters that oppose disrupted Ca2+ handling. It is tempting to speculate that these genes may also enable the use of alternative Ca2+ transport by MCU null neurons. However, it is clear from our findings that such adaptations are insufficient to protect global MCU nulls against ischemic neuronal injury. These results suggest that MCU inhibitors will be best suited to the acute management of ischemic/reperfusion brain injury as therapeutic adjuncts for thrombolytic agents.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the Heart and Stroke Foundation of Canada and American Heart Association (EVP), Heart and Stroke Foundation of Canada and Brain Canada (GSR) and the MS Society of Canada (GSR).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
MN completed all experiments, participated in all data analysis, prepared figures and participated in drafting the manuscript. PE helped in the completion of all isolated mitochondria experiments and associated figures. JW participated in NADH and pyruvate experiments. AT helped in the completion of all isolated mitochondria experiments, assisted in animal surgery and maintained mouse colonies. EP participated in data analysis, figure preparation and drafting the manuscript. GSR participated in data analysis, figure preparation and drafting the manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Di CA. Human and economic burden of stroke. Age Ageing 2009; 38: 4–5. [DOI] [PubMed] [Google Scholar]
- 2.Anderson CS, Carter KN, Brownlee WJ, et al. Very long-term outcome after stroke in Auckland, New Zealand. Stroke 2004; 35: 1920–1924. [DOI] [PubMed] [Google Scholar]
- 3.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 2009; 8: 398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 2011; 14: 1363–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 2006; 7: 437–448. [DOI] [PubMed] [Google Scholar]
- 6.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 2003; 4: 399–415. [DOI] [PubMed] [Google Scholar]
- 7.Bading H. Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci 2013; 14: 593–608. [DOI] [PubMed] [Google Scholar]
- 8.Dezfulian C, Garrett M, Gonzalez NR. Clinical application of preconditioning and postconditioning to achieve neuroprotection. Transl Stroke Res 2013; 4: 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 2012; 13: 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Llorente-Folch I, Rueda CB, Pardo B, et al. The regulation of neuronal mitochondrial metabolism by calcium. J Physiol 2015; 593: 3447–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci 2012; 125: 2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saotome M, Safiulina D, Szabadkai G, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A 2008; 105: 20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutsaeva DR, Carraway MS, Suliman HB, et al. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci 2008; 28: 2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sisalli MJ, Annunziato L, Scorziello A. Novel cellular mechanisms for neuroprotection in ischemic preconditioning: a view from inside organelles. Front Neurol 2015; 6: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuomo O, Vinciguerra A, Cerullo P, et al. Ionic homeostasis in brain conditioning. Front Neurosci 2015; 9: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson GS, Crocker SJ, Nicholson DW, et al. Neuroprotection by the inhibition of apoptosis. Brain Pathol 2000; 10: 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science 2014; 345: 1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005; 434: 652–658. [DOI] [PubMed] [Google Scholar]
- 19.Schinzel AC, Takeuchi O, Huang Z, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005; 102: 12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434: 658–662. [DOI] [PubMed] [Google Scholar]
- 21.Pignataro G, Cuomo O, Vinciguerra A, et al. NCX as a key player in the neuroprotection exerted by ischemic preconditioning and postconditioning. Adv Exp Med Biol 2013; 961: 223–240. [DOI] [PubMed] [Google Scholar]
- 22.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 2006; 34: 232–237. [DOI] [PubMed] [Google Scholar]
- 23.Pan X, Liu J, Nguyen T, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 2013; 15: 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De SD, Raffaello A, Teardo E, et al. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011; 476: 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baughman JM, Perocchi F, Girgis HS, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011; 476: 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Y, Rasmussen TP, Koval OM, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun 2015; 6: 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasmussen TP, Wu Y, Joiner ML, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A 2015; 112: 9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwong JQ, Lu X, Correll RN, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 2015; 12: 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luongo TS, Lambert JP, Yuan A, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 2015; 12: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilkenny C, Browne W, Cuthill IC, et al. Animal research: reporting in vivo experiments–the ARRIVE guidelines. J Cereb Blood Flow Metab 2011; 31: 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol 1960; 36: 1–17. [PMC free article] [PubMed] [Google Scholar]
- 32.Nichols M, Zhang J, Polster BM, et al. Synergistic neuroprotection by epicatechin and quercetin: activation of convergent mitochondrial signaling pathways. Neuroscience 2015; 308: 75–94. [DOI] [PubMed] [Google Scholar]
- 33.Sanderson TH, Reynolds CA, Kumar R, et al. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 2013; 47: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med 2004; 4: 149–177. [DOI] [PubMed] [Google Scholar]
- 35.Llorente-Folch I, Rueda CB, Amigo I, et al. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci 2013; 33: 13957–13971, 13971a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel MS, Nemeria NS, Furey W, et al. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 2014; 289: 16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang S, Nelson JC, Bend EG, et al. Glycolytic enzymes localize to synapses under energy stress to support synaptic function. Neuron 2016; 90: 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wirth C, Brandt U, Hunte C, et al. Structure and function of mitochondrial complex I. Biochim Biophys Acta 2016; 1857: 902–914. [DOI] [PubMed] [Google Scholar]
- 39.Vaseva AV, Marchenko ND, Ji K, et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012; 149: 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mookerjee SA, Goncalves RL, Gerencser AA, et al. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta 2015; 1847: 171–181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.